
Abstract
Background
Calcium ions are involved in several human cellular processes including corticogenesis, transcription, and synaptogenesis. Nevertheless, the relationship between calcium channelopathies (CCs) and intellectual disability (ID)/global developmental delay (GDD) has been poorly investigated. We hypothesised that CCs play a major role in the development of ID/GDD and that both gain- and loss-of-function variants of calcium channel genes can induce ID/GDD. As a result, we performed a systematic review to investigate the contribution of CCs, potential mechanisms underlying their involvement in ID/GDD, advancements in cell and animal models, treatments, brain anomalies in patients with CCs, and the existing gaps in the knowledge. We performed a systematic search in PubMed, Embase, ClinVar, OMIM, ClinGen, Gene Reviews, DECIPHER and LOVD databases to search for articles/records published before March 2021. The following search strategies were employed: ID and calcium channel, mental retardation and calcium channel, GDD and calcium channel, developmental delay and calcium channel.
Main body
A total of 59 reports describing 159 cases were found in PubMed, Embase, ClinVar, and LOVD databases. Variations in ten calcium channel genes including CACNA1A, CACNA1C, CACNA1I, CACNA1H, CACNA1D, CACNA2D1, CACNA2D2, CACNA1E, CACNA1F, and CACNA1G were found to be associated with ID/GDD. Most variants exhibited gain-of-function effect. Severe to profound ID/GDD was observed more for the cases with gain-of-function variants as compared to those with loss-of-function. CACNA1E, CACNA1G, CACNA1F, CACNA2D2 and CACNA1A associated with more severe phenotype. Furthermore, 157 copy number variations (CNVs) spanning calcium genes were identified in DECIPHER database. The leading genes included CACNA1C, CACNA1A, and CACNA1E. Overall, the underlying mechanisms included gain- and/ or loss-of-function, alteration in kinetics (activation, inactivation) and dominant-negative effects of truncated forms of alpha1 subunits. Forty of the identified cases featured cerebellar atrophy. We identified only a few cell and animal studies that focused on the mechanisms of ID/GDD in relation to CCs. There is a scarcity of studies on treatment options for ID/GDD both in vivo and in vitro.
Conclusion
Our results suggest that CCs play a major role in ID/GDD. While both gain- and loss-of-function variants are associated with ID/GDD, the mechanisms underlying their involvement need further scrutiny.
Similar content being viewed by others
Introduction
Intellectual disability (ID) is characterized by limitations in both intellectual functioning and adaptive behaviour, which manifests before the age of 18 [1]. Global developmental delay (GDD) is defined as a profound delay of ≥ 2 standard deviations below the mean in ≥ 2 developmental domains [2]. GDD and ID represent clinically defined and recognized symptoms that are related but not necessarily synonymous. GDD is used for the children aged below 5 years while ID is for those aged 5 years and above [3]. Two-third of the children meriting the diagnosis of GDD in the preschool years, when reassessed later at school age, they continue to meet the diagnostic criteria for ID [4, 5]. Moreover, many older children diagnosed with ID currently, were initially diagnosed with GDD. Therefore, these entities have common features and both represent defects or disorders in learning [6]. Hence, they have a common approach in terms of evaluation and understanding of their etiology. The prevalence of ID/GDD world-wide is 10.37/1000 population according to meta-analysis [7]. It can occur in isolation or in combination with other neurological conditions such as epilepsy, autism spectrum disorders, attention deficit hyperactivity disorder, sensory impairment or congenital malformations. This condition incurs huge cost for the provision of adequate services in the society, and it is associated with stigma, mental and physical complications [7]. Notably, 30% of the cases with ID/GDD have comorbid mental health problems [5]. Neuropsychological tests are used to diagnose ID/GDD, however, the diagnosis is often initially formulated based on clinical judgment rather than on formal standardized assessments especially for the young patients [6] because those tests are limited by the age.
Approximately 65% of the cases with moderate-to-severe ID/GDD have genetic etiologies such as chromosome structural abnormalities, chromosome aneusomies, genomic disorders, and monogenic diseases [8,9,10,11]. With the introduction of next generation sequencing technologies, new ID/GDD genes are now being identified rapidly of which unpin the pathophysiology and provide new targets for treatment. Up to the present time, 450 genes have been implicated in ID/GDD; 400 genes for syndromic ID/GDD and 50 genes for non-syndromic ID [12]. Some of these genes participate in calcium signaling pathway. Prenatally, calcium-facilitated depolarization regulates neural proliferation, migration, and differentiation during the formation of the cerebral cortex [13]. Postnatally, calcium ions modulate cellular excitability by modelling synapses and sensory neural circuits [13]. Calcium ions also contribute to the membrane potential and function as an important signalling molecule [14]. Several processes in humans, including mitochondrial functions [15, 16], transcription, release of neurotransmitters, neurite outgrowth, and activation of some enzymes [14] depend on calcium ions. Noteworthy, mitochondria play a major role in regulating calcium-signalling processes [15, 16]. Besides, ATP is important for regulation of membrane excitability, synaptic transmission, transcription, and apoptosis [15].
Voltage-gated calcium channels belong to the family of 4-domain ion channels. Ten genes encode voltage-gated calcium channels. Each is categorized into one of two major groups: high voltage activated (HVA; R-, P/Q-, N- and L-types), and low voltage activated (LVA; T-type) [14, 17]. The HVA calcium channels are heteromultimeric protein complexes consisting of the pore-forming Cavα1, Cavδ, Cavβ and Cavα2δ subunits [14, 17]. By contrast, low voltage-activated calcium channels are comprised of only the Cavα1. Cav1, Cav2, and Cav3 are subfamilies [14, 17]. All Cavα1 subunits have four major transmembrane domains, and each consist of six membrane-spanning helices (termed S1–S6) [14]. Calcium ions enter into the cell when the channel is open and the opposite happens when it is closed. The movement of calcium ions in and out of the cell is regulated by calcium- and voltage-dependent inactivation of calcium ion channels [14]. S4 segment is positively charged, thus responsible for controlling voltage-dependent activation. The loop between S5 and S6 consists of negatively charged residues (glutamate or aspartate) that form the selectivity filter [14]. Large cytoplasmic linker connects the chief membrane domains regions and are within cytoplasmic N and C termini. Noteworthy, these cytoplasmic domain regions are important for modulating channel function via second messenger and protein–protein interaction [14, 17]. The most common type of mutations in this voltage-gated calcium channels is missense followed by deletion (according to this review). Consequently, mutations in S4 domain between S5 and S6, and in cytoplasmic linker that connects the chief membrane domains can alter the ability of the channels to regulate calcium influx/efflux. Overall, mutations in calcium channels can; (1) decrease channel function (loss-of function) or expression, (2) increase channel function (gain-of function) or expression, and (3) produce a disease without altering the electrophysiological properties of the channels [14]. Table 1 summarizes the general information related to the channels including the name of the gene, type of the current produced, neuronal localization, distribution, and pharmacology.
Calcium channelopathies are associated with several neurological disorders including ID/GDD, epilepsy, migraine, and ataxia [14]. Our hypothesis was that loss-of-function mutations are expected to induce ID/GDD, gain-of-function mutations are expected to enhance learning and memory. Similarly, gain-of-function and not loss-of-function mutations are expected to cause epilepsy. Surprisingly, studies showed that both gain- and loss-of-function mutations in genes encoding calcium channels could lead to epilepsy [18,19,20,21,22,23]. These mutations can dysregulate intrinsic gating processes and cell signalling pathways, which are involved in regulating channel activity and calcium trafficking through the plasma membrane [24]. As a result, these mutations can impair mitochondrial function, neurotransmitter release, and synaptic plasticity.
Our recent review revealed that potassium channelopathies contribute largely to the development of ID/GDD, and both gain- and loss-of-function variants were involved [25]. We observed that potassium channelopathies play an important role in the development of ID/GDD [25]. However, the role of calcium channelopathies in ID/GDD is unknown. Besides, it is unclear whether both gain- and loss-of-function mutations in calcium channel genes can lead to ID/GDD, and what are the possible underlying mechanisms.
Approximately 30% (range, 2–80%) of the cases with ID/GDD have nonspecific brain abnormalities according to magnetic resonance imaging (MRI), and computed tomography [26]. Nevertheless, the contribution of neuroimaging studies in understanding the underlying etiology of this condition range from 0.2 to 2.2% only [27]. Noteworthy, for some genes, brain changes are age-dependent, therefore, normal conventional MRI can be found at an early age [28], however, with an increase of age, malformations can be noticed [29, 30]. Cases with normal conventional MRI can have concealed malformations, which can be detected by advanced brain imaging methods. For instance, functional MRI (fMRI) can detect brain abnormalities that conventional MRI cannot spot in temporal lobe epilepsy and autism [31, 32]. Similarly, proton magnetic resonance spectroscopy (H-MRS) can detect abnormalities in cases diagnosed with neuronopathic Gaucher’s disease (NGD) [33]. Thanks to the advanced technology, the fMRI and H-MRS can also predict the severity of the disease even at an early age of which might be correlated with genotypes. Razek AA et al. revealed in their study that in children with NGD, there is a correlation between choline/creatine ratio and modified disease severity scoring system and genotypes [33]. Although abnormal standardized neuropsychological tests are enough to establish the diagnosis of ID/GDD, those tests can be affected by patient age. Fortunately, some neuroimaging studies can overcome the age limitation; alteration of metabolites revealed by H-MRS and diffusion-weighted magnetic resonance imaging (DWI) can foretell the severity of the cognitive dysfunction. In one study, neuropsychological test results correlated with apparent diffusion coefficient value and metabolic change for the children diagnosed with minimal hepatic encephalopathy with liver cirrhosis signifying that altered metabolic changes and cerebral edema were responsible for cognitive changes [34]. Whether there is a link between mutations in calcium channel genes, brain malformations, metabolic changes, and ID/GDD is yet to be found. Early detection of metabolic and other brain changes can aid in prevention of further cognitive decline.
Based on the important roles of calcium ions in the development of the neural cortex and signalling processes, we hypothesised that calcium channelopathies might contribute to the development of ID/GDD. To prove this, we listed all calcium channel gene variants previously reported in association with ID/GDD. Each mutation was considered in context of the associated degree of severity, current knowledge about possible mechanisms (gain- or loss-of-function), relevant advancements in animal models, treatments, and existing gaps in knowledge. We further aimed to investigate morphological brain anomalies associated with ID/GDD in patients with calcium channelopathies. We also discuss the possible relationship between calcium channelopathies, mitochondria dysfunction, epileptic discharges, cerebellar morphological changes, and ID/GDD. This review will help future studies on the mechanisms of ID/GDD to develop novel treatment strategies for this condition. Although previous narrative reviews summarised the relationship between calcium channelopathies and epilepsy as well as autism spectrum disorder [17, 35,36,37] to the best of our knowledge, this is the first systematic review to explore the relationship between calcium channelopathies and ID/GDD.
Methods
Literature search and selection
The review was conducted according to the Preferred Reporting Items for Systematic Reviews and Meta-Analyses statement [38]. An extensive literature search was conducted in PubMed, Embase, ClinVar, LOVD, OMIM, ClinGen, Gene Reviews and DECIPHER databases to find any relevant study/record published for all years until March 2021. The following search strategies were employed: ID and calcium channel, mental retardation and calcium channel, global developmental delay (GDD) and calcium channel, developmental delay (DD) and calcium channel (Additional file 1). The search strategies were created in consultation with a librarian and were used by three independent reviewers to select papers that met our review objectives.
This review included several kinds of clinical and epidemiological studies such as cohorts, case-controls, cross-sectionals, case series, and case reports. We selected studies that included cases with ID/GDD and calcium-channel gene variants. We excluded papers involving cases of ID/GDD with other types of channelopathies (sodium, potassium, and chloride) or other gene variants. Moreover, we did not include studies that documented patients with calcium channelopathies but no information related to ID/GDD. Lastly, we excluded all non-English papers, abstracts, reviews, patents, book chapters, and conference papers. The reference lists of retrieved studies were hand-searched to identify additional relevant reports.
Data extraction
Two independent reviewers screened the titles and abstracts of candidate papers and subsequently read the entire content of those that apparently met our inclusion criteria. The accuracy of the retrieved information was determined through discussion and consensus among the authors. We collected from articles that met inclusion criteria information related to calcium channel gene variants, phenotype associated with ID/GDD, degree of ID (mild, moderate, severe, and profound), electrophysiological results (gain- or loss-of-function), brain MRI results if reported, and the corresponding references. All identified candidate genes were further researched using the OMIM, ClinVar, Embase, LOVD, and PubMed databases to determine their function, expression profile, any related information gleaned from animal and functional cell studies, available treatments, and how they could contribute to possible mechanisms underlying ID/GDD.
Results
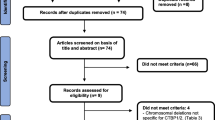
Five thousand eight hundred and seventy articles were retrieved from the initial search. Fifty-nine full-text articles met our inclusion criteria after we excluded non-English papers, abstracts, reviews, patents, book chapters, conference papers, and irrelevant papers on other channelopathies (Fig. 1). Thus, we identified 159 cases documented in 59 papers. Epilepsy was reported in 51.6% (82/159) of the cases. Ten calcium channelopathies related to ID/GDD were identified involving the following genes: CACNA1A [18, 19, 39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66], CACNA1C [67,68,69,70,71,72,73], CACNA1I [74], CACNA1H [75, 76], CACNA2D2 [77,78,79,80], CACNA2D1 [20, 81], CACNA1D [21, 22, 82,83,84], CACNA1E [85], CACNA1F [86], and CACNA1G [23, 87]. The underlying mechanisms included gain- and/ or loss-of-function, alteration in kinetics (activation, inactivation) and dominant-negative effects of truncated forms of alpha1 subunits.

A summary of the steps used for the literature selection
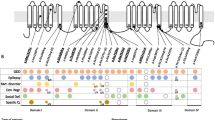
The most common affected calcium genes were CACNA1A, CACNA1E, CACNA1C and CACNA1D. Most variants exhibited gain-of-function effect. Severe to profound ID/GDD was observed more for the cases with gain-of-function variants as compared to those with loss-of-function variants. CACNA1E, CACNA1G, CACNA1F, CACNA2D2 and CACNA1A associated with more severe phenotype (Additional file 2: Table S1). Figures 2, 3, 4, 5, 6, 7 and 8 summarize the effects of genetic aberrations. The S4 transmembrane segment of domain III was the hotspot for CACNA1A-related ID/GDD (Fig. 2), domain I/domain II intracellular interlinker for CACNA1C (Fig. 3), S6 transmembrane segment of domain II for CACNA1E (Fig. 5), and the domain I/domain II intracellular interlinker for CACNA1D (Fig. 4). The detailed genotype–phenotype list can be found in Additional file 3.

