
Abstract
Background
Woodhouse-Sakati syndrome (WSS) is a rare, autosomal recessive genetic disorder with variable clinical manifestations mainly affecting the endocrine and nervous systems. The aim of this study was to systematically review the genetic basis of WSS and report the genetic variants and clinical phenotypes associated with the disease.
Methods
PubMed, Science Direct, Scopus, and Web of Science databases were searched from the time of inception until June 2022. Broad search terms were used to capture the literature describing all genetic variants associated with WSS. The search keywords used are “Woodhouse Sakati” along with the term “mutation” OR “gene” OR “variant” OR “polymorphism”.
Results
Twenty-five eligible studies were included in this study. One hundred and eighty-five patients in 97 families from 12 different countries were diagnosed with WSS. In patients from the Greater Middle East (GME) region, consanguineous marriages were common (67%). Thirteen different DCAF17 variants were associated with WSS development (including 8 identified in the GME region). The most frequent variant was a frameshift deletion variant (c.436delC, p.Ala147Hisfs*9) unique to Arabs that was reported in 11 cases from Tunisia, Kuwait, Qatar, Bahrain, and Saudi Arabia. There were no clear genotype–phenotype correlations for the different variants.
Conclusions
This systematic review highlights the molecular basis and clinical manifestations of WSS globally, including the GME region, where the disease is prevalent due to consanguinity. Additional studies are now needed to understand the genotype–phenotype correlation for different DCAF17 variants and their impact on the phenotypic heterogeneity observed in WSS patients.
Similar content being viewed by others


Background
Woodhouse-Sakati syndrome (WSS), first described in a consanguineous Saudi Arabian family in 1983 [1], is a rare, autosomal recessive genetic disorder [2]. WSS is characterized by a variety of predominantly endocrine and nervous system abnormalities including hypogonadism, diabetes mellitus (DM; in 95% of patients), hypothyroidism, low insulin-like growth factor (IGF-1) levels, deafness, alopecia, and electrocardiographic abnormalities [3, 4]. The prevalence of WSS is estimated to be < 1/1,000,000 of the population [2]. There is no clear age of onset for the disorder, but the different clinical manifestations can present at different times; for example, hypogonadism is often detected around the time of puberty (12–14 years of age); DM and hypothyroidism during adolescence up to the age of 25 years of age; and neurological manifestations between nine and 17 years of age [2].
Therefore, the clinical features of WSS are heterogeneous. On clinical examination, WSS patients characteristically have a flat occiput, triangular face, high forehead, frontal bossing, mild hypertelorism, short and sparse eyebrows, down-slanting palpebral fissures, a prominent nasal root, dental malocclusion, and a high-arched palate [4]. Other clinical features include progressive childhood-onset alopecia (leading to alopecia totalis) and the neurologic findings of progressive extrapyramidal movements (dystonic spasms with dystonic posturing with dysarthria and dysphagia), sensorineural hearing loss, and intellectual disability [5]. Brain MRI performed on some of these patients has revealed diffuse white matter disease and sometimes increased iron accumulation in the basal ganglia [6]. Regardless of the specific features seen in individuals, patients generally suffer severe morbidity and a high risk of early death, and there is currently no effective treatment for the condition.
WSS is caused by the inheritance of mutations in DCAF17 (formerly known as C2orf37), and overexpression of nucleolar DCAF17 protein is associated with the WSS phenotype both clinically and in animal studies [7]. The two transcripts of DCAF17 have unknown function but the encoded proteins localize to the nucleolus, where they are involved in cell cycle regulation. It has been suggested that mutant DCAF17 leads to defective ribosome biogenesis, resulting in cell cycle abnormalities and cellular aging [8]. WSS is also associated with some loss of function mutations that reduce splicing efficiency and truncate the protein. However, there is still no clear genotype–phenotype correlation in WSS, partly due to the marked phenotypic variability and partly because the disease is so rare.
