
Abstract
Objective
To understand the benefit-risk profile for historical and current treatments for MLD.
Methods
A systematic review was conducted on the effectiveness, safety, and costs of MLD treatments: allogeneic haematopoietic stem cell transplantation (HSCT) and atidarsagene autotemcel (arsa-cel) according to best practice.
Results
A total of 6940 titles and abstracts were retrieved from the literature searches and 26 from other sources. From these, 35 manuscripts reporting on a total of 12 studies were selected for inclusion in the review. There were no controlled multi-armed trials. However, we provide observations comparing two interventional therapies (alloHSCT and arsa-cel) and each of these to standard/supportive care (natural history). There were no benefits for survival, gross motor function and cognitive function for LI patients receiving alloHSCT, as patients experienced disease progression similar to LI natural history. For juvenile patients receiving alloHSCT, no differences in survival were observed versus natural history, however stabilisation of cognitive and motor function were reported for some patients (particularly for pre- or minimally-symptomatic LJ patients), while others experienced disease progression. Furthermore, alloHSCT was associated with severe complications such as treatment-related mortality, graft versus host disease, and re-transplantation in both LI and EJ treated patients. Most LI and EJ patients treated with arsa-cel appeared to have normal development, preservation, or slower progression of gross motor function and cognitive function, in contrast to the rapid decline observed in natural history patients. A survival benefit for arsa-cel versus natural history and versus alloHSCT was observed in LI patients.LI and EJ patients treated with arsa-cel had better gross motor function and cognitive function compared to alloHSCT, which had limited effect on motor and cognitive decline. No data has been reported for arsa-cel treatment of LJ patients.
Conclusions
Overall, this systematic review indicates that compared to NHx and HSCT, treatment with arsa-cel results in clinically relevant benefits in LI and EJ MLD patients by preserving cognitive function and motor development in most patients, and increased survival for LI patients. Nevertheless, further research is required to confirm these findings, given they are based on results from non-RCT studies.
Similar content being viewed by others


Background
Metachromatic leukodystrophy (MLD), a rare inherited condition caused by arylsulfatase A (ARSA) deficiency, which results in the accumulation of fats (sulfatides) leading to the destruction of neurons and the protective fatty layer (myelin) surrounding the nerves in the brain and spinal cord [1]. MLD is a progressive disease that results in loss of all previously acquired motor, language, and cognitive skills, dysphagia, seizures, spasticity, and eventually death [2].
There are several subtypes of MLD classified by age at disease onset i.e., late infantile (LI; symptom development from birth to < 30 months of age), juvenile (J; subdivided into early juvenile [EJ; onset from 30 months to < 7 years] and late juvenile [LJ; onset 7 to < 17 years]) and adult onset (onset ≥ 17 years). LI MLD is the most common variant occurring in 50–60% of cases [2]. The clinical course typically begins with a pre-symptomatic phase with normal motor and cognitive development, followed by a period of developmental plateau or the appearance of symptoms and subsequent rapid disease progression ending with premature death of the patient. The initial disease manifestation varies between subtypes with symptoms including abnormal gait, problems with speech, impaired fine motor skills, concentration, and behavioural problems, among others. LI MLD patients experience rapid and homogeneous disease progression after symptom onset resulting in severe motor and cognitive impairment between 2 and 4 years of age [3, 4]. In EJ MLD, disease progression after symptom onset is initially somewhat slower than in LI MLD, but once the ability to walk independently is lost, disease progression is as rapid as that observed in LI MLD. LJ and adult onset MLD often have a more protracted disease course with cognitive and behavioural function affected more than motor function [5].
Besides atidarsagene autotemcel (also referred to as arsa-cel and previously known as compound number OTL-200, trade name Libmeldy™) there are currently no licensed treatment options for MLD; treatment is limited to supportive care (i.e., physiotherapy, muscle relaxants, pain management therapies) aiming to manage disease complications and preserve patients’ health related quality of life (HRQoL). Family and patients can also benefit from counselling sessions [6]. Allogeneic haematopoietic stem cell transplantation (HSCT) has been used as a treatment for MLD, but with limited effect in patients with early-onset MLD (LI and EJ) or those with more advanced symptoms and with better results in LJ or adult MLD patients treated before symptom onset [7, 8].
Atidarsagene autotemcel, a new treatment for MLD, is an ex vivo autologous hematopoietic stem and progenitor cell-based gene therapy that involves extraction of CD34+ stem cells from a patient’s bone marrow or blood, approved in the EU/Norway/Liechtenstein/Iceland and the UK and currently used as an investigational therapy in the US [9]. The stem cells are genetically-modified by a lentiviral vector and following myeloablative conditioning to make space for the genetically-modified cells, returned to the patient by intravenous infusion. The corrected cells can then differentiate and migrate to affected tissues and produce a functional version of the ARSA enzyme. The aim of the treatment is to halt disease progression and/or modify its natural course [1].
The aim of the systematic review was to understand and to summarize the current evidence on the effectiveness and safety of atidarsagene autotemcel, other therapies and standard/supportive care for the treatment of MLD in children (≤ 17 years).