Effects and locations of genetic aberrations for CACNA1A. There is a cluster of four critical residues in S4 transmembrane segment of domain III. Round yellow dots represent gain-of- function variants. Triangular yellow dots represent loss-of-function variants

Location of the identified CACNA1C amino acid substitutions. There is a cluster of four critical residues in the DI/D II intracellular interlinker. Round yellow dots represent gain-of- function variants

Location of the identified CACNA1D amino acid substitutions. There is a cluster of three critical residues in the domain I/domain II intracellular interlinker. Round yellow dots represent gain-of- function variants

Location of the identified CACNA1E amino acid substitutions. There is a cluster of five critical residues important for gating in S6 transmembrane segment of domain II. Round yellow dots represent gain-of- function variants

Location of the identified CACNA1G amino acid substitutions. Round yellow dots represent gain-of- function variants

Location of the identified CACNA1H amino acid substitutions. Round yellow dots represent gain-of- function variants

Location of the identified CACNA2D2 amino acid substitutions. Triangular yellow dots represent loss-of-function variants
Moreover, 157 copy number variations (CNVs) spanning calcium genes were identified in DECIPHER database but it was difficult to include them here due to consent issues. The leading CNVs were those encompassing CACNA1C, CACNA1A, CACNA1E, CACNA1F and CACNA1G. More details can be found in DECIPHER database (decipher@sanger.ac.uk).
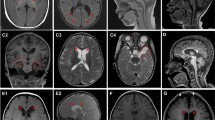
Cerebellar atrophy was reported in 25% (40) of the identified cases, cortical atrophy in 8.8% (14), optic atrophy in 1.3% (2), white matter changes in 5% (8), and other anomalies of the central nervous system in 5% (8). Normal brain magnetic resonance imaging findings accounted for 24.5% (39) of the identified cases and 34.6% (55) of the cases had no information related to brain imaging results. Two cases with gain-of-function variants who underwent muscle biopsy showed mitochondrial dysfunction: decreased mitochondrial complex I and III activity for the case with CACNA1C variant and partial deficits in complexes II and III for the case carrying CACNA1A variant (Additional file 2: Table S1). There is scarcity of cell and animal models for ID/GDD. Several modulators and pathways have been proposed for other calcium channel-related conditions. The commonest involved pathway is apoptotic followed by autophagic (Table 2, 3).
Discussion
Overall, this condition seems to be progressive, however, most primary authors provided less information on the course of the disease. Many of the reported cases with electrophysiological studies had gain-of- function variants. Severe to profound ID/GDD was more predominant for the cases with gain-of-function variants as compared to those with loss-of-function. CACNA1E, CACNA1G, CACNA1F, CACNA2D2 and CACNA1A associated with more severe phenotype. The possible reasons as why these genes associated with more severe phenotype include (1) the neuronal location of the genes; all of them are located in the pre-synaptic membrane, (2) brain distribution; most of them are distributed in the brain cortex and/or hippocampus and/or cerebellum, (3) function of the genes; they all regulate the release of neurotransmitter, and (4) the effect of the variants; most of the reported variants in these genes had gain-of-function property. This review has also revealed some hotspots for future research.
Summary of the clinical features
Calcium channels are widely spread in the human body (Table 1). Therefore, on top of brain, other organs such as eyes, heart, skeletal muscles, endocrine, and kidney can be affected too. In addition to ID/GDD, most cases with CACNA1A variants present with ataxia, epilepsy, attention deficit hyperactive disorder, autism spectrum disorder, dysmorphic features and eye abnormalities such as nystagmus, paroxysmal tonic upgaze, dysmetric saccades, blindness, myoclonus, ocular apraxia, exophthalmos and bilateral esotropia. Schizophrenia, anxiety, depression, hemiplegic migraine, coma, conductive deafness, vertigo attacks, dysarthria, tremors, athetosis, optic nerve glioma, abnormal behaviors such as aggression, sleeping problems can also be noticed. Cases carrying CACNA1C variants mostly present with Timothy syndrome, which is characterized by ID/GDD, autism, facial abnormalities, heart conditions such as atrioventricular block and patent ductus arteriosus, syndactyly and hypoglycemia. However, some cases can present with only ID/GDD, epilepsy, attention deficit hyperactive disorder plus congenital cardiac anomalies and dysmorphic features without autism. Cases with CACNA1E variants mostly present with profound ID/GDD accompanied with spastic dystonic quadriplegia, hypotonia, macrocephaly, and dystonia. ID/GDD, epilepsy, ataxia, and motor impairment, hypotonia, oculomotor apraxia, hyperopia, strabismus and multiple congenital anomalies can be seen for the cases carrying CACNA1G variants. Besides ID/GDD, epilepsy, autism, spastic quadriplegia, cortical blindness, lebers congenital amaurosis, klinefelters and retinitis pigmentosa, congenital nystagmus, rod cone dystrophy and myopia can be observed in those carrying CACNA1F mutations.
Cortical blindness, severe proximal muscular hypotonia, distal muscular hypertonia, epilepsy and ID/GDD can be noticed for the cases carrying CACNA1I variants. ID/GDD and ventral septal defect are the major clinical features for the cases with CACNA1H mutations. Dyskinesia such as choreiform movements, erratic limb movements, tremor, restlessness, sleep disturbance, dysmorphic features, oculo-motor apraxia, strabismus, nystagmus, axial and leg hypertonia, head tonic extension, brisk symmetric reflexes, hyperglycemia, glycosuria, and epilepsy are additional clinical features that can be observed for the cases with ID/GDD and yet carrying CACNA2D2 variants. Whereas, for the CACNA2D1 variants, epilepsy, autism, attention deficit hyperactive disorder, ataxia, facial dysmorphism, clinodactyly, brachymetacarpy, abnormal skin, short stature, transient diabetes with hyperinsulinemia, hearing impairment, aggressiveness, agitation, stereotypic hand movements, primary aldosteronism, heart defects and hypotonia can be seen in additional to ID/GDD.
Brain malformations including cerebellar, cortical and optic nerve atrophy were common in all ID-related calcium channelopathies. For two cases with gain-of-functions variants and underwent muscle biopsy showed evidence of mitochondrial dysfunction.
The pathomechanisms
In normal physiological conditions, calcium ions enter neurons via calcium channels (Cav1, Cav2 and Cav3). Most of the calcium ions enter mitochondria for ATP synthesis, which is crucial for synaptic plasticity. The remaining calcium ions in the cytosol stimulate transcription, facilitate release of neurotransmitters, promote neurite outgrowth, and activate some enzymes, which are important for synaptic plasticity.
Calcium effects on synapses
The calcium channel Cav1.2, which is encoded by CACNA1C, regulates gene expression by activating the cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB) and brain-derived neurotrophic factor (BDNF), both of which are essential for long-term potentiation [88, 89]. The increased expression of Cavα2δ subunit encoded by CACNA2D1 facilitates synapses to make more efficient use of calcium influx to activate neurotransmitter release [90]. In addition, this subunit interacts with big potassium (BK) channels and N-methyl-D-aspartate receptors (NMDARs) [91]. Cav2 channels including Cav2.1 and Cav2.3 encoded by CACNA1A and CACNA1E genes, respectively, form large signalling complexes in the presynaptic nerve terminal, which regulate the calcium entry and in turn facilitate neurotransmitter release and short-term plasticity [92, 93]. There are more than 100 proteins, which interact with Cav2.1 and Cav2.2 channels in presynaptic terminals and are involved in the release of neurotransmitters [92]. Cav1 channels including Cav1.2, Cav1.3, and Cav1.4 encoded by CACNA1C, CACNA1D, and CACNA1F, respectively, form signalling complexes in postsynaptic dendrites as well as dendritic spines, in which calcium entry induces long-term plasticity [92]. Cav3.1 channel that is encoded by CACNA1G is responsible for postsynaptic calcium signaling too and thus contribute to long-term potentiation [94, 95]. They may enhance dendritic depolarization or, on the other hand, can stimulate calcium-activated potassium currents, resulting in membrane hyperpolarizations [96]. The co-activation of Cav3.3 and GluN2B-containing NMDA receptors mediates long-term potentiation at thalamoreticular inputs [97].
Cav2.1 current facilitates short term synaptic plasticity through activation of neuronal calcium sensor proteins (CaS) [92, 98]. It is hypothesised that short-term plasticity is regulated by the SNARE complex that acts as the effector of synaptic vesicle exocytosis [99]. SNARE complex is comprised of SNAP-25, syntaxin, and synaptobrevin, also known as VAMP [100]. These proteins interact with the synaptic protein interaction (synprint) site present on the Cav2.1 calcium channels [101]. In addition, synaptotagmin 1 and 7 (calcium sensor protein), which regulates SNARE function has been implicated in short-term synaptic plasticity [102,103,104]. Cav3.2 current are responsible for retrieval of memory [105], and plays a major role in short-term plasticity [106]. Mutations in the identified calcium genes related to ID/GDD can affect neurotransmitter release [107,108,109,110,111].
Calcium effects on learning and long-term potentiation
Long-term potentiation occurs in two phases. Phase 1 includes increase in the expression of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPAR) while phase 2 involves activation of transcription and protein synthesis (formation of more AMPARs and dendrites for new synapse formation via secretion of growth factors) [112]. Glutamate released from the presynaptic membrane binds to the AMPARs and NMDARs, which are present on the postsynaptic membrane [112]. The binding of glutamate to AMPARs allows sodium ions influx leading to membrane depolarization [112]. When the membrane is depolarized, the magnesium blockage in NMDARs is removed, thereby, allowing the calcium ions to enter into the cell [112]. Calcium influx through NMDARs leads to calmodulin-dependent activation of Ca2+/calmodulin-dependent protein kinase (CaMKII)[112] and Kv4.2 internalization [113]. The activation of CaMKII leads to a rapid surge in the number of AMPARs at synapses [112]. In addition, CaMKII phosphorylates major and auxiliary subunits of AMPARs [114] including serine 831, the carboxyl-terminal of GluA1 [115] stargazin, and transmembrane AMPAR regulatory proteins (TARPs) [116]. CaMKII also plays a role in expanding and consolidating the synapse [114]. The CaMKII/NMDAR complex acts like a switch that regulates synaptic strength [117, 118]. Synaptic plasticity is chiefly facilitated by variations in the number of synaptic AMPARs, which are regulated by auxiliary subunits (stargazin and TARPs) that control channel gating and AMPAR trafficking [116]. Stargazin controls both AMPAR function and calcium channels [116, 119] and its dysfunction affects long-term potentiation [120].
Cav1 channels form signalling complexes in postsynaptic dendrites as well as in dendritic spines, in which calcium influx induces long-term potentiation [92]. CaMKII plays a role in activating the calcium-regulated protein kinase (CaMKIV), which in turn activates the transcription factor CREB [121]. The influx of calcium via Cav1.2 channels leads to transcription, translation, and consequently protein synthesis [122] leading to memory stabilisation [123]. Protein synthesis is very crucial for long-term memory as its inhibitors such as anisomycin, puromycin, acetoxycycloheximide, and cycloheximide were shown to affect long term potential and not initial attainment of task [124]. Beta-2 adrenergic receptors interact with Cav1.2 channels to control the long-term postsynaptic plasticity and the activity of the calcium channel [125, 126]. The Cav3.3 interacts with GluN2B-containing NMDA receptors to induce long-term potentiation [97].
Calcium channels and mitochondria
In this study, we found the underlying mechanisms for the reported variants included gain- and/ or loss-of-function, alteration in kinetics (activation, inactivation) and dominant-negative effects of truncated forms of alpha1 subunits. The contribution of gain- and loss-of-function variants to ID might be attributed to the mitochondrial dysfunction [127]. Mitochondria not only depend on calcium releasing sites such as endoplasmic reticulum (ER) but also interact with calcium channels (Cav2) present on the plasma membrane. Calcium ions can enter through the calcium channels when there is depletion of ER calcium stores [16]. When the cell is activated, there is high accumulation of calcium ions in the mitochondria as compared to the cytosol [15].
Despite the fact that mitochondria are responsible for production of energy, they also regulate cellular signalling (calcium signalling), cell defence, and cell death [16]. Besides, mitochondrial calcium regulates calcium currents in the cell for signalling process, ATP synthesis, and initiation of cell death. Thus, mitochondrial function and calcium homeostasis are entwined processes that regulate each other [16]. Noteworthy, ATP is important for regulation of membrane excitability, synaptic transmission, transcription, and apoptosis [15]. Neurons require ATP to carry out their activity at synapse and the mitochondrial calcium uniporter (MCU) present in these cells allows calcium ions to enter neurons for different activities [15]. Calcium ions can activate the release of BDNF for growth and repair of neurons [128]. Mitochondria can affect synaptic plasticity in several ways; in conjunction with BDNF, they can supply ATP for synaptic connections and they can prune those connections away [128]. Furthermore, mitochondria can also produce neurosteroids that can determine how calcium ions enter the neuron [129].
Calcium overload in the mitochondria activates apoptotic cascade leading to cell death [130, 131]. Figure 9 summarizes the mechanism. Conversely, low levels of calcium in mitochondria can induce autophagy [132]. Figure 10 summarizes the mechanism. Moreover, excessive accumulation of calcium ions in the mitochondria of the neuron can lead to excessive neuronal firing like in epileptic seizure, thereby, leading to neuronal death [129]. Mitochondrial calcium buffering is very important and its impairment can cause several diseases. For example, calcium overload due to failure of the mitochondrial buffering system in skeletal muscles leads to amyotrophic lateral sclerosis [133]. In summary, excitable cells including neurons require proper regulation of calcium and mitochondrial homeostasis. Whereas diminished mitochondrial calcium influx can result in loss of neuronal function, excessive mitochondrial calcium can induce neuronal damage and death. Mitochondria supply energy for proper brain functioning, enhancing synaptic plasticity, production of hormones and signalling molecules, and regulating neurotransmitters, and its dysfunction can lead to several diseases including ID [129]. Thus, we speculate that gain-of-function variants can induce neuronal apoptosis (Fig. 9), whereas, loss-of-function mutations can activate autophagy (Fig. 10) resulting in ID. Nevertheless, this hypothesis needs further studies for confirmation. Noteworthy, for the few cases that received muscle biopsy tests, evidence of mitochondrial dysfunction was noticed for two cases carrying gain-of-function variants; decreased mitochondrial complex I and III activity for the case with CACNA1C variant [73] and partial deficits in complexes II and III for the case carrying CACNA1A variant [63].

