
Abstract
Background
Short-rib polydactyly syndrome (SRPS) refers to a group of lethal skeletal dysplasias that can be difficult to differentiate between subtypes or from other non-lethal skeletal dysplasias such as Ellis-van Creveld syndrome and Jeune syndrome in a prenatal setting. We report the ultrasound and genetic findings of four unrelated fetuses with skeletal dysplasias.
Methods
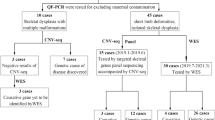
Systemic prenatal ultrasound examination was performed in the second or third trimester. Genetic tests including GTG-banding, single nucleotide polymorphism (SNP) array and exome sequencing were performed with amniocytes or aborted fetal tissues.
Results
The major and common ultrasound anomalies for the four unrelated fetuses included short long bones of the limbs and narrow thorax. No chromosomal abnormalities and pathogenic copy number variations were detected. Exome sequencing revealed three novel variants in the DYNC2H1 gene, namely NM_001080463.2:c.6809G > A p.(Arg2270Gln), NM_001080463.2:3133C > T p.(Gln1045Ter), and NM_001080463.2:c.337C > T p.(Arg113Trp); one novel variant in the IFT172 gene, NM_015662.3:4540-5 T > A; and one novel variant in the WDR19 gene, NM_025132.4:c.2596G > C p.(Gly866Arg). The genotypes of DYNC2H1, IFT172 and WDR19 and the phenotypes of the fetuses give hints for the diagnosis of short-rib thoracic dysplasia (SRTD) with or without polydactyly 3, 10, and 5, respectively.
Conclusion
Our findings expand the mutation spectrum of DYNC2H1, IFT172 and WDR19 associated with skeletal ciliopathies, and provide useful information for prenatal diagnosis and genetic counseling on rare skeletal disorders.
Similar content being viewed by others

Introduction
Short-rib polydactyly syndrome (SRPS) refers to a group of autosomal recessive skeletal dysplasias characterized by markedly short ribs with thoracic hypoplasia, short limbs, and variable presentation of polydactyly and metaphyseal and visceral anomalies. Histopathologically, SRPS, asphyxiating thoracic dysplasia (ATD; also known as Jeune syndrome) and Ellis van Creveld syndrome (EVC) belong to ciliopathies with major skeletal involvement [1]. These disorders exhibit phenotypic overlap, presenting a challenge for clinical diagnosis, especially in the setting of prenatal diagnosis [2, 3].
SRPS, ATD, EVC and Mainzer-Saldino syndrome (MZSDS) are also summarized under the term short-rib thoracic dysplasia (SRTD) with or without polydactyly [4,5,6]. The phenotype-genotype relationships of SRTD types 1–23 can be found in the Online Mendelian Inheritance in Man (OMIM) database (Supplementary Material Table S1).
Cilia are the fine protrusive structures of cell surface that play important roles in development and function of many organs including bones [7]. The movement of substances within cilia, known as intraflagellar transport (IFT), is critical for the assembly and maintenance of cellular structures. The IFT particles responsible for bi-directional transportation within cilia are composed of complex A (IFT-A) and complex B (IFT-B) [8]. Among the ciliary proteins associated with SRTD, WDR35, IFT140, WDR19, IFT43, and TTC21B are components of the complex A; IFT80, IFT172, IFT52, and IFT81 take participation in the complex B; DYNC2H1, WDR60, WDR34, DYNC2LI1, and TCTEX1D2 are IFT-dynein motor proteins; NEK1, CEP120, and KIAA0586 are located at the basal body of primary cilia; and INTU is a ciliogenesis and planar polarity effector (CPLANE) protein [9]. Hence, the genetic architecture of skeletal ciliopathies involves both the structural and regulatory proteins of cilia.
The present study reports four prenatal cases of SRPS/SRTD as implicated by ultrasound findings, which provide useful information regarding the novel variants of ciliopathies-associated genes DYNC2H1, IFT172, and WDR19.
