
Abstract
Background
Craniopharyngioma (CP) and cranial fibrous dysplasia (CFD) are rare embryonic benign cranial diseases that most commonly present during childhood or adolescence. The coexistence of CP and CFD is extremely rare and has not yet been reported.
Methods
We retrospectively reviewed the data of five patients with concomitant CP and CFD treated at Beijing Tiantan Hospital from January 2003 to January 2021 and summarized their clinicopathological features, treatment modalities, and outcomes. We also performed a comprehensive literature review, tested the patients for characteristic GNAS gene mutations related to CFD, and tested the CP specimens for corresponding Gsα protein to explore the potential connection leading to the coexistence of CP and CFD.
Results
The cohort comprised four men and one woman (median age, 39 years). The symptoms mainly included headache, dizziness, fatigue, polyuria/polydipsia, hypogonadism, and blurred vision. CFD most commonly involved the sphenoid bone (n = 4). Four patients underwent surgery to remove the CP (one trans-sphenoidal and three transcranial resections); complete and subtotal resection were achieved in two patients, respectively. The tumor subtype was adamantinomatous in three patients and unknown in one. The common postoperative complications were panhypopituitarism, diabetes insipidus, and hypothyroidism. The mean follow-up duration was 57.2 months. Two patients required postoperative hormone replacement therapy. Three patients underwent genetic study of the tumor specimens; GNAS mutations were not detected, but these patients were positive for Gsα protein.
Conclusions
Although a definite causative relationship has not been proved, the coexistence of CP and CFD means that potential interplay or an atypical fibrous dysplasia course as uncommon manifestations of CP cannot be excluded. It is more challenging to initiate prompt diagnosis and appropriate treatment for concomitant CP and CFD than for solitary CP because of skull base deformations. Current management strategies are aimed at surgical treating the CP and regularly monitoring the CFD.
Similar content being viewed by others


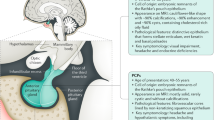
Introduction
Craniopharyngiomas (CPs) are rare epithelial tumors that arise along the path of the craniopharyngeal duct and account for 2%–5% of all primary intracranial neoplasms [1, 2]. The two major histological subtypes of CP are adamantinomatous CP (ACP) and papillary CP, and these subtypes differ in their genesis and age distribution [3, 4]. Although the histological grade of CP is usually low, patients with CP usually have significant morbidity and mortality rates, making it one of the most challenging to treat.
Fibrous dysplasia (FD) is a benign skeletal disorder caused by somatic GNAS-activating mutations [5]. FD accounts for 7% of benign bone tumors and most commonly affects the craniofacial regions [6, 7]. Patients may exhibit involvement of one bone (monostotic FD) or multiple bones (polyostotic FD) or they may have McCune-Albright syndrome (MAS), which is classically defined as the triad of polyostotic FD, café-au-lait skin macules, and endocrinopathies [8].
The disfigurement and thickening of cranial bone caused by coexistent cranial FD (CFD) makes the treatment of CP more complicated. The coexistence of CFD and CP has not yet been reported, and the pathogenesis of such coexistence remains unclear. In the present study, we present our experience with the successful treatment of five patients with concomitant CP and CFD, and discuss the optimal therapeutic strategy and potential links between these two lesions.
Materials and methods
Patients
We retrieved the medical records of all patients diagnosed with both CP and CFD in the Department of Neurosurgery, Beijing Tiantan Hospital, Capital Medical University from January 2003 to November 2021. A total of five patients were diagnosed with concomitant CP and CFD. Their demographic data, clinical manifestations, physical examination, surgery and treatment procedures, and outcomes were verified in the clinical medical reports. Patient follow-up data were obtained via outpatient and telephone follow-up. Endocrine data were measured by laboratory examination of hormone levels, and radiological data were obtained from the Picture Archiving and Communication System in our hospital. All patients underwent contrast-enhanced magnetic resonance (MR) imaging and computed tomography (CT) before and after treatment. This study was approved by the hospital ethics committee, and informed consent was obtained from all included patients at follow-up.