The mechanism of how gain-of-function variants can lead to ID/GDD. Calcium ions can enter into neuronal cell via Cav1.2, Cav1.4, Cavα2δ, Cav2.1, Cav2.2, Cav2.3, Cav3.1, Cav3.2 and Cav3.3. In normal physiology, some of the calcium ions go to the nucleus to initiate gene transcription, translation and protein synthesis essential for learning and memory, some go to mitochondria for ATP synthesis (essential for learning and memory) and some to the endoplasmic reticulum (ER) for storage. Gain-of- function variants can allow excessive influx of calcium ions inside the cells. This will reduce the amount of ATP production while contributing to the accumulation of reactive oxygen species (ROS) and release of cytochrome C that induces apoptosis of neuronal cell. Many red and blue solid circles stand for high calcium levels

A summary of how loss-of- function variants can lead to autophagy. Calcium ions can enter into neuronal cell via Cav1.2, Cav1.4, Cavα2δ, Cav2.1, Cav2.2, Cav2.3, Cav3.1, Cav3.2 and Cav3.3. In normal physiology, some of the calcium ions go to the nucleus to initiate gene transcription, translation and protein synthesis essential for learning and memory, some go to mitochondria for ATP synthesis (essential for learning and memory) and some to the endoplasmic reticulum for storage. Calcium stored in the endoplasmic reticulum (ER) is used when there is minimal/no influx of calcium ions inside the cells. Autophagy occurs when there is metabolic stress such as low ATP and nutrient starvation. Low levels of calcium ions inside the neuronal cell being due to loss-of- function of calcium channels or due to depletion in ER can activate autophagy pathway. Low calcium entrance in the mitochondria will lead to low production of ATP which will activate the AMP-activated protein kinase (AMPK, a sensor of energy levels) and mTOR complex 1 (mTORC1) which in turn induce autophagy. Likewise, low calcium levels from the ER can activate calmodulin-dependent protein kinase kinase β (CaMKKβ) and then AMPK leading to autophagy. Dotted arrows signify low levels. Few red solid circles stand for low calcium ions levels
Excessive calcium influx through NMDARs can induce neurotoxicity via activation of neuronal nitric oxide synthase (nNOS) [127] and neuronal NADPH oxidase-2 (NOX2) pathways [134]. Excessive mitochondrial accumulation of calcium ions in neurons can lead to excessive neuronal hyperexcitabilty, similar to that observed in epilepsy, thus, leading to neuronal death [129].
Calcium channels and epilepsy
We found that more than half of the reported cases had concomitant epilepsy or epileptic encephalopathy. It is unclear whether epileptic activity played a role in the development of ID. Rodent studies have shown that epileptic encephalopathies, frequent seizures, and/or interictal epileptic discharges can lead to synaptic reorganization, abnormal neurogenesis, or disruption of the developing neural circuits, which can cause ID [135]. However, some of the underlying aetiologies for epilepsy can also cause ID independently [135, 136]. Noteworthy, some of the antiepileptic drugs can exacerbate ID [135, 136]. A previous study on sodium channelopathy (SCN1A alleles) revealed that seizure frequency and electroencephalography abnormalities do not correlate with the degree of ID and behavioural disturbances [137]. Besides, Scn1a knockout mice demonstrated cognitive impairment even without seizures [138]. However, no similar study has been performed on calcium channelopathies. Based on our present knowledge about the existing cerebro-cerebellar circuits [139] we speculate that ID and EP might occur independently. Calcium channelopathies can impair the development of the cerebellum leading to abnormalities in different cerebro-cerebellar circuits accounting for ID and EP separately.
Calcium channels and cerebellum
The cerebellum plays a major role in the planning and execution of movement as well as in language and attention [140]. In our study, cerebellar atrophy was observed in 25% of the retrieved cases. There are important cerebro-cerebellar circuits responsible for learning and memory [139]. Calcium channels are crucial for the development of the brain including the cerebellum [23]. Therefore, calcium channelopathies can cause cerebellar morphological changes leading to several neurodevelopmental disorders including ID, autism, epilepsy, and attention deficit hyperactivity disorder [139, 141]. Abnormal cerebellar development and/or early cerebellar damage can affect behaviour via the closed-loop circuits connecting the cerebellum with multiple areas in the cerebral cortex. Behavioural changes depend on the affected cerebro-cerebellar circuits [139]. Six out of 12 cases with CACNA1A variants and ID showed statistically significant association with cerebellar atrophy according to one study [45]. Besides, only two cases in that study presented with epilepsy, thus, questioning the role of epilepsy in developing ID for the cases with CACNA1A variants [45]. In another study, abnormal cerebellar development caused by CACNA1G alleles was hypothesised as the possible cause of cognitive impairment [23]. A rodent study demonstrated the participation of the cerebellum in cognitive function [142]. Our review consolidates evidence for the theory that the cerebellum might be involved in learning and memory, thus, supporting our speculation that calcium channelopathies lead to cerebellar atrophy that can cause ID via abnormal cerebro-cerebellar circuits.
Noteworthy, conventional MRI was used as a method to detect abnormalities for the identified cases. Thus, there is a possibility that other cases were missed due to the limitation of this modality. Advanced imaging methods including the fMRI and H-MRS revealed brain abnormalities (including metabolic changes) that could not be detected with conventional MRI for the cases diagnosed with temporal lobe epilepsy and autism [31, 32], NGD [33], and minimal hepatic encephalopathy with liver cirrhosis [34]. Besides, there was a correlation between choline/creatine ratio and cognitive deficits and genotypes for the cases diagnosed with NGD [33]. Furthermore, abnormal cognitive results correlated with apparent diffusion coefficient value and metabolic changes for the children diagnosed with minimal hepatic encephalopathy with liver cirrhosis signifying that altered metabolic changes and cerebral edema were responsible for cognitive changes [34]. Consequently, fMRI and H-MRS imaging modalities are recommended for the ID/GDD cases. Early detection of metabolic changes can aid clinicians to minimize cognitive decline.
Available animal models, modulators, and pathways for ID/GDD related to calcium channels defects
A comprehensive review of literature was also carried out for animal models of all reported calcium channel genes related to ID. Unfortunately, only a few calcium channel genes have animal models for ID. Many authors focused more to study migraine, ataxia and epilepsy through animal models. There are several interventions used for aforementioned conditions with available animal models. For instance, seizures were inhibited by ethosuximide and valproic acid in Cacna1a Tottering-6j mice [215] and Cacna1a-mutant GRY rat [143]. Acetazolamide could abolish stress-induced ataxia in Cacna1a mice [144] but had no effect on HEK cells carrying mutation for episodic ataxia type 2 [145]. Cav1.2 current could be inhibited by nifedipine [146], diltiazem [147] and miR-135b [148]. Niflumic acid could block Cav3.3 current [149] while 6-prenylnaringenin could block Cav3.2 current [150]. Gabapentin is a ligand of Cav1.3 [151]. The commonest affected pathway for the available studies is the apoptotic pathway [152,153,154,155] followed by oxidative stress pathway [156, 157] and autophagy [158]. More details can be found in Table 2. Therefore, we argue future studies on ID/GDD involving animal models to be conducted. Moreover, studies focused on treatments aiming to mitigate cognitive impairment should be carried out.
Available other functional models, modulators and pathways for ID/GDD related to calcium channels defects
Upon intensive review of literature, we found that tsA-201, HEK293T and mouse chromaffin cells are being used to study ID, epilepsy and autism [74, 82, 84, 85]. Besides, SH-SY5Y human neuroblastoma cells was used for familial hemiplegic migraine type 1 and episodic ataxia [159, 160] and progressive myoclonic epilepsy [161]. Human induced pluripotent stem cell-derived cardiomyocyte (hiPSC-CM) was used for long-QT syndrome phenotype [162]. Induced pluripotent stem cell (iPSC) knockout resource was used for autism spectrum disorder [163]. COS-7 cells was used for spinocerebellar ataxia type 2 [164]. Modulators for Cav2.1 current in functional models include miR-3191-5p [165], SIS-RNAi [160] and barbiturate pentobarbital [166]. Cav1.2 current can be modulated by micro-RNA-137, -221, -153, -103 and -222 [167,168,169,170], nifedipine and benidipine hydrochloride [171], rituximab [172], bay K8644 [173], estradiol [148], roscovitine [174], stac2 and stac3 [175] and azelnidipine [176]. Sipatrigine, eugenol, and lamotrigine can block Cav2.3 current [177, 178]. Endostatin, zonisamide, clozapine, roscovitine, mibefradil, iron and zinc can block Cav3.1 channels [179,180,181,182,183,184,185]. Dearomatized isoprenylated acylphloroglucinol and monoterpenoid, hypatone A could rescue pathological gating properties for spinocerebellar ataxia 42 [186]. KYS-05090S could block Cav3.2 current [187]. Cav1.3 current could be blocked by microRNA-107 [188, 189] and amlodipine [190]. The commonest affected pathway is apoptotic pathway [127, 155, 161, 172, 191,192,193,194,195,196] followed by autophagic pathway [158, 197,198,199], Ras/Raf/MEK-ERK signaling pathway [200,201,202,203] and Wnt pathway [173]. Further details can be found in Table 3.
Evaluation and investigations
The aforementioned clinical features (multisystem abnormalities) should guide clinicians in suspecting mutations in calcium genes. In addition, progressive cerebellar, cerebral and optic atrophy in the brain imaging are important clues. Therefore, we recommend brain imaging and next generation sequencing diagnostic methods to be used whenever there is a suspicion. Since there is an evidence of many CNVs encompassing calcium genes and yet relate to ID/GDD, we suggest microarray tests to be considered for the cases that present with multiple congenital anomalies. We recommend intensive metabolic tests in urine, blood and cerebrospinal fluid. Those tests can check the levels of pyruvate, lactic acid and others as per consensus based expert recommendations for evaluation of mitochondrial disease [204]. Molecular genetic tests for mitochondria can be carried out but muscle biopsy is recommended even if the results becomes negative. Tests that can detect biochemical signs of neurotransmitter abnormalities are recommended. Advanced neuroimaging modalities including fMRI and H-MRS are recommended.
Treatments
Studies on treatment focusing on calcium channelopathies for ID/GDD are not available. Available studies focused more on epilepsy, ataxia and migraine, and those drugs do not have beneficial effects on cognition. Verapamil and acetazolamide are good for migraine [205]. Acetazolamide has been reported to be effective for ataxia to some cases [206] and ineffective for others [207]. Ethosuximide and valproic acid are effective for absence epilepsy [208]. Otherwise, there are some treatable ID/GDD, especially those caused by inborn errors of metabolisms (n = 81). These include 19 disorders of organic acids, lysosomes (n = 12), amino acids (n = 12), hyperhomocysteinemia (n = 7), vitamins/co-factors (n = 8), urea cycle (n = 7), neurotransmission (n = 7), creatine (n = 3), cholesterol and bile acid (n = 2), fatty aldehydes (n = 1), glucose homeostasis and transport (n = 2), metals (n = 3), mitochondria (n = 2), peroxisomes (n = 1), and pyrimidines (n = 2)[209]. Diazoxide choline controlled-release tablets have been reported to be useful in controlling hyperphagia, obesity and aggressive behaviors in cases with Prader-Willi syndrome [210, 211]. This drug has also been reported to prevent the aggravation of the pre-existing ID/GDD for the cases with syndromes known to be accompanied with hyperinsulinaemic hypoglycaemia such as Beckwith-Wiedemann, Sotos, Kabuki and Turner [212]. The early initiation of the diazoxide within 3 months of the onset of symptoms is associated with normal intelligence [213].
Intervention strategies
Upcoming studies should focus on developing animal models for ID/GDD aiming to explore the possible underlying mechanisms for ID/GDD and possible treatment options. If possible, future studies should focus on identifying the effect of calcium blockers and openers both in vivo and in vitro. Additionally, future studies should focus on exploring the relationship between calcium channelopathies, mitochondria, and ID/GDD as well as the role of cerebellar morphological changes in ID/GDD. Lastly, future studies can explore whether epileptic activity in calcium channelopathies can cause ID/GDD or whether ID/GDD and epilepsy occur independently. Expanding the understanding of mechanisms underlying the development of ID/GDD will help to improve the treatment strategies for ID/GDD.
Comparison of our review with other reviews
Previous narrative reviews summarised the relationship between calcium channelopathies and epilepsy as well as autism spectrum disorder [17, 35,36,37]. To the best of our knowledge, this is the first systematic review to explore the relationship between calcium channelopathies and ID/GDD. Our review has revealed variations in ten genes that relate to ID/GDD including CACNA1A, CACNA1C, CACNA1I, CACNA1H, CACNA1D, CACNA2D1, CACNA2D2, CACNA1E, CACNA1F, and CACNA1G. Most variants exhibited gain-of-function effect. Severe to profound ID/GDD was observed more for the cases with gain-of-function variants as compared to those with loss-of-function. CACNA1E, CACNA1G, CACNA1F, CACNA2D2 and CACNA1A associated with more severe phenotype. In another review, both gain- and loss-of-function variants in CACNA1A, gain-of-function variants in CACNA1H, and variants in CACNA1G were linked to epilepsy [35, 36, 214]. Calcium overload resulting to mitochondrial dysfunction, oxidative stress, and cell damage was concluded as a possible pathomechanism important for the development of acquired epilepsies [36]. In the review regarding autism spectrum disorder, CACNA1A, CACNA1B, and CACNA1C (gain-of-function), CACNA1D (gain-of-function), CACNA1E and CACNA1F (gain-of-function), CACNA1G and CACNA1H (loss-of-function), CACNA1I, CACNB1, and CACNB2 (gain-of-function effect) as well as CACNA2D3 and CACNA2D4 (loss-of-function effect) were reported as candidate genes [37]. Our review has highlighted CACNA1C, CACNA1F, CACNA1I, CACNA2D1 and CACNA2D2 as additional genes for epilepsy, and CACNA2D1 for autism spectrum disorder. Furthermore, our review has revealed CACNA1A, CACNA1C and CACNA2D1 as the candidate genes for attention deficit hyperactive disorder.
Merits of the study
This review has revealed that calcium channelopathies contribute to the development of ID/GDD. Variations in 10 genes that relate to ID/GDD including CACNA1A, CACNA1C, CACNA1I, CACNA1H, CACNA1D, CACNA2D1, CACNA2D2, CACNA1E, CACNA1F, and CACNA1G were found, and most variants exhibited gain-of-function effect. It has unveiled that severe to profound ID/GDD is observed more for the cases with gain-of-function variants as compared to those with loss-of-function. Notably, CACNA1E, CACNA1G, CACNA1F, CACNA2D2 and CACNA1A correlated with more severe phenotype. Our review has further revealed variants in CACNA1C, CACNA1F, CACNA1I, CACNA2D1 and CACNA2D2 as additional genes related to epilepsy. Besides, CACNA2D1 is related to autism spectrum disorder while CACNA1A, CACNA1C and CACNA2D1 are candidate genes for attention deficit hyperactive disorder. Our study has showed the existence of the relationship between calcium channelopathies, mitochondria dysfunction, cerebellar morphological changes, and ID/GDD. We have summarized the information related to available animal, and functional cell models, modulators, and pathways, evaluation, investigations, treatments and intervention strategies for ID/GDD related to calcium channels defects. Our review will help future studies on the mechanisms of ID/GDD to develop novel treatment strategies for this condition.