Methods
The systematic review followed recommendations of the Centre for Reviews and Dissemination (CRD) guidance for undertaking reviews in healthcare [10] and the Cochrane Handbook for Systematic Reviews of Interventions [11]. The study was reported according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines [12]. The review protocol was registered on the PROSPERO database (CRD42020192663) prior to study commencement.
The study included randomized controlled trials (RCTs), prospective or retrospective single arm or cohort studies with > 5 participants and any economic evaluation of patients (age ≤ 17 years) with early-onset pre-symptomatic or symptomatic MLD of any type (LI, J, EJ) or LJ. If populations included mixed age groups, only studies where data was reported separately for those with early-onset MLD (i.e., actual or predicted age of onset < 7 years) were included. Case reports and cross-sectional studies were excluded.
The intervention of interest was atidarsagene autotemcel. The comparators are standard care and HSCT. Standard care could also be described as best supportive or usual care and includes any of the following including their combinations: management of dystonia, infections, seizures, or secretions; pain relief/sedative drugs; feeding support including gastrostomy; psychological and social support including specialist schooling; coordination of the multidisciplinary team and community care; genetic advice and planning; and end of life care). Allogeneic HSCT was also included as a comparator. The effectiveness outcomes of interest included mortality; progressive disease; gross motor function; neurological function; cognitive functions (cognitive impairment and language skills); ARSA activity and HRQoL. The safety outcomes (adverse events [AEs]) are presented separately.
MEDLINE, MEDLINE In-Process, MEDLINE Daily Update, MEDLINE Epub Ahead of Print, PubMed, Embase, Cochrane Central Register of Controlled Trials (CENTRAL), Science Citation Index (SCI), Northern Light Life Sciences Conference Abstracts, WORLD Symposium, National Institutes of Health (NIH) ClinicalTrials.gov and Orphanet Clinical trials Search were searched for relevant studies from database inception to July 2021 without language or publication limits. The MEDLINE search strategy is shown in Additional file 1: Appendix 1. The bibliographies of included research and review articles were checked for additional relevant studies. An additional publication of a comparison between an atidarsagene autotemcel trial (Study 201,222 (NCT01560182) plus additional patients recruited through an Expanded Access Framework (EAF)) and a natural history (NHx) cohort published after the searches was also included [13]. This study will be referred to as Fumagalli et al. [13].
Two reviewers independently screened articles for inclusion according to prespecified inclusion criteria (See Additional file 1: Table S1) at title/abstract and full text stage, assessed quality and performed data extraction. Any discrepancies between reviewers were resolved through consensus or consultation with a third reviewer. Data from the included studies were extracted, stored, and analysed using Microsoft Excel. For each study, the background study information, patient baseline characteristics (e.g., age, MLD symptom status), interventions/study arms compared (description of interventions and comparators), outcomes assessed (e.g., definition of outcome, methods of assessment), results (e.g., numbers, percentages, and effect sizes with confidence intervals [CIs, where relevant]) and follow-up time were extracted. The risk of bias in non-randomised studies was assessed using the Joanne Briggs Institute (JBI) Critical Appraisal Checklist for Non-randomised Experimental Studies [14]. RCTs were to be assessed using the Cochrane Risk of Bias Tool for Randomised Controlled Trials [15]. A narrative summary of all included studies was performed across all types of MLD (pre- or symptomatic; LI, J, EJ or LJ). The data are summarised using text and where relevant, accompanying tables and figures.
Results
Study selection and overview of included studies
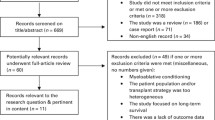
A total of 6940 titles and abstracts were retrieved from the literature searches and 26 from other sources. From these, full papers were obtained for 197 citations. After further review, 162 papers were excluded.
Thirty-five papers reporting on a total of 12 studies were selected for inclusion in the review. The study selection process is detailed in Fig. 1 (Study flowchart). The list of studies excluded at full paper screening are shown in Additional file 1: Appendix 2 (Tables S11 to S16).

Flow diagram
Additional file 1: Table S2 provides details on study designs and Table 1 detail on population characteristics from 13 studies included in the systematic review. The studies were mostly single arm and used retrospective data collection methods. Of those with comparative data, one, Fumagalli et al. [13], compared arsa-cel to NHx, and the other two studies compared HSCT to standard care [16, 17]. Note that Fumagalli et al. [13], whilst having been published in 2022, was based on data that the authors had access to during the conduct of the systematic literature review
The prospective arm of Fumagalli et al. [13] aimed to evaluate the clinical efficacy and safety of atidarsagene autotemcel and a (NHx) arm (n = 31) as a historical control. The atidarsagene autotemcel arm consisted of Study 201,222 (n = 20) plus the so-called Extended Access Framework (EAF) (n = 9), which were similar in design and outcomes, allowing combination of the data [13]. Note that atidarsagene autotemcel was not studied in LJ patients and so results would be restricted to the LI and EJ population.
As expected from the rarity of the condition, the studies included low number of patients and population characteristics were poorly reported. Additional file 1: Table S3 summarises the various treatments, which highlights the lack of information on standard care and variation in HSCT implementation. Due to the differences in the definitions of types of MLD across the studies, comparisons between studies are limited. Therefore, if results from comparative studies are available, any other results are included as appendices.