The diagnosis of WSS can be difficult due to this clinical heterogeneity and spectrum of common symptoms and signs, so molecular genetics is important to secure the diagnosis and for patient management. Moreover, once the pathogenic variant is identified in a family member, further genetic testing and counseling can be conducted to determine the individual risk of passing on/having the condition through carrier testing, prenatal testing, and pre-implementation genetic diagnosis [9]. Here we systematically reviewed the genetics and molecular basis and biology of WSS to shed further light on genotype–phenotype correlations observed globally.
Results
Search outcome
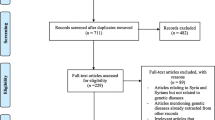
The PRISMA flow chart is shown in Fig. 1. Our search strategy yielded 270 studies, of which 160 remained after removal of duplicates. The remaining articles were subjected to primary screening by title, abstract, and PICOS assessment for eligibility according to the inclusion and exclusion criteria (Table 1). Of the screened articles, 68 were eligible for full assessment. Of these, 44 articles were not genetic studies. Therefore, 25 articles were eligible for inclusion in this review.

PRISMA flow chart detailing the search strategy and study selection process
Quality of the eligible studies
Risk of bias assessments in the four domains of the QUADAS-2 tool are shown in Fig. 2. The patient selection domain had the highest risk of bias, with a high risk of bias in 37.5% of included articles. This was expected, as we included case reports in which participants were preselected. 52.4% of studies were rated as having a low risk of bias, as a full description of the diagnostic threshold and the standardized procedure for the test was given. There was mainly a low risk of bias in the remaining three domains.

Risk of bias assessment using the QUADAS-2 tool
Reported genetic variants
In the 25 included articles, WSS-associated mutations were reported in 185 patients belonging to 97 families. In most cases, genetic testing was performed by a mixture of Sanger and next-generation sequencing approaches. Consanguinity was observed in 67% (n = 65) of families, while only three families were from non-consanguineous marriages. In 29 families (29.8%), consanguinity was not specified.
Thirteen WSS-causing variants were described in the DCAF17 gene. These were reported mainly in the Greater Middle East (GME) countries (Tunisia, Kuwait, Qatar, Bahrain, Saudi Arabia, Pakistan, Iran, Turkey), where consanguinity is common [10], but there were cases in four other countries (Portugal, China, Italy and India) (Table 2). The most frequently identified variant was a deletion mutation (rs797045038, c.436delC p.Ala147Hisfs*9) accounting for 11 cases reported in five countries (Tunisia, Kuwait, Qatar, Bahrain, and Saudi Arabia).
Clinical phenotypes
Additional file 1: Table S1 shows the reported phenotypes in the 185 patients from the 25 included articles. Endocrine manifestations of hypogonadism and DM were seen in all patients, as was ectodermal involvement. The majority of patients showed neurological involvement. Some patients presented with high degree of intellectual disability. Other less common phenotypes included sensorineural hearing loss, dystonia, dysarthria, and other symptoms. Some studies reported the radiological findings from brain MRI, namely iron deposition within the substantia nigra and globus pallidus and subcortical white matter changes.
Genotype–phenotype correlation
Table 3 shows the distribution of genetic variants according to disease phenotypes. No formal statistical analysis of the distribution of genetic variants with clinical manifestations was conducted, as the sample numbers were too low. Figure 3 shows the distribution of the six most common clinical features according to different DECAF17 variants. Intellectual disability was reported for all genetic variants except c.270delA. Alopecia was reported for all variants except c.270dup and c.1A > G. In addition, hypogonadism was reported in all genetic variants reported in this study. Therefore, there was no clear genotype–phenotype correlation identified in these patients.