Immunohistochemistry and genetic analysis
Immunohistochemistry was performed to detect Gsα protein expression in CP specimens. The tissue sections were incubated with primary Gsα antibody (1:100, sc-365855, Santa Cruz). Each stained slide was individually reviewed and independently scored by two neuropathologists.
Genomic DNA was extracted from paraffin-embedded CP specimens using the Wizard Genomic DNA Purification Kit in accordance with the manufacturer’s instructions (Promega, Madison, WI).
Results
Patient characteristics
The clinical, therapeutic, pathological, and outcome characteristics of the five patients are shown in Table 1. The study cohort comprised four men and one woman (median age, 39 years; range, 29–55 years). The mean follow-up time was 57.2 months (range, 6–98 months). The preoperative symptoms included headache (n = 2), dizziness (n = 2), polyuria/polydipsia (n = 2), fatigue (n = 2), visual deterioration (n = 1), and hypogonadism (n = 1). One patient (patient 5) was incidentally diagnosed with CP and CFD during a physical check-up. The mean duration of symptoms was 8.2 months (range, 1.0–24.0 months). None of the patients had received previous radiotherapy or surgery. The bone most commonly affected by FD was the sphenoid bone (n = 4), and none of the patients had previously been diagnosed with FD.
Radiographic characteristics
Table 2 summarizes the radiological characteristics of all five patients. Preoperative and 3-month follow-up MR images are shown in Fig. 1. The CPs were located in the sellar/suprasellar region (n = 5). Four tumors were suprasellar with extension to the third ventricle (patients 1–4), one tumor was purely suprasellar (patient 3), and two tumors had extension into the sellar region (patients 1 and 2). In contrast to CP in the general adult population, the CPs coexisting with CFD were cystic in our study (n = 5). The CPs of patients 1, and 2 showed hypointense on T1-weighted imaging (T1WI) sequence. The CPs of patient 3 showed isointensity on T1WI sequence. The CPs of patients 4, and 5 showed hyperintensity on T1WI sequence. All CPs in the five showed hyperintensity on T2-weighted imaging. After Gd-DTPA administration, the tumors showed inhomogeneous nodular and ring enhancement (n = 3) and ring enhancement (n = 2) on contrast-enhanced T1WI. Additionally, hydrocephalus was found in one patient (patient 4). The CFDs were located in the sphenoid bone (n = 4) and maxilla (n = 1), appearing as thickening of the skull bone with a mixed signal (n = 4). As shown in Fig. 2, CT showed typical CFD in all five patients, comprising mixed radiolucent manifestations in four and “ground-glass” manifestations in one. The sphenoid bone was most commonly affected by FD (n = 4), followed by the maxilla (n = 2), ethmoid (n = 1), and clivus (n = 1). And CT revealed calcifications in CPs of all five patients, including the cyst wall (n = 3), intrasellar region (n = 1), and within the tumor (n = 1).

Pre- and postoperative magnetic resonance imaging of patients. The first line is the preoperative sagittal CE-T1WI MRI sequence. The second line is the sagittal CE-T1WI image obtained 3 months postoperatively, which is blank for patient 5 because the patient did not undergo surgery

Preoperative computed tomography (CT) findings of the five patients. The patient's head CT bone window scan showed significant abnormal skull fibrous dysplasia
Endocrine abnormalities
Three patients had preoperative endocrinological deficits, including hypothyroidism (n = 2) and hypogonadism (n = 1). Patients 2 and 3 had hyperprolactinemia that was thought to be a result of stimulation of the pituitary stalk. Postoperative hormone deficits included panhypopituitarism (n = 2) and hypothyroidism (n = 2). Transient and permanent diabetes insipidus were each observed in one patient.