Risk of bias assessment
The risk of bias in the 12 clinical studies was assessed using the JBI Critical Appraisal Checklist for Non-randomised Experimental Studies and summarised in Table 2. The majority of studies had at least one assessment criterion judged at high risk of bias with only one study with no criterion judged high risk of bias [13]. Overall, twelve studies were judged at high risk of bias and one at an unclear risk of bias [13].
Studies were poorly reported. Three studies were reported as abstract only [18,19,20]. Most studies, except Singh 2012 [20], clearly reported the intervention and its effects. A control group was present only in three studies [13, 16, 17]. Six studies reported multiple measurements of the outcome both pre and post intervention [13, 17, 21,22,23,24]. Follow-up was incomplete or differences between groups in terms of their follow-up were not adequately described and analysed in three studies. [16, 17, 21] and unclear in seven studies [13, 18,19,20, 22, 24, 25]. Six studies measured the outcomes in a reliable way [13, 16, 21,22,23, 25]. An appropriate statistical analysis was used in four studies [13, 16, 25, 26].
Clinical effectiveness
Survival
Eleven studies [13, 16,17,18,19,20,21,22,23,24,25] reported data on overall survival including eight studies reporting Kaplan Meier estimates [13, 16, 17, 21,22,23,24,25]. The summary of survival data for patients treated with atidarsagene autotemcel, HSCT or receiving best supportive care is provided in Table 3.
Five studies reported retrospective or mixed retrospective and prospective data on survival in untreated (natural history) standard care late infantile (LI) to juvenile (J) patients (n = 131) with percentage survival ranging from 18.8% in 16 LI patients (follow-up not reported) to 100% at 5 years in 41 EJ patients.
Eight studies (n = 172) reported survival in patients undergoing HSCT. Approximately 5 years after HSCT survival ranged from 57 to 74.1% regardless of MLD disease subtype [23, 24]. Five to six years after HSCT, survival in LI patients [21,22,23] ranged from 50 to 60% and in juvenile (J) MLD patients [16, 21, 22, 27] from 59 to 82.4%, with little difference between EJ (80%) and LJ (79%).
One study reported survival in atidarsagene autotemcel treated (n = 29) and NHx patients (n = 31). After a median follow-up of 3.0 years (range 1.0–7.5 years), there were no deaths with seven surviving to age 6 in pre-symptomatic LI MLD patients treated with atidarsagene autotemcel in Fumagalli et al. 2022. In contrast, among the 19 LI MLD NHx patients, five had died by the age of 6 years (70.8% survival). In the EJ population, at the age of 9 years, estimated survival was 75·5% and 100% for the treated and NHx respectively. At 10 and 11 years of age, survival was 75·5% for treated EJ patients compared with 88·9% and 76·2%, respectively for NHx patients.
Progressive disease
The number of patients experiencing disease progression was reported by five studies [16, 18,19,20, 23] and the number of patients dying due to disease progression has been reported by three studies [13, 17, 21]. The summary of disease progression data for patients treated with atidarsagene autotemcel, HSCT or receiving best supportive care is provided in Table 4. Definitions of ‘disease progression’ substantially varied across the studies of HSCT and standard of care.
Ten years after HSCT, eight out of 20 (40%) patients with J MLD had disease progression resulting in a low level of gross motor function or loss of language compared to 28 out of 41 (68%) in non-transplanted J MLD patients. Two out of 24 (8.3%) J MLD children died of rapid MLD progression after HSCT in comparison to 11 out of 41 (26.8%) in an untreated control group [16]. In comparison one out of two (50%) LI MLD and one out of five (20%) J MLD patients died due to disease progression within 1 year of HSCT in another comparator study (n = 7), but outcome data were not reported for the untreated control arm [17].
For patients receiving atidarsagene autotemcel, disease progression was reported mostly within different disease facets, such as brain MRI or neuropsychological outcomes (performance IQ), and result are presented in the sections below. For LI MLD patients treated with atidarsagene autotemcel, there were no progressive disease related deaths at the time of interim analysis (median follow-up of 3 years). At the same timepoint, 2 out of 13 (15%) EJ MLD patients treated with atidarsagene autotemcel died due to disease progression (8- and 15-months post-treatment) [13].
Gross motor function
Six studies reported data on gross motor function [4, 13, 16, 17, 21, 25]. Gross motor function was assessed in a variety of ways, but most studies used the Gross Motor Function Measure (GMFM)-88 or the Gross Motor Function Classification (GMFC)-MLD. The summary of gross motor function for patients treated with atidarsagene autotemcel or HSCT versus standard care is provided in Tables 5 and 6 (further non-comparative data on standard care and HSCT are shown in Additional file 1: Tables S4 and S5).
Fumagalli et al. [13] showed a large and statistically significant difference in GMFM-88 in favour of atidarsagene autotemcel compared with NHx patients. Across both MLD subtypes (LI and EJ), patients receiving atidarsagene autotemcel showed better gross motor function at to 3 years follow-up when compared to age-matched NHx patients (Table 5). In contrast, only J type patients receiving HSCT showed any retention of gross motor function vs. non-transplanted (standard care) patients, and in one study the number of patients with no change in GMFM was higher with standard care (100% vs. 21.4%) (Table 6) [21]. Those with LI disease all had declining function (Table 6) [17].