Distribution of the six most common clinical features according to DECAF17 variant
Discussion
To our best knowledge, this is the first comprehensive systematic review of the variants associated with WSS. We retrieved twenty-five published papers representing 185 patients in 97 families with WSS-associated variants from 12 countries globally. This included 171 patients (93%) from eight GME countries, where consanguinity is common. Marriages were consanguineous in the majority (67%) of the WSS families identified, which was expected given that WSS is an autosomal recessive condition and consanguinity increases the likelihood of inheriting two copies of a deleterious allele from a common ancestor (autozygosity). This explains why most cases of WSS are reported in the GME region.
Most of the available data on WSS cases came from case reports and series. As a rare genetic disorder, case reports and case series are a helpful tool, since they provide knowledge that is informative to systematic reviews, especially when data are scarce from randomized controlled trails and observational studies [35].
A study conducted by Abouelhoda et al., identified 259 homozygous mutations in a cohort of 7,000 unrelated patients from Saudi Arabia. They showed that DCAF17 combined carrier frequency is 0.003 ( PMID: 27,124,789) [36]. In our study, we found that the most common variant identified was the c.436delC variant on exon 4 of DCAF17. This variant was found mainly in Arabs and was reported in five Arab countries (Tunisia, Kuwait, Qatar, Bahrain, and Saudi Arabia), mainly in consanguineous families. This variant is in the open reading frame (ORF) of DCAF17 and is predicted to cause a frameshift mutation leading to the production of a prematurely truncated protein (p.Ala147Hisfs*9). The resulting protein is predicted to have 520 amino acids with no significant homology with other proteins [18]. The homozygous form of the c.436delC variant was reported in almost one-half of the reported cases in the Arab population, specifically in the Gulf region, so we consider this variant to be a founder variant in this population [12]. Abouelhoda et al. identified DCAF17 as a founder mutation in Arabic regions [36]. Another exonic variant associated with WSS development was the c.270delA (p.Lys90Asnfs8*) variant on exon 3 of DCAF17, which was identified in a Pakistani family (Table 1). This is a single base pair frameshift deletion affecting both gene transcripts to produce a truncated protein of 96 amino acids [24]. In addition, a deletion mutation (c.1488_1489delAG) and a nonsense mutation (c.1111delA) were reported in Chinese WSS patients, leading to the formation of a premature stop codon and thus predicted to form a truncated protein [3, 32]. Another exonic variant was also reported in India (c.1238delA), which results in a frameshift variant in exon 12, which is predicted to cause premature protein truncation [34]. The truncated DCAF17 protein resulting from these variants lacks the domains and motifs needed for interactions with the DDB1‐CUL4 ubiquitin ligase complex. The inefficient interaction between the truncated DCAF17 protein and other proteins could affect several functions within the nucleoli contributing to WSS [37,38,39].
A loss-of-function variant was also detected in the first start codon of DCAF17 in a Pakistani family suffering from WSS. This variant affects the translation initiation codon of DCAF17, which is predicted to inhibit the translation of its protein [23]. Six different splice site variants (c.127‐3delTAGinsAA, c.1091 + 1G > A, c.321 + 1G > A, c.1423‐1_1425delGACA, c.1091 + 2 T > C, and c.459- 7_499del) leading to exon skipping were found in different populations including patients from Pakistan, Turkey, Iran, Portugal, and India. It is well known that splice site variants contribute to the pathogenesis of different human genetic disorders, as around 15% of such variants affect pre-mRNA splicing [40]. Splice site variants cause inefficient splicing that could lead to exon skipping, intron retention, cryptic splice site activation, and the production of pseudo-exons within an intron [41].