Study limitations
Our study has several limitations. We could not discuss the relationship between ID/GDD and other channelopathies (sodium, potassium and chloride) in detail, as the breadth of content that a review of that scope would provide exceeds the capacity of one article. There is no advanced neuroimaging modalities including fMRI and H-MRS were done for the reported cases; therefore, it is difficult to comment on brain metabolic changes.
Conclusions
In summary, calcium channelopathies can cause ID/GDD. There is a scarcity of animal studies on the mechanisms of ID/GDD in relation to calcium channelopathies. Studies on treatment options for cognitive impairment are lacking. The underlying mechanisms for the reported variants include gain- and/ or loss-of-function, alteration in kinetics (activation, inactivation) and dominant-negative effects of truncated forms of alpha1 subunits. While both gain- and loss-of-function variants are associated with ID/GDD, the mechanisms underlying their involvement are unclear.
Availability of data and materials
All data generated or analysed during this study are included in this published article [and its supplementary information files.
Abbreviations
- cAMP:
-
Cyclic adenosine monophosphate
- ID:
-
Intellectual disability
- GDD:
-
Global developmental delay
- HVA:
-
High voltage activated
- LVA:
-
Low voltage activated
- CACNA1A :
-
Calcium voltage-gated channel subunit alpha1 A
- CACNA1B :
-
Calcium voltage-gated channel subunit alpha1 B
- CACNB1:
-
Calcium voltage-gated channel auxiliary subunit beta 1
- CACNB2:
-
Calcium voltage-gated channel auxiliary subunit beta 1
- CACNA2D3:
-
Calcium voltage-gated channel auxiliary subunit alpha2delta 3
- CACNA2D4:
-
Calcium voltage-gated channel auxiliary subunit alpha2delta 4
- CACNA1C :
-
Calcium voltage-gated channel subunit alpha1 C
- CACNA1D :
-
Calcium voltage-gated channel subunit alpha1 D
- CACNA1E :
-
Calcium voltage-gated channel subunit alpha1 E
- CACNA1F :
-
Calcium voltage-gated channel subunit alpha1 F
- CACNA1H :
-
Calcium voltage-gated channel subunit alpha1 H
- CACNA1I :
-
Calcium voltage-gated channel subunit alpha1 I
- CACNA1G:
-
Calcium voltage-gated channel subunit alpha1
- CACNA2D1 :
-
Calcium voltage-gated channel auxiliary subunit alpha2delta 1
- CACNA2D2 :
-
Calcium voltage-gated channel auxiliary subunit alpha2delta 2
- CREB:
-
Cyclic adenosine monophosphate (cAMP) response element-binding protein
- BDNF:
-
Brain-derived neurotrophic factor
- BK:
-
Big potassium
- NMDARs:
-
N-methyl-D-aspartate receptors
- AMPAR:
-
Alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors
- CaMKII:
-
Calmodulin-dependent activation of Ca2+/calmodulin-dependent protein kinase II
- nNOS:
-
Neuronal nitric oxide synthase
- NOX2:
-
Neuronal NADPH oxidase-2
- MCU:
-
Mitochondrial calcium uniporter
References
van Bokhoven H. Genetic and epigenetic networks in intellectual disabilities. Annu Rev Genet. 2011;45:81–104.
Shevell MI. Present conceptualization of early childhood neurodevelopmental disabilities. J Child Neurol. 2010;25:120–6.
Shevell M, Ashwal S, Donley D, Flint J, Gingold M, Hirtz D, et al. Practice parameter: evaluation of the child with global developmental delay: report of the Quality Standards Subcommittee of the American Academy of Neurology and The Practice Committee of the Child Neurology Society. Neurology. 2003;60:367–80.
Petersen MC, Kube DA, Palmer FB. Classification of developmental delays. Semin Pediatr Neurol. 1998;5:2–14.
Purugganan O. Intellectual disabilities. Pediatr Rev. 2018;39:299–309.
Shevell M. Global developmental delay and mental retardation or intellectual disability: conceptualization, evaluation, and etiology. Pediatr Clin N Am. 2008;55(1071–84):xi.
Maulik PK, Mascarenhas MN, Mathers CD, Dua T, Saxena S. Prevalence of intellectual disability: a meta-analysis of population-based studies. Res Dev Disabil. 2011;32:419–36.
Chelly J, Khelfaoui M, Francis F, Cherif B, Bienvenu T. Genetics and pathophysiology of mental retardation. Eur J Hum Genet. 2006;14:701–13.
Chiurazzi P, Schwartz CE, Gecz J, Neri G. XLMR genes: update 2007. Eur J Hum Genet. 2008;16:422–34.
Patel DR, Greydanus DE, Calles JLJ, Pratt HD. Developmental disabilities across the lifespan. Dis Mon. 2010;56:304–97.
Rauch A, Hoyer J, Guth S, Zweier C, Kraus C, Becker C, et al. Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am J Med Genet A. 2006;140:2063–74.
Srour M, Shevell M. Genetics and the investigation of developmental delay/intellectual disability. Arch Dis Child. 2014;99:386–9.
Smith RS, Walsh CA. Ion channel functions in early brain development. Trends Neurosci. 2020;43:103–14.
Simms BA, Zamponi GW. Neuronal voltage-gated calcium channels: structure, function, and dysfunction. Neuron. 2014;82:24–45.
Rossi A, Pizzo P, Filadi R. Calcium, mitochondria and cell metabolism: a functional triangle in bioenergetics. Biochim Biophys Acta Mol Cell Res. 2019;1866:1068–78.
Bravo-Sagua R, Parra V, López-Crisosto C, Díaz P, Quest AFG, Lavandero S. Calcium transport and signaling in mitochondria. Compr Physiol. 2017;7:623–34.
Rajakulendran S, Hanna MG. The role of calcium channels in epilepsy. Cold Spring Harb Perspect Med. 2016;6:a022723.
Damaj L, Lupien-Meilleur A, Lortie A, Riou E, Ospina LH, Gagnon L, et al. CACNA1A haploinsufficiency causes cognitive impairment, autism and epileptic encephalopathy with mild cerebellar symptoms. Eur J Hum Genet. 2015;23:1505–12.
Jiang X, Raju PK, D’Avanzo N, Lachance M, Pepin J, Dubeau F, et al. Both gain-of-function and loss-of-function de novo CACNA1A mutations cause severe developmental epileptic encephalopathies in the spectrum of Lennox-Gastaut syndrome. Epilepsia. 2019;60:1881–94.
Vergult S, Dheedene A, Meurs A, Faes F, Isidor B, Janssens S, et al. Genomic aberrations of the CACNA2D1 gene in three patients with epilepsy and intellectual disability. Eur J Hum Genet. 2015;23:628–32.
Garza-Lopez E, Lopez JA, Hagen J, Sheffer R, Meiner V, Lee A. Role of a conserved glutamine in the function of voltage-gated Ca(2+) channels revealed by a mutation in human CACNA1D. J Biol Chem. 2018;293:14444–54.
Pinggera A, Mackenroth L, Rump A, Schallner J, Beleggia F, Wollnik B, et al. New gain-of-function mutation shows CACNA1D as recurrently mutated gene in autism spectrum disorders and epilepsy. Hum Mol Genet. 2017;26:2923–32.
Chemin J, Siquier-Pernet K, Nicouleau M, Barcia G, Ahmad A, Medina-Cano D, et al. De novo mutation screening in childhood-onset cerebellar atrophy identifies gain-of-function mutations in the CACNA1G calcium channel gene. Brain. 2018;141:1998–2013.
Simms BA, Zamponi GW. Trafficking and stability of voltage-gated calcium channels. Cell Mol Life Sci Switzerland. 2012;69:843–56.
Kessi M, Chen B, Peng J, Tang Y, Olatoutou E, He F, et al. Intellectual disability and potassium channelopathies: a systematic review [Internet]. Front Genet. 2020;614. Available from: https://www.frontiersin.org/article/https://doi.org/10.3389/fgene.2020.00614.
Bélanger SA, Caron J. Evaluation of the child with global developmental delay and intellectual disability. Paediatr Child Health. 2018;23:403–19.
Moeschler JB, Shevell M. Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics. 2014;134:e903–18.
Saitsu H, Kato M, Mizuguchi T, Hamada K, Osaka H, Tohyama J, et al. De novo mutations in the gene encoding STXBP1 (MUNC18-1) cause early infantile epileptic encephalopathy. Nat Genet. 2008;40:782–8.
Uddin M, Woodbury-Smith M, Chan A, Brunga L, Lamoureux S, Pellecchia G, et al. Germline and somatic mutations in STXBP1 with diverse neurodevelopmental phenotypes. Neurol Genet. 2017;3:e199.
Weckhuysen S, Holmgren P, Hendrickx R, Jansen AC, Hasaerts D, Dielman C, et al. Reduction of seizure frequency after epilepsy surgery in a patient with STXBP1 encephalopathy and clinical description of six novel mutation carriers. Epilepsia. 2013;54:e74-80.
Mankinen K, Ipatti P, Harila M, Nikkinen J, Paakki J-J, Rytky S, et al. Reading, listening and memory-related brain activity in children with early-stage temporal lobe epilepsy of unknown cause-an fMRI study. Eur J Paediatr Neurol EJPN Off J Eur Paediatr Neurol Soc. 2015;19:561–71.
McKechanie AG, Campbell S, Eley SEA, Stanfield AC. Autism in fragile X syndrome; A functional MRI study of facial emotion-processing. Genes (Basel). 2019;10:1052.
Razek AAKA, Abdalla A, Gaber NA, Fathy A, Megahed A, Barakat T, et al. Proton MR spectroscopy of the brain in children with neuronopathic Gaucher’s disease. Eur Radiol. 2013;23:3005–11.
Razek AAKA, Abdalla A, Ezzat A, Megahed A, Barakat T. Minimal hepatic encephalopathy in children with liver cirrhosis: diffusion-weighted MR imaging and proton MR spectroscopy of the brain. Neuroradiology. 2014;56:885–91.
Gambardella A, Labate A. The role of calcium channel mutations in human epilepsy. Prog Brain Res. 2014;213:87–96.
Steinlein OK. Calcium signaling and epilepsy. Cell Tissue Res. 2014;357:385–93.
Breitenkamp AF, Matthes J, Herzig S. Voltage-gated calcium channels and autism spectrum disorders. Curr Mol Pharmacol. 2015;8:123–32.
Moher D, Shamseer L, Clarke M, Ghersi D, Liberati A, Petticrew M, et al. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst Rev. 2015;4:1.
Weyhrauch DL, Ye D, Boczek NJ, Tester DJ, Gavrilova RH, Patterson MC, et al. Whole exome sequencing and heterologous cellular electrophysiology studies elucidate a novel loss-of-function mutation in the CACNA1A-encoded neuronal P/Q-type calcium channel in a child with congenital hypotonia and developmental delay. Pediatr Neurol. 2016;55:46–51.
Kothur K, Holman K, Farnsworth E, Ho G, Lorentzos M, Troedson C, et al. Diagnostic yield of targeted massively parallel sequencing in children with epileptic encephalopathy. Seizure. 2018;59:132–40.
Angelini C, Van Gils J, Bigourdan A, Jouk P-S, Lacombe D, Menegon P, et al. Major intra-familial phenotypic heterogeneity and incomplete penetrance due to a CACNA1A pathogenic variant. Eur J Med Genet. 2019;62:103530.
Reinson K, Oiglane-Shlik E, Talvik I, Vaher U, Ounapuu A, Ennok M, et al. Biallelic CACNA1A mutations cause early onset epileptic encephalopathy with progressive cerebral, cerebellar, and optic nerve atrophy. Am J Med Genet A. 2016;170:2173–6.
Balck A, Hanssen H, Hellenbroich Y, Lohmann K, Munchau A. Adult-onset ataxia or developmental disorder with seizures: two sides of missense changes in CACNA1A. J Neurol. 2017;264:1520–2.
Tantsis EM, Gill D, Griffiths L, Gupta S, Lawson J, Maksemous N, et al. Eye movement disorders are an early manifestation of CACNA1A mutations in children. Dev Med Child Neurol. 2016;58:639–44.
Humbertclaude V, Riant F, Krams B, Zimmermann V, Nagot N, Annequin D, et al. Cognitive impairment in children with CACNA1A mutations. Dev Med Child Neurol. 2020;62:330–7.
Blumkin L, Michelson M, Leshinsky-Silver E, Kivity S, Lev D, Lerman-Sagie T. Congenital ataxia, mental retardation, and dyskinesia associated with a novel CACNA1A mutation. J Child Neurol. 2010;25:892–7.
Fitzsimons RB, Wolfenden WH. Migraine coma. Meningitic migraine with cerebral oedema associated with a new form of autosomal dominant cerebellar ataxia. Brain. 1985;108(Pt 3):555–77.
Kors EE, Terwindt GM, Vermeulen FL, Fitzsimons RB, Jardine PE, Heywood P, et al. Delayed cerebral edema and fatal coma after minor head trauma: role of the CACNA1A calcium channel subunit gene and relationship with familial hemiplegic migraine. Ann Neurol. 2001;49:753–60.
Guerin AA, Feigenbaum A, Donner EJ, Yoon G. Stepwise developmental regression associated with novel CACNA1A mutation. Pediatr Neurol. 2008;39:363–4.
Wada T, Kobayashi N, Takahashi Y, Aoki T, Watanabe T, Saitoh S. Wide clinical variability in a family with a CACNA1A T666m mutation: hemiplegic migraine, coma, and progressive ataxia. Pediatr Neurol. 2002;26:47–50.
de Vries B, Stam AH, Beker F, van den Maagdenberg AMJM, Vanmolkot KRJ, Laan L, et al. CACNA1A mutation linking hemiplegic migraine and alternating hemiplegia of childhood. Cephalalgia. 2008;28:887–91.
Indelicato E, Nachbauer W, Karner E, Eigentler A, Wagner M, Unterberger I, et al. The neuropsychiatric phenotype in CACNA1A mutations: a retrospective single center study and review of the literature. Eur J Neurol. 2019;26:66-e7.