Cognitive function—cognitive impairment and language skills
Five studies reported data on cognitive impairment [13, 17, 21, 23, 26] and six studies on language skills [16, 21, 23, 26, 28, 29]. Because very few data were reported for atidarsagene autotemcel the summary of results for cognitive function for patients treated with HSCT or receiving standard care are provided in Additional file 1: Tables S6–S9.
In general, patients with cognitive impairment at baseline appeared to continue to experience a decline in function after HSCT [21], although some patients (n = 5/14) [23] with borderline or delayed cognitive skills at baseline were reported to continue to gain cognitive skills initially (see Additional file 1: Table S8).
It was reported from Fumagalli et al. 2022 that age-equivalent scores showed normal cognitive skills in 20 of 25 (80%) atidarsagene autotemcel treated patients at similar chronological ages at which NHx patients showed severe cognitive impairment [13].
Intelligence quotient (IQ)
Three studies reported data on IQ scores [16, 17, 21]. The method of IQ assessment was reported in all except one study [17] with the Wechsler Preschool and Primary Scale of Intelligence (WPPSI) and the Wechsler Intelligence Scale for Children (WISC) scales most commonly used. There were no atidarsagene autotemcel studies reporting IQ, although the cognitive age-equivalent scores measured in Fumagalli et al. 2022 are based on the Development Quotient, which is itself derived from IQ [13].
One study of HSCT versus standard care reported that 11/24J MLD patients were more likely to have an IQ of at least 85 when compared with untreated standard care patients [16]. Another study comparing HSCT versus standard care [17], reported that 46.2% of transplanted patients (n = 7; including n = 2 LI and n = 5 J; mean follow-up 4.7 years) did not experience IQ decline (defined as decrease of at least 6 points), whereas all standard care patients (n = 10; mean follow-up 4.6 years) showed a decline in IQ. However, this study also reported that patients with an IQ score below 75 showed no benefit from HSCT [17]. One study of HSCT also reported that there was a trend across all patients (and MLD subtypes) for a gradual decline in VIQ scores (subscore of WISC) after transplant [21].
Neurological function—nerve conduction velocity (NCV) and magnetic resonance imaging (MRI)
Three studies reported data on neurological function assessed using nerve conduction velocity (NCV) [13, 21, 23] and five studies on neurological function assessed using brain magnetic resonance imaging (MRI) [13, 16, 17, 21, 23]. NCV was measured using electroneurography and generally reported using NCV index scores. Brain MRI was used to measure the progression of white matter demyelination and atrophy in the central nervous system and the predominant measure used was the Loes score (Fumagalli et al. [13] used a modified version as described in Sessa et al. 2016). The summary of results for neurological function for patients treated with atidarsagene autotemcel, or HSCT versus standard care is provided in Tables 7, 8, 9 and 10. Only non-comparative NCV data were available for HSCT (see Table 8) (further non-comparative MRI data on HSCT are shown in Additional file 1: Table S10).
Fumagalli et al. [13] compared NCV scores of LI MLD patients treated with atidarsagene autotemcel with age matched natural history (standard care) control patients [13]. For patients with LI MLD, treatment differences in NCV index scores favoured atidarsagene autotemcel versus standard care control at both 2 years (5.8, 95% CI 2.4–9.1; p = 0.004) and 3 years (3.2; 95% CI 1.0–5.3; p = 0.010) post treatment (see Table 7).
Two studies of HSCT [21, 23] reported only the numbers of patients with worsening and stabilising NCV, hampering any comparison with data from the atidarsagene autotemcel studies. For both LI and J MLD subtypes, results showed that, depending on the study, between one and two thirds of patients tend to show worsening NCV after HSCT, whilst the remaining patients showed stabilised NCV (see Table 8).
With respect to MRI, HSCT and standard care studies [16, 17, 21, 23] mostly reported the number of patients with improved, stable, or deteriorating MRI scores, hampering comparisons with the atidarsagene autotemcel studies (see Additional file 1: Table S6). Patients undergoing HSCT appeared to show stabilisation and/or improvement in brain MRI after transplantation. In one study of LI and EJ MLD patients [21], 6/11 (54.5%) and 7/9 (77.8%) of HSCT patients showed the same level of demyelination as the previous timepoint/baseline value 1 and 2 years (respectively) after transplantation. Another study [23] reported that 16 out of 19 children (84.2%) showed an improvement in MRI total scores after transplantation.
Fumagalli et al. [13, 29] suggested that atidarsagene autotemcel may stabilise and prevent MRI deterioration in both LI and EJ patients, when compared to untreated natural history (standard care) patients. For patients with LI MLD, treatment differences in brain MRI (Loes) total scores favoured atidarsagene autotemcel over a natural history (standard care) control patients at both 2 years (− 12.9, 95% CI − 16.2 to − 9.7; p < 0.001) and 3 years (− 18.1, 95% CI − 21.1, − 15.0; p < 0.001) post atidarsagene autotemcel treatment (see Table 9). Corresponding treatment differences in EJ patients compared to standard care were − 8.5, 95% CI − 14.7 to 2.3 (p = 0.010) at 2 years, and − 10.4, 95% CI − 17.0 to − 3.8 (p = 0.004) favouring atidarsagene autotemcel treatment) at 3 years post atidarsagene autotemcel treatment.