DCAF17 is known to produce two main transcripts (alpha and beta isoforms) whose protein products localize to the nucleolus to exert as yet unknown functions in the human cell cycle. Both DCAF17 transcripts are ubiquitously expressed in adult human tissues, so the predominance of ectodermal, endocrine, and neurological involvement in WSS is not fully understood [12]. Alazami and colleagues suggested that WSS is caused by a nucleolar defect resulting from aberrant DCAF17 gene function that results in lymphoblast hypersensitivity to transcriptional blockade [18]. This study also observed that, in WSS, DCAF17 gene expression is high in the brain, skin, and liver, further confirming the pleiotropic nature of the disorder [18]. Similar to WSS, variants in other nucleolar proteins have been associated with a multisystem disorder of multiple endocrinopathies, alopecia, and hypogonadism [42]. Therefore, the nucleolus is likely to play an important role in the pathophysiology of these disorders due to its important role in several physiological processes [43]. All known variants reported in DCAF17 and found to be associated with WSS development possessed a full mutational spectrum covering the entire coding region. However, the nature and severity of phenotypes observed in WSS patients were not correlated with the expected length of the produced protein. Therefore, an intact full-length protein is probably needed for proper functionality, with the resulting protein sensitive to disruption of even a few amino acids [29]. Furthermore, since the length of the truncated protein in WSS patients does not seem to explain the clinical presentation, nonsense‐mediated decay (NMD) of mRNA and inefficient interactions of the truncated protein with other partners could explain the range of phenotypes observed [24]. The genotype–phenotype correlation in WSS still needs to be established.
This study has several limitations, mainly a lack of published peer-reviewed genetic studies related to WSS. Therefore, conducting a meta-analyses to ascertain the concordance of variants between patients was not possible.
Conclusions
In conclusion, this systematic review, reports 13 variants associated with WSS development and its pathogenesis. We consider this systematic review to be the first to discuss the variants associated with WSS globally, including in the GME region, where the condition seems to be most prevalent due to consanguinity. The genotype–phenotype correlation in WSS still needs to be established, as most reported cases displayed marked phenotypic variability. This lack of correlation indicates the presence of unknown modifier genes or epigenetic processes that modify the disease course. Further studies are needed to understand the mechanisms underlying the DCAF17 variants and their implication for the phenotypic heterogeneity observed in WSS patients. Ultimately, examining the genetic and molecular basis of WSS will improve our understanding of the disorder, nucleolar function, and improve the management of patients and their families.
Methods
Search strategy
The PubMed, Science Direct, Web of Science, and Scopus databases were searched from their dates of inception until June 2022. An extensive search of WSS studies globally was conducted using keywords constructed to include our primary or secondary outcomes. The primary outcome of this study was to report the genetic variants associated with WSS globally. The secondary outcome was to report the clinical phenotype variability observed in WSS patients (Table 1). Therefore, the search keywords included: “Woodhouse Sakati” along with the term “mutation” OR “gene” OR “variant” OR “polymorphism”. Initial screening (based on the abstract and title of the study) was performed on all the retrieved articles. Articles that met the inclusion criteria were fully evaluated and were included in this review.
Study selection
Retrieved records were assessed by two researchers, and the senior author resolved any disagreement by consensus. The total number of hits from each database was recorded. Research papers that met the following inclusion criteria were chosen for full assessment: (1) articles from peer-reviewed journals; and (2) the articles discussed genetic variants associated with WSS. Research papers were excluded if at least one of the following exclusion criteria was met: (1) articles lacking genetic information on WSS; and (2) the published material was a review, book, protocol, or guideline or animal studies (Table 1). After removal of duplicates, the titles and abstracts of the remaining articles were assessed. All records not matching our inclusion criteria were removed. Secondary selection included assessment of the entire full text to collect relevant records. Two researchers performed the assessment and a senior researcher helped to solve any disagreement. Figure 1 is a PRISMA flow chart showing the screening and selection process.
Quality control assessment and data extraction
Two researchers evaluated the quality of eligible articles for risk of bias using the Quality Assessment of Diagnostic Accuracy Studies-2 (QUADAS-2) tool [44]. This tool assigns a risk rating to four sections: patient selection, index test, reference standard, and flow and timing. Each section was rated as “low risk”, “high risk”, or “unclear risk”. In case of discrepancy, a senior opinion was sought.