Luo X, Rosenfeld JA, Yamamoto S, Harel T, Zuo Z, Hall M, et al. Clinically severe CACNA1A alleles affect synaptic function and neurodegeneration differentially. PLoS Genet. 2017;13:e1006905.
Epperson MV, Haws ME, Standridge SM, Gilbert DL. An atypical rett syndrome phenotype due to a novel missense mutation in CACNA1A. J Child Neurol. 2018;33:286–9.
Meloche J, Brunet V, Gagnon P-A, Lavoie M-E, Bouchard J-B, Nadaf J, et al. Exome sequencing study of partial agenesis of the corpus callosum in men with developmental delay, epilepsy, and microcephaly. Mol Genet Genomic Med. 2020;8:e992.
Ohba C, Osaka H, Iai M, Yamashita S, Suzuki Y, Aida N, et al. Diagnostic utility of whole exome sequencing in patients showing cerebellar and/or vermis atrophy in childhood. Neurogenetics. 2013;14:225–32.
Bertholon P, Chabrier S, Riant F, Tournier-Lasserve E, Peyron R. Episodic ataxia type 2: unusual aspects in clinical and genetic presentation. Special emphasis in childhood. J Neurol Neurosurg Psychiatry. 2009;80:1289–92.
Myers CT, McMahon JM, Schneider AL, Petrovski S, Allen AS, Carvill GL, Zemel M, Saykally JE, LaCroix AJ, Heinzen EL, Hollingsworth G. De Novo mutations in SLC1A2 and CACNA1A are important causes of epileptic encephalopathies. Am J Hum Genet. 2016;99:287–98.
Bahamonde MI, Serra SA, Drechsel O, Rahman R, Marcé-Grau A, Prieto M, et al. A single amino acid deletion (ΔF1502) in the S6 segment of CaV2.1 domain III associated with congenital ataxia increases channel activity and promotes Ca2+ influx. PLoS ONE. 2015;10:e0146035.
Yamamoto T, Imaizumi T, Yamamoto-Shimojima K, Lu Y, Yanagishita T, Shimada S, et al. Genomic backgrounds of Japanese patients with undiagnosed neurodevelopmental disorders. Brain Dev. 2019;41:776–82.
Mantuano E, Romano S, Veneziano L, Gellera C, Castellotti B, Caimi S, et al. Identification of novel and recurrent CACNA1A gene mutations in fifteen patients with episodic ataxia type 2. J Neurol Sci. 2010;291:30–6.
Freilinger T, Ackl N, Ebert A, Schmidt C, Rautenstrauss B, Dichgans M, et al. A novel mutation in CACNA1A associated with hemiplegic migraine, cerebellar dysfunction and late-onset cognitive decline. J Neurol Sci. 2011;300:160–3.
Garcia Segarra N, Gautschi I, Mittaz-Crettol L, Kallay Zetchi C, Al-Qusairi L, Van Bemmelen MX, et al. Congenital ataxia and hemiplegic migraine with cerebral edema associated with a novel gain of function mutation in the calcium channel CACNA1A. J Neurol Sci. 2014;342:69–78.
Vahedi K, Denier C, Ducros A, Bousson V, Levy C, Chabriat H, et al. CACNA1A gene de novo mutation causing hemiplegic migraine, coma, and cerebellar atrophy. Neurology. 2000;55:1040–2.
Kashimada A, Hasegawa S, Nomura T, Shiraku H, Moriyama K, Suzuki T, et al. Genetic analysis of undiagnosed ataxia-telangiectasia-like disorders. Brain Dev. 2019;41:150–7.
Naik S, Pohl K, Malik M, Siddiqui A, Josifova D. Early-onset cerebellar atrophy associated with mutation in the CACNA1A gene. Pediatr Neurol. 2011;45:328–30.
Dufendach KA, Timothy K, Ackerman MJ, Blevins B, Pflaumer A, Etheridge S, et al. Clinical outcomes and modes of death in timothy syndrome: a multicenter international study of a rare disorder. JACC Clin Electrophysiol. 2018;4:459–66.
Boczek NJ, Miller EM, Ye D, Nesterenko VV, Tester DJ, Antzelevitch C, et al. Novel Timothy syndrome mutation leading to increase in CACNA1C window current. Hear Rhythm. 2015;12:211–9.
Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, et al. Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell. 2004;119:19–31.
Diep V, Seaver LH. Long QT syndrome with craniofacial, digital, and neurologic features: Is it useful to distinguish between Timothy syndrome types 1 and 2? Am J Med Genet A. 2015;167A:2780–5.
Kosaki R, Ono H, Terashima H, Kosaki K. Timothy syndrome-like condition with syndactyly but without prolongation of the QT interval. Am J Med Genet A. 2018;176:1657–61.
Bozarth X, Dines JN, Cong Q, Mirzaa GM, Foss K, Lawrence Merritt II J, et al. Expanding clinical phenotype in CACNA1C related disorders: from neonatal onset severe epileptic encephalopathy to late-onset epilepsy. Am J Med Genet A. 2018;176:2733–9.
Hennessey JA, Boczek NJ, Jiang Y-H, Miller JD, Patrick W, Pfeiffer R, et al. A CACNA1C variant associated with reduced voltage-dependent inactivation, increased CaV12 channel window current, and arrhythmogenesis. PLoS ONE. 2014;9:e106982.
El Ghaleb Y, Schneeberger PE, Fernández-Quintero ML, Geisler SM, Pelizzari S, Polstra AM, et al. CACNA1I gain-of-function mutations differentially affect channel gating and cause neurodevelopmental disorders. Brain. 2021. https://doi.org/10.1093/brain/awab101.
Han JY, Jang JH, Park J, Lee IG. Targeted next-generation sequencing of Korean patients with developmental delay and/or intellectual disability. Front Pediatr. 2018;6:391.
Han JY, Jang W, Park J, Kim M, Kim Y, Lee IG. Diagnostic approach with genetic tests for global developmental delay and/or intellectual disability: Single tertiary center experience. Ann Hum Genet. 2019;83:115–23.
Edvardson S, Oz S, Abulhijaa FA, Taher FB, Shaag A, Zenvirt S, et al. Early infantile epileptic encephalopathy associated with a high voltage gated calcium channelopathy. J Med Genet. 2013;50:118–23.
Butler KM, Holt PJ, Milla SS, da Silva C, Alexander JJ, Escayg A. Epileptic encephalopathy and cerebellar atrophy resulting from compound heterozygous CACNA2D2 variants. Case Rep Genet. 2018. https://doi.org/10.1155/2018/6308283.
Pippucci T, Parmeggiani A, Palombo F, Maresca A, Angius A, Crisponi L, et al. A novel null homozygous mutation confirms CACNA2D2 as a gene mutated in epileptic encephalopathy. PLoS ONE. 2013;8:e82154.
Punetha J, Karaca E, Gezdirici A, Lamont RE, Pehlivan D, Marafi D, et al. Biallelic CACNA2D2 variants in epileptic encephalopathy and cerebellar atrophy. Ann Clin Transl Neurol. 2019;6:1395–406.
Siddique A, Willoughby J, McNeill A. A 7q21.11 microdeletion presenting with apparent intellectual disability without epilepsy. Am J Med Genet A. 2017;178:1128–30.
Pinggera A, Lieb A, Benedetti B, Lampert M, Monteleone S, Liedl KR, et al. CACNA1D de novo mutations in autism spectrum disorders activate Cav1.3 L-type calcium channels. Biol Psychiatry. 2015;77:816–22.
Flanagan SE, Vairo F, Johnson MB, Caswell R, Laver TW, Lango Allen H, et al. A CACNA1D mutation in a patient with persistent hyperinsulinaemic hypoglycaemia, heart defects, and severe hypotonia. Pediatr Diabetes. 2017;18:320–3.
Hofer NT, Tuluc P, Ortner NJ, Nikonishyna YV, Fernándes-Quintero ML, Liedl KR, et al. Biophysical classification of a CACNA1D de novo mutation as a high-risk mutation for a severe neurodevelopmental disorder. Mol Autism. 2020;11:4.
Helbig KL, Lauerer RJ, Bahr JC, Souza IA, Myers CT, Uysal B, et al. De Novo pathogenic variants in CACNA1E cause developmental and epileptic encephalopathy with contractures, macrocephaly, and dyskinesias. Am J Hum Genet. 2018;103:666–78.
Hope CI, Sharp DM, Hemara-Wahanui A, Sissingh JI, Lundon P, Mitchell EA, et al. Clinical manifestations of a unique X-linked retinal disorder in a large New Zealand family with a novel mutation in CACNA1F, the gene responsible for CSNB2. Clin Exp Ophthalmol. 2005;33:129–36.
Barresi S, Dentici ML, Manzoni F, Bellacchio E, Agolini E, Pizzi S, et al. Infantile-onset syndromic cerebellar ataxia and CACNA1G mutations. Pediatr Neurol. 2020;104:40–5.
Moon AL, Haan N, Wilkinson LS, Thomas KL, Hall J. CACNA1C: association with psychiatric disorders, behavior, and neurogenesis. Schizophr Bull. 2018;44:958–65.
Lu B, Nagappan G, Lu Y. BDNF and synaptic plasticity, cognitive function, and dysfunction. Handb Exp Pharmacol. 2014;220:223–50.
Hoppa MB, Lana B, Margas W, Dolphin AC, Ryan TA. α2δ expression sets presynaptic calcium channel abundance and release probability. Nature. 2012;486:122–5.
Dolphin AC. Voltage-gated calcium channel α (2)δ subunits: an assessment of proposed novel roles. F1000Research [Internet]. F1000 Research Limited; 2018;7:F1000 Faculty Rev-1830. Available from: https://pubmed.ncbi.nlm.nih.gov/30519455.
Nanou E, Catterall WA. Calcium channels, synaptic plasticity, and neuropsychiatric disease. Neuron. 2018;98:466–81.
Serra SA, Gene GG, Elorza-Vidal X, Fernandez-Fernandez JM. Cross talk between beta subunits, intracellular Ca(2+) signaling, and SNAREs in the modulation of CaV 2.1 channel steady-state inactivation. Physiol Rep. 2018;6:e13557.
Ly R, Bouvier G, Schonewille M, Arabo A, Rondi-Reig L, Léna C, et al. T-type channel blockade impairs long-term potentiation at the parallel fiber-Purkinje cell synapse and cerebellar learning. Proc Natl Acad Sci U S A. 2013;110:20302–7.
Isope P, Hildebrand ME, Snutch TP. Contributions of T-type voltage-gated calcium channels to postsynaptic calcium signaling within Purkinje neurons. Cerebellum. 2012;11:651–65.
Wolfart J, Roeper J. Selective coupling of T-type calcium channels to SK potassium channels prevents intrinsic bursting in dopaminergic midbrain neurons. J Neurosci. 2002;22:3404–13.
Astori S, Lüthi A. Synaptic plasticity at intrathalamic connections via CaV3.3 T-type Ca2+ channels and GluN2B-containing NMDA receptors. J Neurosci. 2013;33:624–30.
Mochida S, Few AP, Scheuer T, Catterall WA. Regulation of presynaptic Ca(V)2.1 channels by Ca2+ sensor proteins mediates short-term synaptic plasticity. Neuron. 2008;57:210–6.
Rettig J, Neher E. Emerging roles of presynaptic proteins in Ca++-triggered exocytosis. Science (80+). 2002;298:781–5.
Yoon T-Y, Munson M. SNARE complex assembly and disassembly. Curr Biol. 2018;28:R397-401.
Weiss N, Zamponi GW. Regulation of voltage-gated calcium channels by synaptic proteins. Adv Exp Med Biol. 2012;740:759–75.
Jackman SL, Turecek J, Belinsky JE, Regehr WG. The calcium sensor synaptotagmin 7 is required for synaptic facilitation. Nature. 2016;529:88–91.
Jackman SL, Regehr WG. The mechanisms and functions of synaptic facilitation. Neuron. 2017;94:447–64.
Sheng ZH, Yokoyama CT, Catterall WA. Interaction of the synprint site of N-type Ca2+ channels with the C2B domain of synaptotagmin I. Proc Natl Acad Sci U S A. 1997;94:5405–10.
Chen C-C, Shen J-W, Chung N-C, Min M-Y, Cheng S-J, Liu IY. Retrieval of context-associated memory is dependent on the Ca(v)3.2 T-type calcium channel. PLoS ONE. 2012;7:e29384.
Uebachs M, Schaub C, Perez-Reyes E, Beck H. T-type Ca2+ channels encode prior neuronal activity as modulated recovery rates. J Physiol. 2006;571:519–36.
Kaja S, Van de Ven RCG, Broos LAM, Frants RR, Ferrari MD, Van den Maagdenberg AMJM, et al. Severe and progressive neurotransmitter release aberrations in familial hemiplegic migraine type 1 Cacna1a S218L knock-in mice. J Neurophysiol. 2010;104:1445–55.
Kaja S, van de Ven RCG, Broos LAM, Frants RR, Ferrari MD, van den Maagdenberg AMJM, et al. Characterization of acetylcholine release and the compensatory contribution of non-Ca(v)2.1 channels at motor nerve terminals of leaner Ca(v)2.1-mutant mice. Neuroscience. 2007;144:1278–87.
Heck J, Parutto P, Ciuraszkiewicz A, Bikbaev A, Freund R, Mitlöhner J, et al. Transient confinement of Ca(V)2.1 Ca(2+)-channel splice variants shapes synaptic short-term plasticity. Neuron. 2019;103:66–79.
Mortensen OV. MKP3 eliminates depolarization-dependent neurotransmitter release through downregulation of L-type calcium channel Cav1.2 expression. Cell Calcium. 2013;53:224–30.
Hoppa MB, Lana B, Margas W, Dolphin AC, Ryan TA. alpha2delta expression sets presynaptic calcium channel abundance and release probability. Nature. 2012;486:122–5.
Herring BE, Nicoll RA. Long-term potentiation: from CaMKII to AMPA receptor trafficking. Annu Rev Physiol. 2016;78:351–65.
Kim J, Jung S-C, Clemens AM, Petralia RS, Hoffman DA. Regulation of dendritic excitability by activity-dependent trafficking of the A-type K+ channel subunit Kv4.2 in hippocampal neurons. Neuron. 2007;54:933–47.
Lisman J, Yasuda R, Raghavachari S. Mechanisms of CaMKII action in long-term potentiation. Nat Rev Neurosci. 2012;13:169–82.
Mammen AL, Kameyama K, Roche KW, Huganir RL. Phosphorylation of the alpha-amino-3-hydroxy-5-methylisoxazole4-propionic acid receptor GluR1 subunit by calcium/calmodulin-dependent kinase II. J Biol Chem. 1997;272:32528–33.
Nicoll RA, Tomita S, Bredt DS. Auxiliary subunits assist AMPA-type glutamate receptors. Science. 2006;311:1253–6.