In one study [17], standard care (untreated) LI to J patients showed a deterioration (increase) in MRI Loes scores compared to patients undergoing HSCT. Similarly, non-transplanted J MLD standard care patients had significantly higher MRI severity scores compared to transplanted patients (p = 0.06) and a significant increase in severity scores from early to late disease stage (p < 0.001), compared to transplanted patients pre- to post-HSCT.
ARSA activity
Three studies reported data on ARSA activity [13, 19, 21].
Data on ARSA activity after HSCT (n = 39) was limited and was not reported as mean change from baseline hampering any comparison with atidarsagene autotemcel treated patients. However, one retrospective study [21] reported that at last follow-up (mean 3.5 years post-HSCT) 2/3 (66.7%) of LI patients and 17/24 (63.0%) of J patients (63.0%) had ‘100% ARSA activity’ (no details reported).
All atidarsagene autotemcel treated patients in Fumagalli et al. 2022 showed ARSA activity in peripheral blood mononuclear cells within or above normal range from 3 months post-treatment onward, which was significantly increased above baseline 2 years post-treatment by a mean 18·sevenfold (95% confidence interval CI 8.3–42.2; p < 0.001) and 5·sevenfold (95% CI 2.6–12.4; p < 0.001) in late-infantile and early-juvenile patients, respectively. Mean ARSA activity in cerebrospinal fluid (CSF) was above the level of quantification by the first post-baseline measurement at 3 months post-treatment, reached normal levels by 6–12 months, and remained within normal range throughout available follow-up (Year 3 early-juvenile; Year 5 late-infantile).
Health-related quality of life (HRQoL)
Data on the HRQoL of patients with MLD was very limited and only one study assessing HSCT reported data for mix of LI and J patients [21]. There was no assessment of the change in HRQoL from baseline (pre-treatment) and long-term follow up (duration unclear) was measured using the Cornell-Brown Scale that is often used in dementia patients [30]. There were no atidarsagene autotemcel studies reporting IQ.
Amongst the included patients with evaluable data (n = 12) the Cornell-Brown Scale numerical scores were mostly greater than zero suggesting a favourable HRQoL after HSCT. The only LI MLD patient had a score of 4; the mean score across nine J MLD patients was 10.2 (range − 13 to 23), with only one patient having a negative score (− 13).
Safety
Six included studies reported on AEs [13, 16, 17, 19, 21, 23]. Rates of fatal AEs for HSCT are shown in Table 11.
Five studies reported limited data mostly relating to the number of fatal AE after HSCT [16, 17, 19, 21, 23]. In Boucher 2015 [21] 17/48 (35.4%) patients experienced fatal events after HSCT (follow-up not reported) including 13 J MLD patients (hepatic veno-occlusive disease n = 1; Graft versus Host Disease (GvHD) n = 2; acute GvHD n = 1; sepsis n = 5; MLD progression n = 2; multi-system organ failure n = 1; and thrombotic thrombocytopenic purpura n = 1) and two LI MLD patients (cause unknown n = 1; and hepatic veno-occlusive disease n = 1). Treatment related fatal events within weeks post HSCT were experienced by four out of 24 (16.7%) J MLD patients in Groeschel [16]; all died of infections including two with bacterial, infections, one invasive fungal infection and one viral interstitial pneumonitis associated in part with graft rejection. Martin [23] reported that a median of 5.1 years after HSCT 25.9% (7/27) of patients experienced a fatal AE; causes of death included multiple organ failure (n = 1); respiratory failure after chronic lung disease (n = 2); viral infections/malignancy (n = 3); and disease progression and infection (n = 1). Bohringer [19] reported no case of transplant-related mortality or no chronic GvHD.
In Fumagalli et al. [13] all 39 atidarsagene autotemcel treated patients experienced at least one Grade 3+ adverse event: three were fatal (two AEs of dysphagia due to disease progression, one AE of ischaemic stroke, all unrelated to treatment) and the only treatment-related adverse event was the transient development of anti-ARSA antibodies in four patients, which was reported to have not impacted clinical outcomes.
Discussion
Summary of findings
To our knowledge, this systematic review is the first to evaluate treatments (atidarsagene autotemcel and HSCT) for children (age ≤ 17 years) with MLD and to compare it to the natural progression of the disease. The previous systematic review by Musolino [31] focused on the effects of HSCT in the treatment of patients with leukodystrophies. The authors searched the literature up to June 2012 and included evidence beyond the scope of this review (case reports, case series and other). Out of 698 patients included in the review, only 114 were MLD patients with the results focusing on safety and the characteristics of HSCT procedures. The systematic review by Mahmood [32], reported alongside the case report of triplets with MLD, identified studies reporting on new cases with definite diagnosis of MLD up to June 2006. Any study reporting exclusively on patients receiving transplants were excluded. The authors focused on patients’ survival.