Materials and methods
Study participants
Four pregnant women with fetal ultrasound abnormalities including thoracic dysplasia, short ribs, and short long bones with or without organ anomalies were included in the present study.
SNP array for CNV detection
Preparation of genomic DNA from amniocytes or fetal tissue was performed using QIAamp DNA Blood Mini Kit or QIAamp DNA Mini Kit (QIAGEN, Germany). The purified DNA was then processed using CytoScan 750 K reagent kit and subsequently loaded onto the CytoScan 750 K array for hybridization following the manufacturer’s instructions (Applied Biosystems, Thermo Fisher Scientific). The CytoScan 750 K array was preloaded with 200,000 SNPs and 550,000 non-polymorphic probes for copy number analysis. The Chromosome Analysis Suite (ChAS) software (available at https://www.thermofisher.com/) was used to analyze the raw data. CNVs ≥ 100 kb were chosen for blast analysis and annotated with the reference databases including DGV (http://dgv.tcag.ca/), DECIPHER (https://decipher.sanger.ac.uk/), OMIM, UCSC (https://genome.ucsc.edu/hg19), ClinVar and PubMed.
Exome sequencing
Novaseq6000 platform (Illumina, San Diego, USA) with 150 bp pair-end reads was used for sequencing the genomic DNA from the fetus and parents. Qualified genomic DNA was sheared into around 150 bp fragments, then blunt-ended and added with deoxyadenosine (dA) to the 5’ tails, followed by adding adaptors to the ends of DNA double strands. The library was amplified by polymerase chain reaction (PCR) and then hybridized with a pool of biotin-labeled oligo probes specific for exons. DNA-probes hybrids were captured using streptavidin magnetic beads and a round of PCR was used to amplify the library to sufficient levels for sequencing. Raw image files were processed using CASAVA v1.82 for base calling and generating raw data with sufficient CCDS coverage (95.87%–98.88%, for depth ≥ 20). The sequencing reads were aligned to the human reference genome (hg19/GRCh37) using Burrows–Wheeler Aligner tool and PCR duplicates were removed by using Picard v1.57 (http://picard.sourceforge.net/). The interpretation of sequence variants were conducted, referring to the American College of Medical Genetics and Genomics (ACMG) guidelines [10, 11] and the Enliven® Variants Annotation Interpretation System authorized by Berry Genomics (Beijing, China). Variants with a frequency > 1% in the databases including 1000 Genomes (http://browser.1000genomes.org), Exome Aggregation Consortium (ExAC) and Genome Aggregation Database (gnomAD, http://gnomad.broadinstitute.org/), and those that are thought to have no functional impact (e.g. synonymous mutations, non-coding mutations) were excluded. Sequence variants were further screened based on pathogenicity prediction using online tools SIFT (http://sift.jcvi.org), Polyphen2 (http://genetics.bwh.harvard.edu/pph2/), Mutation Taster (https://www.mutationtaster.org/), and Combined Annotation Dependent Depletion (CADD, https://cadd.gs.washington.edu), clinical phenotypes, inheritance and literature reports. Validation of candidate variants was performed by Sanger sequencing.
Results
Clinical presentation
The clinical features of the prenatal cases were given in Table 1 and Figs. 1, 2, 3 and 4, The major and common ultrasound anomalies for the four unrelated fetuses included short long bones of the limbs and narrow thorax.

Ultrasound findings of the fetus in patient 1. Narrow thorax (a), shortened femur (b), bilateral pyelectasis (c), pleural effusion (d), and peritoneal effusion (e) are indicated by red arrows

Ultrasound features of the fetus in patient 2. Short femurs (a) and humeri (b) and high echo spot in gallbladder crystals (c) are indicated with red arrows

Ultrasound features of the fetus in patient 3. Narrow thorax (a), short bell-shaped ribs (b), and enlarged kidneys and enhanced renal parenchymal echo (c) are indicated with red arrows

Ultrasound features of the fetus in patient 4. Narrow abdominal circumference is revealed (a). Short femurs (b) and humeri (c) are indicated with red arrows
Genetic findings
Karyotypes were normal, and no pathogenic CNVs were detected by SNP array in the four unrelated fetuses. The top 10 candidate variants for individual fetuses as revealed by ES are listed in Supplementary Material Table S2.