Data items were collected from the tables and text of eligible articles and collated in a Microsoft Excel spreadsheet. Two researchers reviewed the collected data for accuracy. The data variables extracted included the name and function of the gene, sample size, genetic test used, number of patients tested and proportion with the causative variant, zygosity, consanguinity, gender, and phenotypic information. All captured variants were checked in PubMed (SNP), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), Exome Variant Server (EVS; http://evs.gs.washington.edu/EVS/), Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk/ac/index.php), and Google Scholar to obtain further insights on the variants identified.
References
Woodhouse N, Sakati NA. A syndrome of hypogonadism, alopecia, diabetes mellitus, mental retardation, deafness, and ECG abnormalities. J Med Genet. 1983;20:216–9.
Bohlega SA, Abusrair A, et al. Woodhouse-Sakati syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Gripp KW, et al., editors. GeneReviews®. Seattle: University of Washington; 1993.
Zhou M, Shi N, Zheng J, Chen Y, Wang S, Xiao K, et al. Case report: a Chinese family of woodhouse-Sakati syndrome with diabetes mellitus, with a novel biallelic deletion mutation of the DCAF17 gene. Front Endocrinol (Lausanne). 2021;12:770871.
Agopiantz M, Corbonnois P, Sorlin A, Bonnet C, Klein M, Hubert N, et al. Endocrine disorders in Woodhouse-Sakati syndrome: a systematic review of the literature. J Endocrinol Invest. 2014;37:1–7.
Gonzalez-Latapi P, Sousa M, Lang AE. Movement disorders associated with hypogonadism. Mov Disord Clin Pract. 2021;8:997–1011.
Salomão RPA, Pedroso JL, Gama MTD, Dutra LA, Maciel RH, Godeiro-Junior C, et al. A diagnostic approach for neurodegeneration with brain iron accumulation: clinical features, genetics and brain imaging. Arq Neuropsiquiatr. 2016;74:587–96.
Gurbuz F, Desai S, Diao F, Turkkahraman D, Wranitz F, Wood-Trageser M, et al. Novel inactivating mutations of the DCAF17 gene in American and Turkish families cause male infertility and female subfertility in the mouse model. Clin Genet. 2018;93:853–9.
Pandey A, Yadav SK, Vishvkarma R, Singh B, Maikhuri JP, Rajender S, et al. The dynamics of gene expression during and post meiosis sets the sperm agenda. Mol Reprod Dev. 2019;86:1921–39.
Malik SD, Al-Shafai M, Abdallah AM. The special features of prenatal and preimplantation genetic counseling in Arab countries. Genes. 2022;13:167.
Tadmouri GO, Nair P, Obeid T, Al Ali MT, Al Khaja N, Hamamy HA. Consanguinity and reproductive health among Arabs. Reprod Health. 2009;6:17.
Hdiji O, Turki E, Bouzidi N, Bouchhima I, Damak M, Bohlega S, et al. Woodhouse-Sakati syndrome: report of the first tunisian family with the C2orf37 gene mutation. J Mov Disord. 2016;9:120–3.
Almeqdadi M, Kemppainen JL, Pichurin PN, Gavrilova RH. Phenotypic variability of c.436delC DCAF17 gene mutation in Woodhouse-Sakati syndrome. Am J Case Rep. 2018;19:347–53.
Nanda A, Pasternack SM, Mahmoudi H, Ishorst N, Grimalt R, Betz RC. Alopecia and hypotrichosis as characteristic findings in Woodhouse-Sakati syndrome: report of a family with mutation in the C2orf37 gene. Pediatr Dermatol. 2014;31:83–7.
Ben-Omran T, Ali R, Almureikhi M, Alameer S, Al-Saffar M, Walsh CA, et al. Phenotypic heterogeneity in Woodhouse-Sakati syndrome: two new families with a mutation in the C2orf37 gene. Am J Med Genet A. 2011;155A:2647–53.