Overall, this is the first systematic review of atidarsagene autotemcel, HSCT and standard care control patients in MLD and confirmed that the condition is associated with significant mortality and morbidity in children and adolescents. Comparability between treatments was hampered by lack of randomised controlled trials, which is typical for rare diseases. In LI patients, survival for atidarsagene autotemcel appeared longer than for NHx and HSCT as observed in other studies. Survival for EJ patients appeared to be similar between atidarsagene autotemcel treated, HSCT recipients and the NHx cohort. However, no LI and only 15% of EJ patients treated with atidarsagene autotemcel were observed to have died due to disease progression over about 3 years, in contrast to 50% of LI and 20% of J patients treated with HSCT within 1 year [13, 16, 17]. Gross motor function was improved with atidarsagene autotemcel versus with no treatment for both LI and EJ, which contrasted with HSCT, where fewer J patients and no LI patients retained function [21]. HSCT seemed to make little difference to cognitive decline [21], whereas normal acquisition of cognitive skills in the majority of patients with atidarsagene autotemcel treatment [13]. HSCT appeared to be associated with treatment related deaths [16, 17, 19, 21, 23], unlike atidarsagene autotemcel, where no treatment related deaths were observed [13].
Strengths and limitations
The content of any systematic review is dependent on the methods used and quality of the included research. The review was prepared according to the systematic review methodologies recommended by the Cochrane Collaboration. This review used the best available evidence found through extensive literature searches and other sources with no restrictions.
MLD is a rare genetic disease which poses great challenges to researchers in terms of patient recruitment and comparing treatments to relevant control arms. Most of the studies included in the review had small sample size and included only retrospective, single arm data without control group. A sample size of at least 5 patients was considered sufficient to reduce uncertainty and ensure generalizability of results.
Given the limited patient population and the poorly reported population characteristics, there is potential for overlap of study populations across studies. This is particularly a concern for those studies based in populations from Germany and those using data from the LEUKONET database [16, 18, 19, 26]. Moreover, the data were reported differently across the studies depending on whether patients were assessed from disease onset, diagnosis or with regard to age, making any comparisons between studies difficult.
Overall, outcomes were not well defined across studies. The section ‘progressive disease’ reports on studies that assessed outcome described as ‘progressive disease’ or patients dying with ‘progressive disease’ only. Disease progression has also been reported within different disease facets, such as gross motor function, brain MRI or neuropsychological outcomes (performance IQ).
All included studies were judged as high or unclear risk of bias with poor reporting of the results. Again, small number of patients, and lack of control arm for some studies, limits the reliability of findings. The comparisons between atidarsagene autotemcel, HSCT and standard care is also limited by high heterogeneity in study populations, follow-up time and outcome measures. Due to improvements in HSCT technologies and standard of care of patients with MLD, the results of the studies might not be relevant to current practice. Moreover, studies of HSCT used different sources of stem cells e.g., umbilical cord cells [17, 24] or bone marrow [19]. The comparison with atidarsagene autotemcel studies is limited as this technology involved bone marrow and blood and was not yet applied to all sources of stem cells i.e., umbilical cord cells.
No quantitative analysis was possible due to the lack of comparability of the evidence.
Research recommendations
The authors identified several gaps in the evidence base for MLD treatments. Firstly, there is lack of prospective studies comparing atidarsagene autotemcel with HSCT. The available evidence does not focus on patient and caregiver HRQoL. Future studies should focus on methodological rigour with more emphasis on the methods of data collection, measurement of outcomes, and the reporting of baseline patient characteristics to ensure the reliability of the results. Any potential of overlap between patient populations in similar studies should be clearly stated and references to relevant studies provided. To aid any future meta-analyses, an agreed set of standardised outcomes measured across all MLD studies should be available: currently, high heterogeneity between studies and outcomes reported hampers any comparisons. An adequate follow-up of MLD patients should also be clearly described.
Conclusions
Consistent with other research into treatments for rare diseases, there is limited low-quality evidence on which to base an assessment of the clinical effectiveness and cost effectiveness of treatments for MLD in children. Heterogeneity between studies with respect to study designs, populations, follow-up, and outcome measures limits any comparison, along with a lack of direct concurrent control data. Studies were generally small and reliant on retrospective data, particularly with respect to HSCT and standard of care patients. However, studies of the natural history and standard care of the disease in children and adolescents showed that MLD has significant effects on patient mortality and morbidity, including debilitating effects on cognitive and gross motor function. Evidence of the effects of HSCT were unclear especially in EJ patients, and longer-term follow-up suggested that any improvement or stabilisation in cognitive and gross motor function was not likely to be maintained. Data suggested that HSCT may only show potential benefits in LJ. Initial data on patients treated with atidarsagene autotemcel, albeit in only one trial plus EAF, showed promising results particularly when used in patients treated before symptoms appear (pre-symptomatic). This included apparent improvements in survival, as well as reduced rate of decline in cognitive function, and gross motor function. The treatment also appeared well tolerated with no serious or treatment related effects; adverse effects observed appeared mainly due to pre-treatment procedures (conditioning treatment) and MLD disease progression. However, further data is required to confirm these findings and further follow-up of existing studies is ongoing.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
References
NIHR Innovation Observatory: OTL-200 for metachromatic leukodystrophy: health technology briefing, June 2019 [Internet]. Leeds: NIHR, 2019. Accessed 6 Nov 2019.