Sanhueza M, Fernandez-Villalobos G, Stein IS, Kasumova G, Zhang P, Bayer KU, et al. Role of the CaMKII/NMDA receptor complex in the maintenance of synaptic strength. J Neurosci. 2011;31:9170–8.
Lisman J, Raghavachari S. Biochemical principles underlying the stable maintenance of LTP by the CaMKII/NMDAR complex. Brain Res. 2015;1621:51–61.
Letts VA, Felix R, Biddlecome GH, Arikkath J, Mahaffey CL, Valenzuela A, et al. The mouse stargazer gene encodes a neuronal Ca2+-channel gamma subunit. Nat Genet. 1998;19:340–7.
Tomita S, Stein V, Stocker TJ, Nicoll RA, Bredt DS. Bidirectional synaptic plasticity regulated by phosphorylation of stargazin-like TARPs. Neuron. 2005;45:269–77.
Cohen SM, Li B, Tsien RW, Ma H. Evolutionary and functional perspectives on signaling from neuronal surface to nucleus. Biochem Biophys Res Commun. 2015;460:88–99.
Flavell SW, Greenberg ME. Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu Rev Neurosci. 2008;31:563–90.
Mayford M, Siegelbaum SA, Kandel ER. Synapses and memory storage. Cold Spring Harb Perspect Biol. 2012;4:a005751.
Davis HP, Squire LR. Protein synthesis and memory: a review. Psychol Bull. 1984;96:518–59.
Patriarchi T, Qian H, Di Biase V, Malik ZA, Chowdhury D, Price JL, et al. Phosphorylation of Cav1.2 on S1928 uncouples the L-type Ca2+ channel from the beta2 adrenergic receptor. EMBO J. 2016;35:1330–45.
Qian H, Patriarchi T, Price JL, Matt L, Lee B, Nieves-Cintron M, et al. Phosphorylation of Ser 1928 mediates the enhanced activity of the L-type Ca2+ channel Cav1.2 by the β2-adrenergic receptor in neurons. Sci Signal. 2017;10:eaaf9659.
Stanika RI, Villanueva I, Kazanina G, Andrews SB, Pivovarova NB. Comparative impact of voltage-gated calcium channels and NMDA receptors on mitochondria-mediated neuronal injury. J Neurosci. 2012;32:6642–50.
Markham A, Bains R, Franklin P, Spedding M. Changes in mitochondrial function are pivotal in neurodegenerative and psychiatric disorders: how important is BDNF? Br J Pharmacol. 2014;171:2206–29.
Kramer P, Bressan P. Our (mother’s) mitochondria and our mind. Perspect Psychol Sci J Assoc Psychol Sci. 2018;13:88–100.
Bravo-Sagua R, Rodriguez AE, Kuzmicic J, Gutierrez T, Lopez-Crisosto C, Quiroga C, et al. Cell death and survival through the endoplasmic reticulum-mitochondrial axis. Curr Mol Med. 2013;13:317–29.
Naon D, Scorrano L. At the right distance: ER-mitochondria juxtaposition in cell life and death. Biochim Biophys Acta. 2014;1843:2184–94.
Bootman MD, Chehab T, Bultynck G, Parys JB, Rietdorf K. The regulation of autophagy by calcium signals: do we have a consensus? Cell Calcium. 2018;70:32–46.
Zhou J, Yi J, Fu R, Liu E, Siddique T, Ríos E, et al. Hyperactive intracellular calcium signaling associated with localized mitochondrial defects in skeletal muscle of an animal model of amyotrophic lateral sclerosis. J Biol Chem. 2010;285:705–12.
Minnella AM, Zhao JX, Jiang X, Jakobsen E, Lu F, Wu L, et al. Excitotoxic superoxide production and neuronal death require both ionotropic and non-ionotropic NMDA receptor signaling. Sci Rep. 2018;8:17522.
Nickels KC, Zaccariello MJ, Hamiwka LD, Wirrell EC. Cognitive and neurodevelopmental comorbidities in paediatric epilepsy. Nat Rev Neurol. 2016;12:465–76.
Nickels KC, Wirrell EC. Cognitive and social outcomes of epileptic encephalopathies. Semin Pediatr Neurol. 2017;24:264–75.
Nabbout R, Chemaly N, Chipaux M, Barcia G, Bouis C, Dubouch C, et al. Encephalopathy in children with Dravet syndrome is not a pure consequence of epilepsy. Orphanet J Rare Dis. 2013;8:176.
Bender AC, Natola H, Ndong C, Holmes GL, Scott RC, Lenck-Santini P-P. Focal Scn1a knockdown induces cognitive impairment without seizures. Neurobiol Dis. 2013;54:297–307.
Stoodley CJ. The cerebellum and neurodevelopmental disorders. Cerebellum. 2016;15:34–7.
Buckner RL. The cerebellum and cognitive function: 25 years of insight from anatomy and neuroimaging. Neuron. 2013;80:807–15.
Tavano A, Grasso R, Gagliardi C, Triulzi F, Bresolin N, Fabbro F, et al. Disorders of cognitive and affective development in cerebellar malformations. Brain. 2007;130:2646–60.
Shipman ML, Green JT. Cerebellum and cognition: does the rodent cerebellum participate in cognitive functions? Neurobiol Learn Mem. 2019;106996.
Tokuda S, Kuramoto T, Tanaka K, Kaneko S, Takeuchi IK, Sasa M, et al. The ataxic groggy rat has a missense mutation in the P/Q-type voltage-gated Ca2+ channel alpha1A subunit gene and exhibits absence seizures. Brain Res. 2007;1133:168–77.
Salvi J, Bertaso F, Mausset-Bonnefont A-L, Metz A, Lemmers C, Ango F, et al. RNAi silencing of P/Q-type calcium channels in Purkinje neurons of adult mouse leads to episodic ataxia type 2. Neurobiol Dis. 2014;68:47–56.
Spacey SD, Hildebrand ME, Materek LA, Bird TD, Snutch TP. Functional implications of a novel EA2 mutation in the P/Q-type calcium channel. Ann Neurol. 2004;56:213–20.
Calorio C, Gavello D, Guarina L, Salio C, Sassoè-Pognetto M, Riganti C, et al. Impaired chromaffin cell excitability and exocytosis in autistic Timothy syndrome TS2-neo mouse rescued by L-type calcium channel blockers. J Physiol. 2019;597:1705–33.
Fujioka Y, Nishide S, Ose T, Suzuki T, Kato I, Fukuhara H, et al. A sialylated voltage-dependent Ca(2+) channel binds hemagglutinin and mediates influenza a virus entry into mammalian cells. Cell Host Microbe. 2018;23:809–18.
Chu Q, Li A, Chen X, Qin Y, Sun X, Li Y, et al. Overexpression of miR-135b attenuates pathological cardiac hypertrophy by targeting CACNA1C. Int J Cardiol. 2018;269:235–41.
Balderas E, Ateaga-Tlecuitl R, Rivera M, Gomora JC, Darszon A. Niflumic acid blocks native and recombinant T-type channels. J Cell Physiol. 2012;227:2542–55.
Sekiguchi F, Fujita T, Deguchi T, Yamaoka S, Tomochika K, Tsubota M, et al. Blockade of T-type calcium channels by 6-prenylnaringenin, a hop component, alleviates neuropathic and visceral pain in mice. Neuropharmacology. 2018;138:232–44.
Warnier M, Roudbaraki M, Derouiche S, Delcourt P, Bokhobza A, Prevarskaya N, et al. CACNA2D2 promotes tumorigenesis by stimulating cell proliferation and angiogenesis. Oncogene. 2015;34:5383–94.
Lau FC, Frank TC, Nahm S-S, Stoica G, Abbott LC. Postnatal apoptosis in cerebellar granule cells of homozygous leaner (tg1a/tg1a) mice. Neurotox Res. 2004;6:267–80.
Kawaida M, Abe T, Nakanishi T, Miyahara Y, Yamagishi H, Sakamoto M, et al. A case of Timothy syndrome with adrenal medullary dystrophy. Pathol Int. 2016;66:587–92.
Blaich A, Pahlavan S, Tian Q, Oberhofer M, Poomvanicha M, Lenhardt P, et al. Mutation of the calmodulin binding motif IQ of the L-type Ca(v)1.2 Ca2+ channel to EQ induces dilated cardiomyopathy and death. J Biol Chem. 2012;287:22616–25.
Wang M-X, Liu X, Li J-M, Liu L, Lu W, Chen G-C. Inhibition of CACNA1H can alleviate endoplasmic reticulum stress and reduce myocardial cell apoptosis caused by myocardial infarction. Eur Rev Med Pharmacol Sci. 2020;24:12887–95.
Michels S, Ganjam GK, Martins H, Schratt GM, Wöhr M, Schwarting RKW, et al. Downregulation of the psychiatric susceptibility gene Cacna1c promotes mitochondrial resilience to oxidative stress in neuronal cells. Cell Death Discov. 2018;4:54.
Qi F, Zhang R, Chen J, Zhao F, Sun Y, Du Z, et al. Down-regulation of Cav1.3 in auditory pathway promotes age-related hearing loss by enhancing calcium-mediated oxidative stress in male mice. Aging (Albany NY). 2019;11:6490–502.
Li S, Hao M, Li B, Chen M, Chen J, Tang J, et al. CACNA1H downregulation induces skeletal muscle atrophy involving endoplasmic reticulum stress activation and autophagy flux blockade. Cell Death Dis. 2020;11:279.
Veneziano L, Guida S, Mantuano E, Bernard P, Tarantino P, Boccone L, et al. Newly characterised 5’ and 3’ regions of CACNA1A gene harbour mutations associated with Familial Hemiplegic Migraine and Episodic Ataxia. J Neurol Sci. 2009;276:31–7.
Tsou W-L, Soong B-W, Paulson HL, Rodríguez-Lebrón E. Splice isoform-specific suppression of the Cav2.1 variant underlying spinocerebellar ataxia type 6. Neurobiol Dis. 2011;43:533–42.
Sun J, Sun X, Li Z, Ma D, Lv Y. An elongated tract of polyQ in the carboxyl-terminus of human α1A calcium channel induces cell apoptosis by nuclear translocation. Oncol Rep. 2020;44:156–64.
Estes SI, Ye D, Zhou W, Dotzler SM, Tester DJ, Bos JM, et al. Characterization of the CACNA1C-R518C missense mutation in the pathobiology of long-QT syndrome using human induced pluripotent stem cell cardiomyocytes shows action potential prolongation and L-type calcium channel perturbation. Circ Genomic Precis Med. 2019;12:e002534.
Deneault E, White SH, Rodrigues DC, Ross PJ, Faheem M, Zaslavsky K, et al. Complete disruption of autism-susceptibility genes by gene editing predominantly reduces functional connectivity of isogenic human neurons. Stem Cell Rep. 2018;11:1211–25.
Wan J, Khanna R, Sandusky M, Papazian DM, Jen JC, Baloh RW. CACNA1A mutations causing episodic and progressive ataxia alter channel trafficking and kinetics. Neurology. 2005;64:2090–7.
Miyazaki Y, Du X, Muramatsu S-I, Gomez CM. An miRNA-mediated therapy for SCA6 blocks IRES-driven translation of the CACNA1A second cistron. Sci Transl Med. 2016;8:347ra94.
Schober A, Sokolova E, Gingrich KJ. Pentobarbital inhibition of human recombinant alpha1A P/Q-type voltage-gated calcium channels involves slow, open channel block. Br J Pharmacol. 2010;161:365–83.
Jiang Y, Xu B, Chen J, Sui Y, Ren L, Li J, et al. Micro-RNA-137 inhibits tau hyperphosphorylation in Alzheimer’s disease and targets the CACNA1C gene in transgenic mice and human neuroblastoma SH-SY5Y cells. Med Sci Monit Int Med J Exp Clin Res. 2018;24:5635–44.
Binas S, Knyrim M, Hupfeld J, Kloeckner U, Rabe S, Mildenberger S, et al. miR-221 and -222 target CACNA1C and KCNJ5 leading to altered cardiac ion channel expression and current density. Cell Mol Life Sci. 2020;77:903–18.
Xu H, Abuhatzira L, Carmona GN, Vadrevu S, Satin LS, Notkins AL. The Ia-2β intronic miRNA, miR-153, is a negative regulator of insulin and dopamine secretion through its effect on the Cacna1c gene in mice. Diabetologia. 2015;58:2298–306.
Sun Z, Cao X, Hu Z, Zhang L, Wang H, Zhou H, et al. MiR-103 inhibits osteoblast proliferation mainly through suppressing Cav1.2 expression in simulated microgravity. Bone. 2015;76:121–8.
Li H, Zhang L-K, Li S-F, Zhang S-F, Wan W-W, Zhang Y-L, et al. Calcium channel blockers reduce severe fever with thrombocytopenia syndrome virus (SFTSV) related fatality. Cell Res. 2019;29:739–53.
Zhang J-Y, Zhang P-P, Zhou W-P, Yu J-Y, Yao Z-H, Chu J-F, et al. L-type Cav 1.2 calcium channel-α-1C regulates response to rituximab in diffuse large B-cell lymphoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2019;25:4168–78.
Fei D, Zhang Y, Wu J, Zhang H, Liu A, He X, et al. Ca(v) 1.2 regulates osteogenesis of bone marrow-derived mesenchymal stem cells via canonical Wnt pathway in age-related osteoporosis. Aging Cell. 2019;18:e12967.
Yazawa M, Dolmetsch RE. Modeling Timothy syndrome with iPS cells. J Cardiovasc Transl Res. 2013;6:1–9.
Polster A, Perni S, Bichraoui H, Beam KG. Stac adaptor proteins regulate trafficking and function of muscle and neuronal L-type Ca2+ channels. Proc Natl Acad Sci U S A. 2015;112:602–6.
Nasu F, Obara Y, Okamoto Y, Yamaguchi H, Kurakami K, Norota I, et al. Azelnidipine treatment reduces the expression of Ca(v)1.2 protein Life Sci. 2021;269:119043.
Hainsworth AH, McNaughton NCL, Pereverzev A, Schneider T, Randall AD. Actions of sipatrigine, 202W92 and lamotrigine on R-type and T-type Ca2+ channel currents. Eur J Pharmacol. 2003;467:77–80.
Chung G, Rhee JN, Jung SJ, Kim JS, Oh SB. Modulation of CaV2.3 calcium channel currents by eugenol. J Dent Res. 2008;87:137–41.
Zhang Y, Zhang J, Jiang D, Zhang D, Qian Z, Liu C, et al. Inhibition of T-type Ca2+ channels by endostatin attenuates human glioblastoma cell proliferation and migration. Br J Pharmacol. 2012;166:1247–60.