Sheridan MB, Wohler E, Batista DAS, Applegate C, Hoover-Fong J. The use of high-density SNP array to map homozygosity in consanguineous families to efficiently identify candidate genes: application to Woodhouse-Sakati syndrome. Case Rep Genet. 2015;2015:169482–169482.
Al-Khawaga S, Khalifa A, Hussain K. Woodhouse-Sakati Syndrome: Clinical and Molecular Study on a Qatari Family with C2orf37 Gene Mutation. European Society for Paediatric Endocrinology; 2018.
Ali R, Al-Dewik N, Mohammed S, Elfituri M, Agouba S, Musa S, et al. Expanding on the phenotypic spectrum of Woodhouse-Sakati syndrome due to founder pathogenic variant in DCAF17: report of 58 additional patients from Qatar and literature review. Am J Med Genet A. 2022;188:116–29.
Alazami AM, Al-Saif A, Al-Semari A, Bohlega S, Zlitni S, Alzahrani F, et al. Mutations in C2orf37, encoding a nucleolar protein, cause hypogonadism, alopecia, diabetes mellitus, mental retardation, and extrapyramidal syndrome. Am J Hum Genet. 2008;83:684–91.
Alharbi MS. Woodhouse-Sakati syndrome (WSS): a case report of 3 Saudi sisters with urogenital anomalies. Saudi Med J. 2021;42:1237–42.
Bohlega S, Abusrair AH, Al-Ajlan FS, Alharbi N, Al-Semari A, Bohlega B, et al. Patterns of neurological manifestations in Woodhouse-Sakati Syndrome. Parkinsonism Relat Disord. 2019;69:99–103.
Abusrair A, AlHamoud I, Bohlega S. Multimodal evoked potential profiles in Woodhouse-Sakati syndrome. J Clin Neurophysiol. 2020;5:89.
Alderson J, Ghosh PS. Clinical reasoning: seven-year-old girl with progressive gait difficulties. Neurology. 2020;94:364.
Shah K, Jan A, Ahmad F, Basit S, Ramzan K, Ahmad W. Woodhouse-Sakati syndrome in a family is associated with a homozygous start loss mutation in the DCAF17 gene. Clin Exp Dermatol. 2020;45:159–64.
Ali RH, Shah K, Nasir A, Steyaert W, Coucke PJ, Ahmad W. Exome sequencing revealed a novel biallelic deletion in the DCAF17 gene underlying Woodhouse Sakati syndrome. Clin Genet. 2016;90:263–9.
Habib R, Basit S, Khan S, Khan MN, Ahmad W. A novel splice site mutation in gene C2orf37 underlying Woodhouse-Sakati syndrome (WSS) in a consanguineous family of Pakistani origin. Gene. 2011;490:26–31.
Kurnaz E, Türkyılmaz A, Yaralı O, Demir B, Çayır A. A novel DCAF17 homozygous mutation in a girl with Woodhouse-Sakati syndrome and review of the current literature. J Pediatr Endocrinol Metab. 2019;32:1287–93.
Haeri G, Akhoundi FH, Alavi A, Abdi S, Rohani M. Endocrine abnormalities in a case of neurodegeneration with brain iron accumulation. Mov Disord Clin Pract. 2020;7:706–7.
Sendur SN, Oguz S, Utine GE, Dagdelen S, Oguz KK, Erbas T, et al. A case of Woodhouse-Sakati syndrome with pituitary iron deposition, cardiac and intestinal anomalies, with a novel mutation in DCAF17. Eur J Med Genet. 2019;62:103687.
Alazami AM, Schneider SA, Bonneau D, Pasquier L, Carecchio M, Kojovic M, et al. C2orf37 mutational spectrum in Woodhouse-Sakati syndrome patients. Clin Genet. 2010;78:585–90.
Fozia F, Shah K, Nazli R, Khan SA, Ahmad I, Mohammad N, et al. Novel splicing-site mutation in DCAF17 gene causing Woodhouse-Sakati syndrome in a large consanguineous family. J Clin Lab Anal. 2022;36:e24127–e24127.