Surgical approach and complications
Four of five patients underwent surgical resection of CPs (three transcranial and one trans-sphenoidal). Patient 5 refused surgical treatment because the lesion was found incidentally and there was no discomfort; the patient is still under close observation and regular follow-up. Patient 4 received a ventriculo-peritoneal shunt for hydrocephalus before tumor resection. Patient 3 underwent FD resection with optic nerve decompression; her vision was normal preoperatively, but was impaired postoperatively even after surgical repair. Extent of resection was confirmed both by intraoperative impression and immediate postoperative imaging in all cases. The extent of resection was considered gross total in two patients and subtotal in two patients.
Postoperative complications included panhypopituitarism (n = 2), diabetes insipidus (n = 2), visual loss (n = 1), hypothyroidism (n = 2), meningitis (n = 1), hypernatremia (n = 1), central fever (n = 1), and venous thrombus of the lower extremity (n = 1). No patient received any adjuvant therapy postoperatively.
Histopathologic findings
The CP subtype was ACP in three patients and unidentified in one (Fig. 3). Patients 2 and 3 underwent DNA sequencing of the CP specimens, but no GNAS mutations were detected (Additional file 1: Figure S1). However, immunohistochemical examinations revealed a strong positive Gsα expression (Fig. 4).

Pathological hematoxylin–eosin staining of the tumor specimens from patients 1–4. Three tumors were adamantinomatous craniopharyngiomas (ACPs) and one was a craniopharyngioma of an unknown subtype. A Histopathological examination reveals an ACP characterized by squamous epithelium arranged in a trabecular pattern as well as nodules of wet keratin in patient 1. B Postoperative pathology shows an ACP in patient 2. C Postoperative pathological examination shows a craniopharyngioma in patient 3; however, the specific subtype cannot be accurately identified. D An ACP that invaded and infiltrated the normal brain tissue in patient 4

Immunohistochemical analysis of the Gsα expression of the craniopharyngiomas in patients 1–3. Gsα is strongly positively expressed in all three patients
Follow-up and outcome
The median follow-up period was 57.2 months (range, 6–98 months). During the follow-up period, one patient (patient 1) experienced radiological recurrence and received long-term hormone replacement therapy for permanent panhypopituitarism. The CP in patient 4 was obviously calcified, closely related to blood vessels, and pathologically showed brain invasion (Additional file 2: Figure S2), making the resection very difficult. Postoperatively, the hypothalamic/pituitary function of patient 4 was severely damaged, causing water and electrolyte imbalance, lower extremity venous thrombosis, and eventually death from pulmonary embolism caused by thrombosis at 6 months postoperatively.
Discussion
To the best of our knowledge, this is the first reported case series of concomitant CP and CFD that highlights the diagnostic and management challenges. In addition to summarizing the clinicopathologic features, treatment modalities, and outcomes of these patients, we also reviewed the literature and discussed possible links between these two conditions based on genetic mutation detection and immunohistochemical studies.
The incidence of the coexistence of primary brain tumors of different pathologies is low and the most common association is meningioma and glioma, followed by meningioma and pituitary tumor, as these are the most common primary intracranial tumors [9]. The definitive pathogenesis of such coexistence of primary brain tumors remains unknown, except in patients with familial or hereditary diseases such as neurofibromatosis type 2 or multiple endocrine neoplasia type 1. Controversial hypotheses that have been advocated to explain the coexistence of primary brain tumors of different pathologies include pure coincidence, surgical trauma, radiation and/or chemical exposure, and genetic predisposition [10,11,12].
Because the sellar region undergoes a complex process of embryonic development, it is easy for primary intracranial tumors of different cell types to coexist in this region at the same time. However, it is extremely rare for CPs to coexist with other central nervous system neoplasms. To the best of our knowledge, there have only been a few reported cases of CPs coexisting with other central nervous system tumors, and the most common concomitant tumors are pituitary adenomas and meningiomas. The latest review reported that only 22 patients with CP (median age 47.0 years; 15 men, seven women) had an associated pituitary adenoma, and the CP was an ACP in 19 of these 22 patients [13]. Liu et al. reported eight patients (median age 62.0 years; five men, three women) with concomitant CP and meningioma, and five of these eight patients had ACPs [14]. Similarly, in our series (median age 39 years; five men, one woman), three of four operative patients had ACPs. These findings suggest that such coexistence is more likely to occur in adults with ACPs, with an obvious male predominance. In contrast, ACP that occurs alone has no sex orientation and is more common in adolescents [15]. Therefore, there may be some underlying reason for the coexistence of CPs with other central nervous system diseases rather than pure chance, and some factors may stimulate ACP cells to induce a secondary tumor formation, which is quite different from papillary CPs.