Oguri A, Tanaka T, Iida H, Meguro K, Takano H, Oonuma H, et al. Involvement of CaV3.1 T-type calcium channels in cell proliferation in mouse preadipocytes. Am J Physiol Cell Physiol. 2010;298:C1414-23.
Traboulsie A, Chemin J, Chevalier M, Quignard J-F, Nargeot J, Lory P. Subunit-specific modulation of T-type calcium channels by zinc. J Physiol. 2007;578:159–71.
Yarotskyy V, Elmslie KS. Roscovitine inhibits CaV3.1 (T-type) channels by preferentially affecting closed-state inactivation. J Pharmacol Exp Ther. 2012;340:463–72.
Matar N, Jin W, Wrubel H, Hescheler J, Schneider T, Weiergräber M. Zonisamide block of cloned human T-type voltage-gated calcium channels. Epilepsy Res. 2009;83:224–34.
Lopin KV, Gray IP, Obejero-Paz CA, Thévenod F, Jones SW. Fe2+ block and permeation of CaV3.1 (α1G) T-type calcium channels: candidate mechanism for non-transferrin-mediated Fe2+ influx. Mol Pharmacol. 2012;82:1194–204.
Choi K-H, Rhim H. Inhibition of recombinant Ca(v)3.1 (alpha(1G)) T-type calcium channels by the antipsychotic drug clozapine. Eur J Pharmacol. 2010;626:123–30.
Ye Y-S, Li W-Y, Du S-Z, Yang J, Nian Y, Xu G. Congenetic hybrids derived from dearomatized isoprenylated acylphloroglucinol with opposite effects on Ca(v)3.1 low voltage-gated Ca(2+) channel. J Med Chem. 2020;63:1709–16.
M’Dahoma S, Gadotti VM, Zhang F-X, Park B, Nam JH, Onnis V, et al. Effect of the T-type channel blocker KYS-05090S in mouse models of acute and neuropathic pain. Pflugers Arch. 2016;468:193–9.
Huang C, Wang Z, Zhang K, Dong Y, Zhang A, Lu C, et al. MicroRNA-107 inhibits proliferation and invasion of laryngeal squamous cell carcinoma cells by targeting CACNA2D1 in vitro. Anticancer Drugs. 2020;31:260–71.
Ruan J, Liu X, Xiong X, Zhang C, Li J, Zheng H, et al. miR-107 promotes the erythroid differentiation of leukemia cells via the downregulation of Cacna2d1. Mol Med Rep. 2015;11:1334–9.
Shiozaki A, Katsurahara K, Kudou M, Shimizu H, Kosuga T, Ito H, et al. Amlodipine and verapamil, voltage-gated Ca(2+) channel inhibitors, suppressed the growth of gastric cancer stem cells. Ann Surg Oncol. 2021.
Ishikawa K, Fujigasaki H, Saegusa H, Ohwada K, Fujita T, Iwamoto H, et al. Abundant expression and cytoplasmic aggregations of [alpha]1A voltage-dependent calcium channel protein associated with neurodegeneration in spinocerebellar ataxia type 6. Hum Mol Genet. 1999;8:1185–93.
Fujikawa I, Ando T, Suzuki-Karasaki M, Suzuki-Karasaki M, Ochiai T, Suzuki-Karasaki Y. Aspirin induces mitochondrial Ca(2+) remodeling in tumor cells via ROS-Depolarization-Voltage-Gated Ca(2+) Entry. Int J Mol Sci. 2020;21:4771.
Toyota M, Ho C, Ohe-Toyota M, Baylin SB, Issa JP. Inactivation of CACNA1G, a T-type calcium channel gene, by aberrant methylation of its 5’ CpG island in human tumors. Cancer Res. 1999;59:4535–41.
Dziegielewska B, Brautigan DL, Larner JM, Dziegielewski J. T-type Ca2+ channel inhibition induces p53-dependent cell growth arrest and apoptosis through activation of p38-MAPK in colon cancer cells. Mol Cancer Res. 2014;12:348–58.
Ohkubo T, Yamazaki J. T-type voltage-activated calcium channel Cav3.1, but not Cav32, is involved in the inhibition of proliferation and apoptosis in MCF-7 human breast cancer cells. Int J Oncol. 2012;41:267–75.
Carboni GL, Gao B, Nishizaki M, Xu K, Minna JD, Roth JA, et al. CACNA2D2-mediated apoptosis in NSCLC cells is associated with alterations of the intracellular calcium signaling and disruption of mitochondria membrane integrity. Oncogene. 2003;22:615–26.
Buddell T, Friedman V, Drozd CJ, Quinn CC. An autism-causing calcium channel variant functions with selective autophagy to alter axon targeting and behavior. PLoS Genet. 2019;15:e1008488.
Barceló C, Sisó P, Maiques O, García-Mulero S, Sanz-Pamplona R, Navaridas R, et al. T-type calcium channels as potential therapeutic targets in vemurafenib-resistant BRAF(V600E) melanoma. J Invest Dermatol. 2020;140:1253–65.
Tian X, Gala U, Zhang Y, Shang W, Nagarkar Jaiswal S, di Ronza A, et al. A voltage-gated calcium channel regulates lysosomal fusion with endosomes and autophagosomes and is required for neuronal homeostasis. PLoS Biol. 2015;13:e1002103.
Choi J, Park J-H, Kwon OY, Kim S, Chung JH, Lim DS, et al. T-type calcium channel trigger p21ras signaling pathway to ERK in Cav3.1-expressed HEK293 cells. Brain Res. 2005;1054:22–9.
Mor M, Beharier O, Levy S, Kahn J, Dror S, Blumenthal D, et al. ZnT-1 enhances the activity and surface expression of T-type calcium channels through activation of Ras-ERK signaling. Am J Physiol Cell Physiol. 2012;303:C192-203.
Duran P, Sandoval A, González-Ramírez R, Zarco N, Felix R. Regulation of the Ca(2+) channel α(2)δ-1 subunit expression by epidermal growth factor via the ERK/ELK-1 signaling pathway. Am J Physiol Endocrinol Metab. 2020;319:E232-44.
Zhang Y, Zhao W, Li S, Lv M, Yang X, Li M, et al. CXCL11 promotes self-renewal and tumorigenicity of α2δ1(+) liver tumor-initiating cells through CXCR3/ERK1/2 signaling. Cancer Lett. 2019;449:163–71.
Parikh S, Goldstein A, Koenig MK, Scaglia F, Enns GM, Saneto R, et al. Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2015;17:689–701.
Di Stefano V, Rispoli MG, Pellegrino N, Graziosi A, Rotondo E, Napoli C, et al. Diagnostic and therapeutic aspects of hemiplegic migraine. J Neurol Neurosurg Psychiatry. 2020;91:764–71.
Orsucci D, Raglione LM, Mazzoni M, Vista M. Therapy of episodic ataxias: case report and review of the literature. Drugs Context. 2019;8:212576.
Algahtani H, Shirah B, Algahtani R, Al-Qahtani MH, Abdulkareem AA, Naseer MI. A novel mutation in CACNA1A gene in a Saudi female with episodic ataxia type 2 with no response to acetazolamide or 4-aminopyridine. Intractable Rare Dis Res. 2019;8:67–71.
Glauser TA, Cnaan A, Shinnar S, Hirtz DG, Dlugos D, Masur D, et al. Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy. N Engl J Med. 2010;362:790–9.
van Karnebeek CDM, Stockler S. Treatable inborn errors of metabolism causing intellectual disability: a systematic literature review. Mol Genet Metab. 2012;105:368–81.
Bischof JM, Wevrick R. Chronic diazoxide treatment decreases fat mass and improves endurance capacity in an obese mouse model of Prader-Willi syndrome. Mol Genet Metab. 2018;123:511–7.
Kimonis V, Surampalli A, Wencel M, Gold J-A, Cowen NM. A randomized pilot efficacy and safety trial of diazoxide choline controlled-release in patients with Prader-Willi syndrome. PLoS ONE. 2019;14:e0221615.
Kostopoulou E, Dastamani A, Güemes M, Clement E, Caiulo S, Shanmugananda P, et al. Syndromic forms of hyperinsulinaemic hypoglycaemia-A 15-year follow-up study. Clin Endocrinol (Oxf). 2021;94:399–412.
Grant DB, Dunger DB, Burns EC. Long-term treatment with diazoxide in childhood hyperinsulinism. Acta Endocrinol Suppl (Copenh). 1986;279:340–5.
Zamponi GW, Lory P, Perez-Reyes E. Role of voltage-gated calcium channels in epilepsy. Pflugers Arch. 2010;460:395–403.
Adams ME, Myers RA, Imperial JS, Olivera BM. Toxityping rat brain calcium channels with omega-toxins from spider and cone snail venoms. Biochemistry. 1993;32:12566–70.
Nimmrich V, Gross G. P/Q-type calcium channel modulators. Br J Pharmacol. 2012;167:741–59.
Randall A, Tsien RW. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J Neurosci. 1995;15:2995–3012.
Bunda A, LaCarubba B, Akiki M, Andrade A. Tissue- and cell-specific expression of a splice variant in the II-III cytoplasmic loop of Cacna1b. FEBS Open Bio. 2019;9:1603–16.
Catterall WA, Striessnig J, Snutch TP, Perez-Reyes E. International Union of Pharmacology. XL. Compendium of voltage-gated ion channels: calcium channels. Pharmacol Rev. 2003;55:579–81.
Teleb M, Rizk OH, Zhang F-X, Fronczek FR, Zamponi GW, Fahmy H. Synthesis of some new C2 substituted dihydropyrimidines and their electrophysiological evaluation as L-/T-type calcium channel blockers. Bioorg Chem. 2019;88:102915.
Schlick B, Flucher BE, Obermair GJ. Voltage-activated calcium channel expression profiles in mouse brain and cultured hippocampal neurons. Neuroscience. 2010;167:786–98.
Bourinet E, Stotz SC, Spaetgens RL, Dayanithi G, Lemos J, Nargeot J, et al. Interaction of SNX482 with domains III and IV inhibits activation gating of alpha(1E) (Ca(V)2.3) calcium channels. Biophys J. 2001;81:79–88.
Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev. 2003;83:117–61.
Perez-Reyes E, Van Deusen AL, Vitko I. Molecular pharmacology of human Cav3.2 T-type Ca2+ channels: block by antihypertensives, antiarrhythmics, and their analogs. J Pharmacol Exp Ther. 2009;328:621–7.
Mallmann RT, Elgueta C, Sleman F, Castonguay J, Wilmes T, van den Maagdenberg A, et al. Ablation of Ca(V)2.1 voltage-gated Ca(2)(+) channels in mouse forebrain generates multiple cognitive impairments. PLoS ONE. 2013;8:e78598.
Pastor PDH, Du X, Fazal S, Davies AN, Gomez CM. Targeting the CACNA1A IRES as a treatment for spinocerebellar ataxia type 6. Cerebellum. 2018;17:72–7.
Rose SJ, Kriener LH, Heinzer AK, Fan X, Raike RS, van den Maagdenberg AMJM, et al. The first knockin mouse model of episodic ataxia type 2. Exp Neurol. 2014;261:553–62.
Fioretti B, Catacuzzeno L, Sforna L, Gerke-Duncan MB, van den Maagdenberg AMJM, Franciolini F, et al. Trigeminal ganglion neuron subtype-specific alterations of Ca(V)2.1 calcium current and excitability in a Cacna1a mouse model of migraine. J Physiol. 2011;589:5879–95.
van den Maagdenberg AMJM, Pietrobon D, Pizzorusso T, Kaja S, Broos LAM, Cesetti T, et al. A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron. 2004;41:701–10.
Hullugundi SK, Ansuini A, Ferrari MD, van den Maagdenberg AMJM, Nistri A. A hyperexcitability phenotype in mouse trigeminal sensory neurons expressing the R192Q Cacna1a missense mutation of familial hemiplegic migraine type-1. Neuroscience. 2014;266:244–54.
Tsou W-L, Hosking RR, Burr AA, Sutton JR, Ouyang M, Du X, et al. DnaJ-1 and karyopherin α3 suppress degeneration in a new Drosophila model of Spinocerebellar Ataxia Type 6. Hum Mol Genet. 2015;24:4385–96.
Yamamura S, Hoshikawa M, Dai K, Saito H, Suzuki N, Niwa O, et al. ONO-2506 inhibits spike-wave discharges in a genetic animal model without affecting traditional convulsive tests via gliotransmission regulation. Br J Pharmacol. 2013;168:1088–100.
Jansen NA, Schenke M, Voskuyl RA, Thijs RD, van den Maagdenberg AMJM, Tolner EA. Apnea associated with brainstem seizures in Cacna1a (S218L) mice is caused by medullary spreading depolarization. J Neurosci. 2019;39:9633–44.
Saito H, Okada M, Miki T, Wakamori M, Futatsugi A, Mori Y, et al. Knockdown of Cav2.1 calcium channels is sufficient to induce neurological disorders observed in natural occurring Cacna1a mutants in mice. Biochem Biophys Res Commun. 2009;390:1029–33.
Kim TY, Maki T, Zhou Y, Sakai K, Mizuno Y, Ishikawa A, et al. Absence-like seizures and their pharmacological profile in tottering-6j mice. Biochem Biophys Res Commun. 2015;463:148–53.
Kim TY, Niimi K, Takahashi E. Protein expression pattern in cerebellum of Cav2.1 mutant, tottering-6j mice. Exp Anim. 2016;65:207–14.
Marinelli S, Eleuteri C, Vacca V, Strimpakos G, Mattei E, Severini C, et al. Effects of age-related loss of P/Q-type calcium channels in a mice model of peripheral nerve injury. Neurobiol Aging. 2015;36:352–64.
Oda S-I, Lee KJ, Arii T, Imoto K, Hyun B-H, Park IS, et al. Differential regulation of Purkinje cell dendritic spines in rolling mouse Nagoya (tg/tg), P/Q type calcium channel (α1(A)/Ca(v)2.1) mutant. Anat Cell Biol. 2010;43:211–7.
Braun MD, Kisko TM, Vecchia DD, Andreatini R, Schwarting RKW, Wöhr M. Sex-specific effects of Cacna1c haploinsufficiency on object recognition, spatial memory, and reversal learning capabilities in rats. Neurobiol Learn Mem. 2018;155:543–55.
Koppe G, Mallien AS, Berger S, Bartsch D, Gass P, Vollmayr B, et al. CACNA1C gene regulates behavioral strategies in operant rule learning. PLoS Biol. 2017;15:e2000936.
Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Muller J, et al. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci. 2005;25:9883–92.