MLD Support Association UK: About MLD. http://www.mldsupportuk.org.uk/about-mld/.
Kehrer C, Elgun S, Raabe C, Bohringer J, Beck-Wodl S, Bevot A, Kaiser N, Schols L, Krageloh-Mann I, Groeschel S. Association of age at onset and first symptoms with disease progression in patients with metachromatic leukodystrophy. Neurology. 2021;96(2):e255–66.
Kehrer C, Blumenstock G, Gieselmann V, Krageloh-Mann I. The natural course of gross motor deterioration in metachromatic leukodystrophy. Dev Med Child Neurol. 2011;53(9):850–5.
Kehrer C, Kustermann-Kuhn B, Raabe C, Krageloh-Mann I. Natural history of metachromatic leucodystrophy (MLD): clinical course. Eur J Pediatr. 2008;167(3):374.
Noyes J, Godfrey C, Beecham J. Resource use and service costs for ventilator-dependent children and young people in the UK. Health Soc Care Commun. 2006;14(6):508–22.
Tan EY, Boelens JJ, Jones SA, Wynn RF. Hematopoietic stem cell transplantation in inborn errors of metabolism. Front Pediatr. 2019;7:6.
Beschle J, Döring M, Kehrer C, Raabe C, Bayha U, Strölin M, Böhringer J, Bevot A, Kaiser N, Bender B. Early clinical course after hematopoietic stem cell transplantation in children with juvenile metachromatic leukodystrophy. Mol Cell Pediatr. 2020;7(1):1–9.
Orchard Therapeutics: orchard Therapeutics announces publication in The Lancet of long-term clinical outcomes with Libmeldy for the treatment of children with early-onset MLD (Press Release). https://ir.orchard-tx.com/news-releases/news-release-details/orchard-therapeutics-announces-publication-lancet-long-term.
Centre for Reviews and Dissemination: Systematic reviews: CRD’s guidance for undertaking reviews in health care. York: University of York, 2009. Accessed 23 Mar 2011.
Higgins JPT, Green S, eds: Cochrane handbook for systematic reviews of interventions. Version 5.1.0 [updated March 2011]: The Cochrane Collaboration, 2011. http://handbook.cochrane.org/. Accessed 23 Mar 2011.
PRISMA Group: PRISMA (preferred reporting items for systematic reviews and meta-analyses) statement. http://prisma-statement.org/.
Fumagalli F, Calbi V, Sora MGN, Sessa M, Baldoli C, Rancoita PMV, Ciotti F, Sarzana M, Fraschini M, Zambon AA, Acquati S, Redaelli D, Attanasio V, Miglietta S, Mattia FD, Barzaghi F, Ferrua F, Migliavacca M, Tucci F, Gallo V, Carro UD, Canale S, Spiga I, Lorioli L, Recupero S, Fratini ES, Morena F, Silvani P, Calvi MR, Facchini M, et al. Lentiviral haematopoietic stem-cell gene therapy for early-onset metachromatic leukodystrophy: long-term results from a non-randomised, open-label, phase 1/2 trial and expanded access. Lancet. 2022;399(10322):372–83.
The Joanna Briggs Institute: checklist for cohort studies: The Joanna Briggs Institute critical appraisal tools for use in JBI systematic reviews. Adelaide: JBI, 2017. Accessed 19 Nov 2019.
Higgins JP, Altman DG, Gotzsche PC, Juni P, Moher D, Oxman AD, Savovic J, Schulz KF, Weeks L, Sterne JA. The cochrane collaboration’s tool for assessing risk of bias in randomised trials. BMJ. 2011;343:d5928.
Groeschel S, Kuhl JS, Bley AE, Kehrer C, Weschke B, Doring M, Bohringer J, Schrum J, Santer R, Kohlschutter A, Krageloh-Mann I, Muller I. Long-term outcome of allogeneic hematopoietic stem cell transplantation in patients with juvenile metachromatic leukodystrophy compared with nontransplanted control patients. JAMA Neurol. 2016;73(9):1133–40.
van Rappard DF, Boelens JJ, van Egmond ME, Kuball J, van Hasselt PM, Oostrom KJ, Pouwels PJ, van der Knaap MS, Hollak CE, Wolf NI. Efficacy of hematopoietic cell transplantation in metachromatic leukodystrophy: the Dutch experience. Blood. 2016;127(24):3098–101.
Bley A, Muller I, Lobel U, Schrum J, Santer R, Hartmann M, Kohlschutter A. Hematopoietic stem cell transplantation (HSCT) in nine patients with juvenile MLD. Neuropediatrics. 2013;44(2):PS14_1097.
Bohringer J, Kustermann-Kuhn B, Gieseke F, Erbacher A, Doring M, Kehrer C, Lang P, Krageloh-Mann I, Handgretinger R, Muller I. Hematopoietic stem cell therapy in eight patients with metachromatic leukodystrophy relevance of post-transplant medication [Abstract 3727]. Presented at American Society of Hematology Annual Meeting and Exposition 2010; 4 Dec 2010; Orlando: US. 2010.