FD can also occur in association with other primary brain tumors under two conditions. The first is purely accidental and involves the coexistence of FD with the most common brain tumor (meningioma), which has been reported in only a few cases [16, 17]. The second is the coexistence of FD with pituitary adenoma (possibly in accordance with MAS), which has been reported in dozens of cases. As a postzygotic disease, the clinical spectrum and severity of FD varies depending on the timing of the mutation [18]. In addition to the classic triad, MAS may have a broad spectrum or atypical presentations [19]. Furthermore, MAS may be associated with gastrointestinal disease and breast cancer [20, 21]. Although no GNAS mutations were detected in the CP specimens in our series, we should not completely exclude the possibility of an atypical FD/MAS course, as immunohistochemical staining showed a high expression of Gsα protein in the three CP specimens. Further studies are needed to clarify this issue.
Most CPs or CFDs are diagnosed correctly based on the typical clinical symptoms and imaging features. However, it is difficult to accurately and promptly diagnose the coexistence of CP and CFD before surgery because the imaging features of FDs and CPs are variable and mutually influenced [22, 23]. Deformation and thickening of the skull base might make the situation more confusing, especially when the two lesions are adjacent to each other. However, it is important when devising a treatment strategy preoperatively to confirm which lesion is responsible for the clinical symptoms. In our case series, the four patients who underwent surgical treatment were accurately diagnosed preoperatively.
There are currently no established treatment strategies for concomitant CP and CFD. The current standard treatment for CP is surgical resection via a transcranial or extended endonasal endoscopic approach, followed by adjuvant radiation therapy if required for residual tumor [24]. Given the benign nature of CFD and the fact that the patients are adults, observation is considered adequate for asymptomatic quiescent FD [25]. However, all the primary symptoms (such as headache, fatigue, polydipsia/polyuria, and visual impairment) of all patients in our study were considered to be caused by CPs; thus, our management strategy was to surgically resect the CP and regularly monitor the FD. With the progress of microsurgery and endoscope technology, the surgical obstacles and risks caused by FD-related skull hyperplasia and deformation will be gradually reduced. Our results also show that there were no complications directly related to surgery, except for vision deterioration in patient 3 because of preventive decompression of the optic canal encased by FD. It should also be noted that FD radiotherapy may produce adverse effects of malignant transformation [26].
The outcome of collision tumors composed of CP and CFD are undefined because of the small number of reported cases. In our experience, the outcome of such tumors is similar to that of sporadic CPs, and long-term morbidity is associated with tumor- and treatment-related risk factors.
Our study has some limitations. First, due to the extreme rarity of concomitant CP and CFD, the total number of cases was small; more cases are needed to strengthen the reliability of data. Second, the mechanism of the coexistence of CP and CFD still needs further exploration and verification; technologies such as whole exome sequencing can be used to study the mechanism of the coexistent relationship between CP and CFD in future research.
Conclusion
We reported a very rare case series of concomitant CP and CFD and provided a detailed description of the clinicopathological features, treatment, and outcome. A comprehensive assessment of this rare and complicated situation is warranted to determine the appropriate treatment strategy. The clinical course of our cases show that an individual treatment plan for CP or CFD is required and can lead to a good outcome. The pathogenesis of the coexistence of the two tumor types is still undefined and requires investigation in further studies.
Availability of data and materials
The datasets generated and/or analysed during the current study are not publicly available due to individual privacy of the patients included but are available from the corresponding author on reasonable request.