Louro P, Durães J, Oliveira D, Paiva S, Ramos L, Macário MC. Woodhouse-Sakati syndrome: first report of a portuguese case. Am J Med Genet. 2019;179:2237–40.
Chen G, Zhou L, Chen Q, Wang J, Jiang P, Shen R, et al. Case report: a deletion variant in the DCAF17 gene underlying Woodhouse-Sakati syndrome in a chinese consanguineous family. Front Genet. 2021;12:741323.
Steindl K, Alazami A, Bhatia K, Wuerfel J, Petersen D, Cartolari R, et al. A novel C2orf37 mutation causes the first Italian cases of Woodhouse Sakati syndrome. Clin Genet. 2010;78:594–7.
Abdulla MC, Alazami AM, Alungal J, Koya JM, Musambil M. Novel compound heterozygous frameshift mutations of C2orf37 in a familial Indian case of Woodhouse-Sakati syndrome. J Genet. 2015;94:489–92.
Nambiema A, Sembajwe G, Lam J, Woodruff T, Mandrioli D, Chartres N, et al. A protocol for the use of case reports/studies and case series in systematic reviews for clinical toxicology. Front Med. 2021;8:708380.
Abouelhoda M, Sobahy T, El-Kalioby M, Patel N, Shamseldin H, Monies D, et al. Clinical genomics can facilitate countrywide estimation of autosomal recessive disease burden. Genet Med. 2016;18:1244–9.
Kerr L, Birse-Archbold J-L, Short D, McGregor A, Heron I, Macdonald D, et al. Nucleophosmin is a novel Bax chaperone that regulates apoptotic cell death. Oncogene. 2007;26:2554–62.
Boisvert F-M, van Koningsbruggen S, Navascués J, Lamond AI. The multifunctional nucleolus. Nat Rev Mol Cell Biol. 2007;8:574–85.
Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequences. Hum Genet. 1992;90:41–54.
Nakai K, Sakamoto H. Construction of a novel database containing aberrant splicing mutations of mammalian genes. Gene. 1994;141:171–7.
Aldahmesh MA, Abu-Safieh L, Khan AO, Al-Hassnan ZN, Shaheen R, Rajab M, et al. Allelic heterogeneity in inbred populations: the Saudi experience with Alström syndrome as an illustrative example. Am J Med Genet A. 2009;149:662–5.
Nousbeck J, Spiegel R, Ishida-Yamamoto A, Indelman M, Shani-Adir A, Adir N, et al. Alopecia, neurological defects, and endocrinopathy syndrome caused by decreased expression of RBM28, a nucleolar protein associated with ribosome biogenesis. Am J Hum Genet. 2008;82:1114–21.
Dousset T, Wang C, Verheggen C, Chen D, Hernandez-Verdun D, Huang S. Initiation of nucleolar assembly is independent of RNA polymerase I transcription. Mol Biol Cell. 2000;11:2705–17.
Whiting PF. QUADAS-2: a revised tool for the quality assessment of diagnostic accuracy studies. Ann Intern Med. 2011;155:529.
Acknowledgements
Not applicable
Funding
Open Access funding provided by the Qatar National Library.
Author information
Authors and Affiliations
Contributions
Conceptualization: AK and MAS, methodology: AK, AMA and MAS and data curation: AK and KH, original draft preparation: AK, data analysis, figures and writing of the first draft: AK and MAS, review, and editing: KH, and AMA, supervision: MAS, project administration: MAS. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1
. The clinical features associated with the genetic variants in DCAF17 reported in the literature.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kohil, A., Abdallah, A.M., Hussain, K. et al. Genetic epidemiology of Woodhouse-Sakati Syndrome in the Greater Middle East region and beyond: a systematic review. Orphanet J Rare Dis 18, 22 (2023). https://doi.org/10.1186/s13023-023-02614-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-023-02614-8