The fetus 1 carried a heterozygous DYNC2H1 variant NM_001080463.2:c.3133C > T p.(Gln1045Ter) in one allele and a heterozygous DYNC2H1 variant NM_001080463.2:c.6809G > A p.(Arg2270Gln) in the second allele, which are maternally and paternally inherited, respectively (Fig. 5). The two variants were not found in public databases HGMD professional and Varsome (accessed on Nov 11th, 2023). According to the ACMG guidelines, the c.3133C > T variant was regarded as likely pathogenic, whereas the c.6809G > A variant was of uncertain significance (Table 1). The identified genotype and ultrasonic features of the fetus 1 support a diagnosis of SRTD3.

Sanger sequencing confirmed the candidate causative mutations in individual fetuses
The fetus 2 was biallelic heterozygous for IFT172 NM_015662.2:c.4540-5 T > A and NM_015662.2:c.1513C > T p.(Arg505Trp) (Fig. 5). The c.4540-5 T > A variant had not been indexed by HGMD professional and Varsome, whereas the c.1513C > T variant was previously reported by our team [12]. Based on the ACMG guidelines, the variants c.4540-5 T > A and c.1513C > T are considered as VUS (Table 1). The identified genotype and ultrasonic features of the fetus 2 support a diagnosis of SRTD10.
The heterozygous variants WDR19 NM_025132.4:c.2596G > C p.(Gly866Arg) and NM_025132.4:c.2363 + 1G > A, which are of maternal and paternal origins, respectively, were identified in the fetus 3 (Fig. 5). The c.2596G > C variant was not documented in the databases HGMD professional and Varsome. The c.2363 + 1G > A variant is seen recurrently in publications [9, 13,14,15,16]. In terms of the ACMG guidelines, the c.2363 + 1G > A variant is considered as likely pathogenic (PVS1 + PM2-p), the c.2596G > C variant is of uncertain significance (VUS) (Table 1). Notably, the c.2363 + 1G > A variant is classified as pathogenic/likely pathogenic in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/, Variation ID: 280,765). The c.2597G > A variant of WDR19, with identical amino acid change (p.Gly866Arg) to the c.2596G > C variant, has been documented in ClinVar (Variation ID: 1,494,663) and classified as VUS. Mutations in WDR19 are associated with autosomal recessive cranioectodermal dysplasia 4 (MIM 614378), SRTD5 (MIM 4376), nephronophthisis 13 (MIM 614377) and Senior-Loken syndrome (MIM 616307). The genotype and phenotype of the fetus 3 suggests a diagnosis of SRTD5.
Heterozygous variants DYNC2H1 NM_001080463.2:c.1390C > T p.(Arg464Ter) and NM_001080463.2:c.337C > T p.(Arg113Trp) were identified in the fetus 4 (Fig. 5). The c.1390C > T variant was not found in public databases HGMD professional and Varsome, and the c.337C > T variant was previously reported [9]. Based on the ACMG standards, the c.1390C > T variant is classified as likely pathogenic (PVS1 + PM2-p). It has been previously reported that the c.337 C > T variant is in trans position to the c.3353del (p.Ser1118IlefsTer46) variant in an individual diagnosed with ATD [9]. Therefore, the c.337C > T variant is classified as VUS (Table 1). In addition, the c.337C > T variant is graded as disease-causing mutation (DM) in HGMD. The genotype and phenotype of the fetus 4 supports a diagnosis of SRTD3.