Singh J, Simmons L, Chakrapani A, Wassmer E. Metachromatic leukodystrophy: mortality data to support counselling of parents. J Inherit Metab Dis. 2012;35(1 Suppl 1):S107.
Boucher AA, Miller W, Shanley R, Ziegler R, Lund T, Raymond G, Orchard PJ. Long-term outcomes after allogeneic hematopoietic stem cell transplantation for metachromatic leukodystrophy: the largest single-institution cohort report. Orphanet J Rare Dis. 2015;10:94.
van den Broek BTA, Page K, Paviglianiti A, Hol J, Allewelt H, Volt F, Michel G, Diaz MA, Bordon V, O’Brien T, Shaw PJ, Kenzey C, Al-Seraihy A, van Hasselt PM, Gennery AR, Gluckman E, Rocha V, Ruggeri A, Kurtzberg J, Boelens JJ. Early and late outcomes after cord blood transplantation for pediatric patients with inherited leukodystrophies. Blood Adv. 2018;2(1):49–60.
Martin HR, Poe MD, Provenzale JM, Kurtzberg J, Mendizabal A, Escolar ML. Neurodevelopmental outcomes of umbilical cord blood transplantation in metachromatic leukodystrophy. Biol Blood Marrow Transplant. 2013;19(4):616–24.
Prasad VK, Mendizabal A, Parikh SH, Szabolcs P, Driscoll TA, Page K, Lakshminarayanan S, Allison J, Wood S, Semmel D, Escolar ML, Martin PL, Carter S, Kurtzberg J. Unrelated donor umbilical cord blood transplantation for inherited metabolic disorders in 159 pediatric patients from a single center: influence of cellular composition of the graft on transplantation outcomes. Blood. 2008;112(7):2979–89.
Fumagalli F, Zambon AA, Rancoita PMV, Baldoli C, Canale S, Spiga I, Medaglini S, Penati R, Facchini M, Ciotti F, Sarzana M, Lorioli L, Cesani M, Natali Sora MG, Del Carro U, Cugnata F, Antonioli G, Recupero S, Calbi V, Di Serio C, Aiuti A, Biffi A, Sessa M. Metachromatic leukodystrophy: a single-center longitudinal study of 45 patients. J Inherit Metab Dis. 2021;44(5):1151–64.
Kehrer C, Groeschel S, Kustermann-Kuhn B, Burger F, Kohler W, Kohlschutter A, Bley A, Steinfeld R, Gieselmann V, Krageloh-Mann I, German L. Language and cognition in children with metachromatic leukodystrophy: onset and natural course in a nationwide cohort. Orphanet J Rare Dis. 2014;9:18.
Muhlstein A, Gelperina S, Shipulo E, Maksimenko O, Kreuter J. Arylsulfatase A bound to poly(butyl cyanoacrylate) nanoparticles for enzyme replacement therapy–physicochemical evaluation. Pharmazie. 2014;69(7):518–24.
Orchard Therapeutics: A phase I/II clinical trial of haematopoietic stem cell gene therapy for the treatment of metachromatic leukodystrophy (Interim Report No. 2.2). Clinical study report {201222/v.2.2} [PDF provided by Orchard]. Orchard Therapeutics, 30th September 2019. Aaccessed 14 April 2020.
Orchard Therapeutics. Clinical study report for expanded access programmes (EAPs) for haematopoietic stem cell gene therapy OTL-200-F (formerly known as GSK2696274) in early onset metachromatic leukodystrophy (MLD) patients. Hospital exemption (HE) protocol 20529 and compassionate use program (CUP) 206258. 205029–206258 vl.0 [PDF provided by Orchard]. Orchard Therapeutics, 1st October 2019. Accessed 14 April 2020.
Ready RE, Ott BR, Grace J, Fernandez I. The Cornell-Brown scale for quality of life in dementia. Alzheimer Dis Assoc Disord. 2002;16(2):109–15.
Musolino PL, Lund TC, Pan J, Escolar ML, Paker AM, Duncan CN, Eichler FS. Hematopoietic stem cell transplantation in the leukodystrophies: a systematic review of the literature. Neuropediatrics. 2014;45(3):169–74.
Mahmood A, Berry J, Wenger DA, Escolar M, Sobeih M, Raymond G, Eichler FS. Metachromatic leukodystrophy: a case of triplets with the late infantile variant and a systematic review of the literature. J Child Neurol. 2010;25(5):572–80.
Acknowledgements
Not applicable.
Funding
Orchard Therapeutics (Europe) Limited funded the project.
Author information
Authors and Affiliations
Contributions
NA and CN were involved in the acquisition, analysis and interpretation of data and drafting of the manuscript. AO and FP were involved in the conception, and interpretation of the review and drafting the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
AO and FP are employees of Orchard Therapeutics (Europe) Limited. NA and CN work for KSR Ltd, which received funding from Orchard Therapeutics (Europe) Limited for the project.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. Supplementary appendices.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Armstrong, N., Olaye, A., Noake, C. et al. A systematic review of clinical effectiveness and safety for historical and current treatment options for metachromatic leukodystrophy in children, including atidarsagene autotemcel. Orphanet J Rare Dis 18, 248 (2023). https://doi.org/10.1186/s13023-023-02814-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-023-02814-2