References
Martinez-Barbera JP, Andoniadou CL. Biological behaviour of craniopharyngiomas. Neuroendocrinology. 2020;110(9–10):797–804.
Bunin GR, Surawicz TS, Witman PA, Preston-Martin S, Davis F, Bruner JM. The descriptive epidemiology of craniopharyngioma. J Neurosurg. 1998;89(4):547–51.
Sekine S, Shibata T, Kokubu A, Morishita Y, Noguchi M, Nakanishi Y, Sakamoto M, Hirohashi S. Craniopharyngiomas of adamantinomatous type harbor beta-catenin gene mutations. Am J Pathol. 2002;161(6):1997–2001.
Brastianos PK, Taylor-Weiner A, Manley PE, Jones RT, Dias-Santagata D, Thorner AR, Lawrence MS, Rodriguez FJ, Bernardo LA, Schubert L, Sunkavalli A, Shillingford N, Calicchio ML, Lidov HG, Taha H, Martinez-Lage M, Santi M, Storm PB, Lee JY, Palmer JN, Adappa ND, Scott RM, Dunn IF, Laws ER Jr, Stewart C, Ligon KL, Hoang MP, Van Hummelen P, Hahn WC, Louis DN, Resnick AC, Kieran MW, Getz G, Santagata S. Exome sequencing identifies BRAF mutations in papillary craniopharyngiomas. Nat Genet. 2014;46(2):161–5.
Weinstein LS. G(s)alpha mutations in fibrous dysplasia and McCune-Albright syndrome. J Bone Miner Res. 2006;21(Suppl 2):P120–4.
Diah E, Morris DE, Lo LJ, Chen YR. Cyst degeneration in craniofacial fibrous dysplasia: clinical presentation and management. J Neurosurg. 2007;107(3):504–8.
Riminucci M, Liu B, Corsi A, Shenker A, Spiegel AM, Robey PG, Bianco P. The histopathology of fibrous dysplasia of bone in patients with activating mutations of the Gs alpha gene: site-specific patterns and recurrent histological hallmarks. J Pathol. 1999;187(2):249–58.
Tafti D, Cecava ND, Fibrous Dysplasia, StatPearls, Treasure Island (FL), 2021.
Maiuri F, Cappabianca P, Iaconetta G, Esposito F, Messina A. Simultaneous presentation of meningiomas with other intracranial tumours. Br J Neurosurg. 2005;19(4):368–75.
Singh DK, Singh N, Parihar A, Singh R. Craniopharyngioma and epidermoid tumour in same child: a rare association, BMJ Case Rep 2013 (2013).
Takei H, Powell SZ. Tumor-to-tumor metastasis to the central nervous system. Neuropathology. 2009;29(3):303–8.
Amit A, Achawal S, Dorward N. Pituitary macro adenoma and vestibular schwannoma: a case report of dual intracranial pathologies. Br J Neurosurg. 2008;22(5):695–6.
Hasegawa H, Jentoft ME, Young WF Jr, Lakomkin N, Van Gompel JJ, Link MJ, Atkinson JL, Meyer FB. Collision of Craniopharyngioma and pituitary adenoma: comprehensive review of an extremely rare Sellar condition. World Neurosurg. 2021;149:e51–62.
Liu G, Su L, Xiang Y, Liu Y, Zhang S. Coexistence of craniopharyngioma and meningioma: two rare cases and literature review. Medicine (Baltimore). 2020;99(50):e23183.
Pekmezci M, Louie J, Gupta N, Bloomer MM, Tihan T. Clinicopathological characteristics of adamantinomatous and papillary craniopharyngiomas: University of California, San Francisco experience 1985–2005. Neurosurgery. 2010;67(5):1341–9 (discussion 1349).
Ghosal N, Furtado SV, Santosh V, Sridhar M, Hegde AS. Co-existing fibrous dysplasia and atypical lymphoplasmacyte-rich meningioma. Neuropathology. 2007;27(3):269–72.