Discussion
In the present study, differential diagnosis of ultrasound-indicated fetal skeletal dysplasias in four unrelated fetuses was achieved by using ES, suggesting that ES is an efficient and cost-effective method for prenatal diagnosis of rare genetic skeletal disorders.
Compound heterozygous (or homozygous) mutations in DYNC2H1 or digenic biallelic mutations in DYNC2H1 and NEK1 have previously been identified as a genetic cause for SRPS type III, the most common type of SRPS [17, 18]. DYNC2H1 encodes cytoplasmic dynein 2 heavy chain 1, which acts as a motor for intraflagellar retrograde transport and take participation in cilia biogenesis [19]. In a previous study to screen causal genes for ATD, EVC and SRPS spectrum in 152 unrelated families by using exome sequencing, mutations in DYNC2H1 were found in 43 SRPS families (40 with SRPS type III), and 110 different pathogenic mutations in DYNC2H1 were identified, two thirds of which were missense mutations and mainly clustered in the N-terminal tail, the AAA2-4 ATPase domains and the conserved C-terminal domain (C domain) [9]. In the present study, the p.Arg2270Gln, p. Gln1045Ter and p.Arg464Ter mutations are located in the AAA3 domain, the interval region between the DHC_N1 and DHC_N2 domains and the DHC_N1 domain, respectively. Interestingly, the biallelic heterozygous mutations in DYNC2H1 in the two unrelated fetuses both comprise a loss-of-function (nonsense) mutation and a missense mutation, in line with a previous report that biallelic loss of function of DYNCH2H1 might be embryo lethal [9].
The IFT172 gene has been associated with autosomal recessive Bardet-Biedl syndrome [20], non-syndromic retinitis pigmentosa [21] and SRTD10 [22]. In a previous study, homozygous or compound heterozygous mutations in IFT172 were detected in 12 families with affected individuals diagnosed with asphyxiating thoracic dystrophy (ATD; also known as Jeune syndrome) or Mainzer-Saldino syndrome (MZSDS). The patients were characterized by abnormalities of the thorax and/or long bones with involvement of other organs such as kidney, liver, or retina. In the present study, no remarkable visceral anomalies were observed in the fetus 2.
The WD domain repeat 19 (WDR19) gene, also known as IFT144, belongs to the WD (tryptophan-aspartic acid) repeat family. Mutations in WDR19 have been associated with a broad spectrum of ciliopathies, such as Sensenbrenner syndrome (cranioectodermal dysplasia), Jeune syndrome, Senior-Loken syndrome (nephronophthisis and pigmentary retinopathy), and nonsyndromic asthenoteratospermia [13, 23, 24]. In our study, renal involvement was observed in the fetus 3. This finding is in line with the fact that kidney disease is frequently seen in WDR19-related ciliopathies.
At prenatal stage, the information regarding the clinical phenotype of fetus might be incomplete due to later stage-specific onset of certain anomalies or limitations from ultrasound examination. Genetic analysis would contribute to understand the etiology and predict prognosis of the affected fetus, as well as guide the next pregnancy. Moreover, proper genetic counseling for the affected family is essential in the case of rare genetic diseases, since parental genetic screening/diagnosis is the best strategy for managing these diseases currently having no therapy [25,26,27]. Reporting additional cases on fetal skeletal ciliopathies and causal genes would help identify the genotype–phenotype correlations and lead to clinical trials in the future [28].
Collectively, our case series provide information regarding the novel variants of ciliopathies-associated genes DYNC2H1, IFT172, and WDR19, and emphasize the importance of both clinical and genetic findings in the setting of prenatal diagnosis for skeletal disorders.
Availability of data and materials
The data that support the findings of this study are available via the BioProject database (Accession: PRJNA878618) at https://ncbi.nlm.nih.gov/bioproject/.
References
Mortier GR, Cohn DH, Cormier-Daire V, Hall C, Krakow D, Mundlos S, et al. Nosology and classification of genetic skeletal disorders: 2019 revision. Am J Med Genet A. 2019;179(12):2393–419.
Naki MM, Gur D, Zemheri E, Tekcan C, Kanadikirik F, Has R. Short rib-polydactyly syndrome. Arch Gynecol Obstet. 2005;272(2):173–5.
Eleftheriades M, Iavazzo C, Manolakos E, Hassiakos D, Botsis D, Petersen M, et al. Recurrent short rib polydactyly syndrome. J Obstet Gynaecol. 2013;33(1):14–6.
Schmidts M. Clinical genetics and pathobiology of ciliary chondrodysplasias. J Pediatr Genet. 2014;3(2):46–94.
Chen CP, Ko TM, Chang TY, Chern SR, Chen SW, Lai ST, et al. Prenatal diagnosis of short-rib polydactyly syndrome type III or short-rib thoracic dysplasia 3 with or without polydactyly (SRTD3) associated with compound heterozygous mutations in DYNC2H1 in a fetus. Taiwan J Obstet Gynecol. 2018;57(1):123–7.
Yakar O, Tatar A. INTU-related oral-facial-digital syndrome XVII: Clinical spectrum of a rare disorder. Am J Med Genet A. 2022;188(2):590–4.
Anvarian Z, Mykytyn K, Mukhopadhyay S, Pedersen LB, Christensen ST. Cellular signalling by primary cilia in development, organ function and disease. Nat Rev Nephrol. 2019;15(4):199–219.
Liem KF Jr, Ashe A, He M, Satir P, Moran J, Beier D, et al. The IFT-A complex regulates Shh signaling through cilia structure and membrane protein trafficking. J Cell Biol. 2012;197(6):789–800.
Zhang W, Taylor SP, Ennis HA, Forlenza KN, Duran I, Li B, et al. Expanding the genetic architecture and phenotypic spectrum in the skeletal ciliopathies. Hum Mutat. 2018;39(1):152–66.
Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19(2):249–55.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Peng Y, Yang S, Huang X, Pang J, Liu J, Hu J, et al. Whole Exome Sequencing Analysis in Fetal Skeletal Dysplasia Detected by Ultrasonography: An Analysis of 38 Cases. Front Genet. 2021;12:728544.
Bredrup C, Saunier S, Oud MM, Fiskerstrand T, Hoischen A, Brackman D, et al. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am J Hum Genet. 2011;89(5):634–43.
Halbritter J, Porath JD, Diaz KA, Braun DA, Kohl S, Chaki M, et al. Identification of 99 novel mutations in a worldwide cohort of 1,056 patients with a nephronophthisis-related ciliopathy. Hum Genet. 2013;132(8):865–84.
Westerfield LE, Stover SR, Mathur VS, Nassef SA, Carter TG, Yang Y, et al. Reproductive genetic counseling challenges associated with diagnostic exome sequencing in a large academic private reproductive genetic counseling practice. Prenat Diagn. 2015;35(10):1022–9.
Daoud H, Luco SM, Li R, Bareke E, Beaulieu C, Jarinova O, et al. Next-generation sequencing for diagnosis of rare diseases in the neonatal intensive care unit. CMAJ. 2016;188(11):E254–60.
Thiel C, Kessler K, Giessl A, Dimmler A, Shalev SA, von der Haar S, et al. NEK1 mutations cause short-rib polydactyly syndrome type majewski. Am J Hum Genet. 2011;88(1):106–14.
Dagoneau N, Goulet M, Genevieve D, Sznajer Y, Martinovic J, Smithson S, et al. DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib-polydactyly syndrome, type III. Am J Hum Genet. 2009;84(5):706–11.
Tsurumi Y, Hamada Y, Katoh Y, Nakayama K. Interactions of the dynein-2 intermediate chain WDR34 with the light chains are required for ciliary retrograde protein trafficking. Mol Biol Cell. 2019;30(5):658–70.