Tasar M, Ors F, Yetiser S, Ugurel MS, Ucoz T. Multiple globoid meningiomas associated with craniomandibular fibrous dysplasia: case report. Clin Imaging. 2004;28(1):20–2.
Shenker A, Weinstein LS, Moran A, Pescovitz OH, Charest NJ, Boney CM, Van Wyk JJ, Merino MJ, Feuillan PP, Spiegel AM. Severe endocrine and nonendocrine manifestations of the McCune-Albright syndrome associated with activating mutations of stimulatory G protein GS. J Pediatr. 1993;123(4):509–18.
Spencer T, Pan KS, Collins MT, Boyce AM. The Clinical SPECTRUM of McCune–Albright syndrome and its management. Horm Res Paediatr. 2019;92(6):347–56.
Robinson C, Estrada A, Zaheer A, Singh VK, Wolfgang CL, Goggins MG, Hruban RH, Wood LD, Noe M, Montgomery EA, Guthrie LC, Lennon AM, Boyce AM, Collins MT. Clinical and radiographic gastrointestinal abnormalities in McCune-Albright syndrome. J Clin Endocrinol Metab. 2018;103(11):4293–303.
Majoor BC, Boyce AM, Bovee JV, Smit VT, Collins MT, Cleton-Jansen AM, Dekkers OM, Hamdy NA, Dijkstra PS, Appelman-Dijkstra NM. Increased risk of breast cancer at a young age in women with fibrous dysplasia. J Bone Miner Res. 2018;33(1):84–90.
Florenzano P, Pan KS, Brown SM, Paul SM, Kushner H, Guthrie LC, de Castro LF, Collins MT, Boyce AM. Age-related changes and effects of bisphosphonates on bone turnover and disease progression in fibrous dysplasia of bone. J Bone Miner Res. 2019;34(4):653–60.
Zada G, Lin N, Ojerholm E, Ramkissoon S, Laws ER. Craniopharyngioma and other cystic epithelial lesions of the sellar region: a review of clinical, imaging, and histopathological relationships. Neurosurg Focus. 2010;28(4):E4.
Jensterle M, Jazbinsek S, Bosnjak R, Popovic M, Zaletel LZ, Vesnaver TV, Kotnik BF, Kotnik P. Advances in the management of craniopharyngioma in children and adults. Radiol Oncol. 2019;53(4):388–96.
Lee JS, FitzGibbon EJ, Chen YR, Kim HJ, Lustig LR, Akintoye SO, Collins MT, Kaban LB. Clinical guidelines for the management of craniofacial fibrous dysplasia. Orphanet J Rare Dis. 2012;7(Suppl 1):S2.
Li Z, Wang Z, Qian H. Malignant transformation of craniofacial fibrous dysplasia: a systematic review of overall survival. Neurosurg Rev. 2020;43(3):911–21.
Acknowledgements
Not applicable.
Funding
No.
Author information
Authors and Affiliations
Contributions
YHF analyzed and interpreted the patient data and was a major contributor in writing the manuscript. LZ designed the study and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethic approval and consent to participate
This study was approved by Institutional Review Board of Beijing Tiantan Hospital, Capital Medical University. Due to the retrospective nature of our study, the board waived the need for written consent.
Consent for publication
All the patients included signed the consent for publication.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
DNA sequencing of GNAS in the craniopharyngiomas of patients 2 and 3. The mutations in codon 227 and 201 of GNAS are characteristic of fibrous dysplasia. No mutations were found in the craniopharyngioma samples of patients 2 and 3. The specimen from patient 1 could not be adequately assessed because the craniopharyngioma had little cystic wall tissue.
Additional file 2: Figure S2.
Head computed tomographic angiography of patient 4 shows that the tumor is closely associated with the blood vessels, increasing the difficulty of the operation
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fan, YH., Li, Z. Coexistence of craniopharyngioma and cranial fibrous dysplasia: a case series of clinicopathological study. Orphanet J Rare Dis 17, 126 (2022). https://doi.org/10.1186/s13023-022-02281-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-022-02281-1