Schaefer E, Stoetzel C, Scheidecker S, Geoffroy V, Prasad MK, Redin C, et al. Identification of a novel mutation confirms the implication of IFT172 (BBS20) in Bardet-Biedl syndrome. J Hum Genet. 2016;61(5):447–50.
Bujakowska KM, Zhang Q, Siemiatkowska AM, Liu Q, Place E, Falk MJ, et al. Mutations in IFT172 cause isolated retinal degeneration and Bardet-Biedl syndrome. Hum Mol Genet. 2015;24(1):230–42.
Mhatre S, Muranjan M, Karande S. Short rib thoracic dysplasia without polydactyly due to novel variant in IFT172 gene. J Postgrad Med. 2020;66(4):224–5.
Fehrenbach H, Decker C, Eisenberger T, Frank V, Hampel T, Walden U, et al. Mutations in WDR19 encoding the intraflagellar transport component IFT144 cause a broad spectrum of ciliopathies. Pediatr Nephrol. 2014;29(8):1451–6.
Ni X, Wang J, Lv M, Liu C, Zhong Y, Tian S, et al. A novel homozygous mutation in WDR19 induces disorganization of microtubules in sperm flagella and nonsyndromic asthenoteratospermia. J Assist Reprod Genet. 2020;37(6):1431–9.
Alfadhel M, Umair M, Almuzzaini B, Alsaif S, AlMohaimeed SA, Almashary MA, et al. Targeted SLC19A3 gene sequencing of 3000 Saudi newborn: a pilot study toward newborn screening. Ann Clin Transl Neurol. 2019;6(10):2097–103.
Alyafee Y, Al Tuwaijri A, Alam Q, Umair M, Haddad S, Alharbi M, et al. Next Generation Sequencing Based Non-invasive Prenatal Testing (NIPT): First Report From Saudi Arabia. Front Genet. 2021;12:630787.
Alyafee Y, Alam Q, Tuwaijri AA, Umair M, Haddad S, Alharbi M et al: Next-Generation Sequencing-Based Pre-Implantation Genetic Testing for Aneuploidy (PGT-A): First Report from Saudi Arabia. Genes (Basel) 2021, 12(4).
Alyafee Y, Al Tuwaijri A, Umair M, Alharbi M, Haddad S, Ballow M, et al. Non-invasive prenatal testing for autosomal recessive disorders: A new promising approach. Front Genet. 2022;13:1047474.
Acknowledgements
We thank the parents of the fetuses for their cooperation and continued support for this study.
Funding
This research was supported by The National Key Research and Development Program of China(2021YFC1005305), The Major Scientific and Technological Projects for Collaborative Prevention and Control of Birth Defects in Hunan Province (2019SK1013 and 2019SK1015), and Natural Science Foundation of Hunan Province (2018JJ3274).
Author information
Authors and Affiliations
Contributions
YP and LZ recruited the patients and collected the clinical data. JC, XH and JP performed the karyotype and SNP array analysis. JL, WT and SY performed WES interpreted the data. YP and CL designed the study. YP and WX wrote the manuscript daft and all authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study was approved by the Ethics Committee of Hunan Provincial Maternal and Child Health Care Hospital. All methods were performed in accordance with the Declaration of Helsinki. Written informed consent was obtained from patients or patients’ legal representative for participating in the study.
Consent for publication
Not applicable.
Competing interests
All authors declare that they have no commercial or other conflicting interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Supplementary Table S1.
The phenotype-genotype relationships of short-rib thoracic dysplasia (SRTD) with or without polydactyly types 1-23.
Additional file 2: Supplementary Table S2.
Top 10 candidate mutations as revealed by WES.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Peng, Y., Zhou, L., Chen, J. et al. Clinical features and genetic analysis of a case series of skeletal ciliopathies in a prenatal setting. BMC Med Genomics 16, 318 (2023). https://doi.org/10.1186/s12920-023-01753-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01753-y