White JA, McKinney BC, John MC, Powers PA, Kamp TJ, Murphy GG. Conditional forebrain deletion of the L-type calcium channel Ca V 1.2 disrupts remote spatial memories in mice. Learn Mem. 2008;15:1–5.
Kabitzke PA, Brunner D, He D, Fazio PA, Cox K, Sutphen J, et al. Comprehensive analysis of two Shank3 and the Cacna1c mouse models of autism spectrum disorder. Genes Brain Behav. 2018;17:4–22.
Bader PL, Faizi M, Kim LH, Owen SF, Tadross MR, Alfa RW, et al. Mouse model of Timothy syndrome recapitulates triad of autistic traits. Proc Natl Acad Sci USA. 2011;108:15432–7.
Cheli VT, Santiago González DA, Zamora NN, Lama TN, Spreuer V, Rasmusson RL, et al. Enhanced oligodendrocyte maturation and myelination in a mouse model of Timothy syndrome. Glia. 2018;66:2324–39.
Dedic N, Pöhlmann ML, Richter JS, Mehta D, Czamara D, Metzger MW, et al. Cross-disorder risk gene CACNA1C differentially modulates susceptibility to psychiatric disorders during development and adulthood. Mol Psychiatry. 2018;23:533–43.
Kisko TM, Braun MD, Michels S, Witt SH, Rietschel M, Culmsee C, et al. Cacna1c haploinsufficiency leads to pro-social 50-kHz ultrasonic communication deficits in rats. Dis Model Mech. 2018;11:dmm034116.
Miranda AS, Cardozo PL, Silva FR, de Souza JM, Olmo IG, Cruz JS, et al. Alterations of calcium channels in a mouse model of Huntington’s disease and neuroprotection by blockage of Ca(V)1 channels. ASN Neuro. 2019;11:1759091419856811.
Kabir ZD, Lee AS, Burgdorf CE, Fischer DK, Rajadhyaksha AM, Mok E, et al. Cacna1c in the prefrontal cortex regulates depression-related behaviors via REDD1. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol. 2017;42:2032–42.
Hsu Y-H, Chen Y-C, Chen Y-W, Chiu T-H, Kuo Y-T, Chen C-H. Far-infrared radiation prevents decline in β-cell mass and function in diabetic mice via the mitochondria-mediated Sirtuin1 pathway. Metabolism. 2020;104:154143.
Banono NS, Gawel K, De Witte L, Esguerra CV. Zebrafish larvae carrying a splice variant mutation in cacna1d: a new model for schizophrenia-like behaviours? Mol Neurobiol. 2020;58:877–94.
Stiglbauer V, Hotka M, Ruiß M, Hilber K, Boehm S, Kubista H. Ca(v) 1.3 channels play a crucial role in the formation of paroxysmal depolarization shifts in cultured hippocampal neurons. Epilepsia. 2017;58:858–71.
Gray EE, Murphy JG, Liu Y, Trang I, Tabor GT, Lin L, et al. Disruption of GpI mGluR-dependent Cav2.3 translation in a mouse model of fragile X syndrome. J Neurosci. 2019;39:7453–64.
Rijkers K, Mescheriakova J, Majoie M, Lemmens E, van Wijk X, Philippens M, et al. Polymorphisms in CACNA1E and Camk2d are associated with seizure susceptibility of Sprague-Dawley rats. Epilepsy Res. 2010;91:28–34.
Gandla J, Lomada SK, Lu J, Kuner R, Bali KK. miR-34c-5p functions as pronociceptive microRNA in cancer pain by targeting Cav2.3 containing calcium channels. Pain. 2017;158:1765–79.
Matthews EA, Bee LA, Stephens GJ, Dickenson AH. The Cav2.3 calcium channel antagonist SNX-482 reduces dorsal horn neuronal responses in a rat model of chronic neuropathic pain. Eur J Neurosci. 2007;25:3561–9.
Dai X, Pang S, Wang J, FitzMaurice B, Pang J, Chang B. Photoreceptor degeneration in a new Cacna1f mutant mouse model. Exp Eye Res. 2019;179:106–14.
Lodha N, Bonfield S, Orton NC, Doering CJ, McRory JE, Mema SC, et al. Congenital stationary night blindness in mice–a tale of two Cacna1f mutants. Adv Exp Med Biol. 2010;664:549–58.
Waldner DM, Giraldo Sierra NC, Bonfield S, Nguyen L, Dimopoulos IS, Sauvé Y, et al. Cone dystrophy and ectopic synaptogenesis in a Cacna1f loss of function model of congenital stationary night blindness (CSNB2A). Channels (Austin). 2018;12:17–33.
An J, Zhang L, Jiao B, Lu F, Xia F, Yu Z, et al. Cacna1f gene decreased contractility of skeletal muscle in rat model with congenital stationary night blindness. Gene. 2015;562:210–9.
Knoflach D, Kerov V, Sartori SB, Obermair GJ, Schmuckermair C, Liu X, et al. Cav1.4 IT mouse as model for vision impairment in human congenital stationary night blindness type 2. Channels (Austin). 2013;7:503–13.
Shin A, Woo J, Kim JE, Kim D. Nodding behavior couples to vigilance fluctuation in a high-calorie diet model of drowsiness. Mol Brain. 2018;11:33.
Hashiguchi S, Doi H, Kunii M, Nakamura Y, Shimuta M, Suzuki E, et al. Ataxic phenotype with altered Ca(V)3.1 channel property in a mouse model for spinocerebellar ataxia 42. Neurobiol Dis. 2019;130:104516.
Park Y-G, Choi JH, Lee C, Kim S, Kim Y, Chang K-Y, et al. Heterogeneity of tremor mechanisms assessed by tremor-related cortical potential in mice. Mol Brain. 2015;8:3.
Chang K-Y, Park Y-G, Park H-Y, Homanics GE, Kim J, Kim D. Lack of CaV3.1 channels causes severe motor coordination defects and an age-dependent cerebellar atrophy in a genetic model of essential tremor. Biochem Biophys Res Commun. 2011;410:19–23.
Kim C-H. Cav3.1 T-type calcium channel modulates the epileptogenicity of hippocampal seizures in the kainic acid-induced temporal lobe epilepsy model. Brain Res. 2015;1622:204–16.
Sharop BR, Boldyriev OI, Batiuk MY, Shtefan NL, Shuba YM. Compensatory reduction of Cav3.1 expression in thalamocortical neurons of juvenile rats of WAG/Rij model of absence epilepsy. Epilepsy Res. 2016;119:10–2.
Sakkaki S, Gangarossa G, Lerat B, Françon D, Forichon L, Chemin J, et al. Blockade of T-type calcium channels prevents tonic-clonic seizures in a maximal electroshock seizure model. Neuropharmacology. 2016;101:320–9.
Wang H, Zhang X, Xue L, Xing J, Jouvin M-H, Putney JW, et al. Low-voltage-activated CaV3.1 calcium channels shape T helper cell cytokine profiles. Immunity. 2016;44:782–94.
Le Quang K, Benito B, Naud P, Qi XY, Shi YF, Tardif J-C, et al. T-type calcium current contributes to escape automaticity and governs the occurrence of lethal arrhythmias after atrioventricular block in mice. Circ Arrhythm Electrophysiol. 2013;6:799–808.
Powell KL, Cain SM, Ng C, Sirdesai S, David LS, Kyi M, et al. A Cav3.2 T-type calcium channel point mutation has splice-variant-specific effects on function and segregates with seizure expression in a polygenic rat model of absence epilepsy. J Neurosci. 2009;29:371–80.
Scanzi J, Accarie A, Muller E, Pereira B, Aissouni Y, Goutte M, et al. Colonic overexpression of the T-type calcium channel Ca(v) 3.2 in a mouse model of visceral hypersensitivity and in irritable bowel syndrome patients. Neurogastroenterol Motil Off J Eur Gastrointest Motil Soc. 2016;28:1632–40.
Garcia-Caballero A, Gadotti VM, Chen L, Zamponi GW. A cell-permeant peptide corresponding to the cUBP domain of USP5 reverses inflammatory and neuropathic pain. Mol Pain. 2016;12:1744806916642444.
Fuller-Bicer GA, Varadi G, Koch SE, Ishii M, Bodi I, Kadeer N, et al. Targeted disruption of the voltage-dependent calcium channel alpha2/delta-1-subunit. Am J Physiol Heart Circ Physiol. 2009;297:H117-24.
Zhou C, Luo ZD. Nerve injury-induced calcium channel alpha-2-delta-1 protein dysregulation leads to increased pre-synaptic excitatory input into deep dorsal horn neurons and neuropathic allodynia. Eur J Pain. 2015;19:1267–76.
Boroujerdi A, Zeng J, Sharp K, Kim D, Steward O, Luo ZD. Calcium channel alpha-2-delta-1 protein upregulation in dorsal spinal cord mediates spinal cord injury-induced neuropathic pain states. Pain. 2011;152:649–55.
Wang X, Whalley BJ, Stephens GJ. The du(2J) mouse model of ataxia and absence epilepsy has deficient cannabinoid CB1 receptor-mediated signalling. J Physiol. 2013;591:3919–33.
Ivanov SV, Ward JM, Tessarollo L, McAreavey D, Sachdev V, Fananapazir L, et al. Cerebellar ataxia, seizures, premature death, and cardiac abnormalities in mice with targeted disruption of the Cacna2d2 gene. Am J Pathol. 2004;165:1007–18.
Saegusa H, Wakamori M, Matsuda Y, Wang J, Mori Y, Zong S, et al. Properties of human Cav2.1 channel with a spinocerebellar ataxia type 6 mutation expressed in Purkinje cells. Mol Cell Neurosci. 2007;34:261–70.
Du X, Wang J, Zhu H, Rinaldo L, Lamar K-M, Palmenberg AC, et al. Second cistron in CACNA1A gene encodes a transcription factor mediating cerebellar development and SCA6. Cell. 2013;154:118–33.
Yang J, Liu H, Sun H, Wang Z, Zhang R, Liu Y, et al. Construction of induced pluripotent stem cell line (ZZUi0017-A) from the fibroblast cells of a female patient with CACNA1A mutation by unintegrated reprogramming approach. Stem Cell Res. 2020;48:101946.
Du X, Wei C, Hejazi Pastor DP, Rao ER, Li Y, Grasselli G, et al. α1ACT is essential for survival and early cerebellar programming in a critical neonatal window. Neuron. 2019;102:770–85.
Choi CSW, Souza IA, Sanchez-Arias JC, Zamponi GW, Arbour LT, Swayne LA. Ankyrin B and Ankyrin B variants differentially modulate intracellular and surface Cav2.1 levels. Mol Brain. 2019;12:75.
Mark MD, Maejima T, Kuckelsberg D, Yoo JW, Hyde RA, Shah V, et al. Delayed postnatal loss of P/Q-type calcium channels recapitulates the absence epilepsy, dyskinesia, and ataxia phenotypes of genomic Cacna1a mutations. J Neurosci. 2011;31:4311–26.
Vinueza Veloz MF, Zhou K, Bosman LWJ, Potters J-W, Negrello M, Seepers RM, et al. Cerebellar control of gait and interlimb coordination. Brain Struct Funct. 2015;220:3513–36.
Izquierdo-Serra M, Martínez-Monseny AF, López L, Carrillo-García J, Edo A, Ortigoza-Escobar JD, et al. Stroke-like episodes and cerebellar syndrome in phosphomannomutase deficiency (PMM2-CDG): evidence for hypoglycosylation-driven channelopathy. Int J Mol Sci. 2018;19:619.
Serra SA, Cuenca-León E, Llobet A, Rubio-Moscardo F, Plata C, Carreño O, et al. A mutation in the first intracellular loop of CACNA1A prevents P/Q channel modulation by SNARE proteins and lowers exocytosis. Proc Natl Acad Sci U S A. 2010;107:1672–7.
Garza-López E, González-Ramírez R, Gandini MA, Sandoval A, Felix R. The familial hemiplegic migraine type 1 mutation K1336E affects direct G protein-mediated regulation of neuronal P/Q-type Ca2+ channels. Cephalalgia. 2013;33:398–407.
Yang T, Qin J, Zhang Q, Sun H, Wang Z, Yang J, et al. Generation of induced pluripotent stem cell line (ZZUi0018-A) from a patient with spinocerebellar ataxia type 6. Stem Cell Res. 2020;44:101777.
Rajakulendran S, Graves TD, Labrum RW, Kotzadimitriou D, Eunson L, Davis MB, et al. Genetic and functional characterisation of the P/Q calcium channel in episodic ataxia with epilepsy. J Physiol. 2010;588:1905–13.
Chen H, Piedras-Rentería ES. Altered frequency-dependent inactivation and steady-state inactivation of polyglutamine-expanded alpha1A in SCA6. Am J Physiol Cell Physiol. 2007;292:C1078-86.
Condliffe SB, Fratangeli A, Munasinghe NR, Saba E, Passafaro M, Montrasio C, et al. The E1015K variant in the synprint region of the CaV2.1 channel alters channel function and is associated with different migraine phenotypes. J Biol Chem. 2013;288:33873–83.
Sutphin BS, Boczek NJ, Barajas-Martínez H, Hu D, Ye D, Tester DJ, et al. Molecular and functional characterization of rare CACNA1C variants in sudden unexplained death in the young. Congenit Heart Dis. 2016;11:683–92.
Zhou H-M, Zhou X-Q, Lu J-Z, Jia W-W, Kang J-H. An induced pluripotent stem cell line (SHEHi002-A (5426)) from a patient of long QT syndrome type 8 with c.2573G>A mutation in the gene CACNA1C. Stem Cell Res. 2020;43:101731.
Chavali NV, Kryshtal DO, Parikh SS, Wang L, Glazer AM, Blackwell DJ, et al. Patient-independent human induced pluripotent stem cell model: a new tool for rapid determination of genetic variant pathogenicity in long QT syndrome. Hear Rhythm. 2019;16:1686–95.
Yang X, Mao X, Xu G, Xing S, Chattopadhyay A, Jin S, et al. Estradiol up-regulates L-type Ca(2+) channels via membrane-bound estrogen receptor/phosphoinositide-3-kinase/Akt/cAMP response element-binding protein signaling pathway. Hear Rhythm. 2018;15:741–9.
Ortner NJ, Pinggera A, Hofer NT, Siller A, Brandt N, Raffeiner A, et al. RBP2 stabilizes slow Cav1.3 Ca(2+) channel inactivation properties of cochlear inner hair cells. Pflugers Arch. 2020;472:3–25.
Kim S, Rhim H. Effects of amyloid-β peptides on voltage-gated L-type Ca(V)1.2 and Ca(V)1.3 Ca(2+) channels. Mol Cells. 2011;32:289–94.
Peloquin JB, Rehak R, Doering CJ, McRory JE. Functional analysis of congenital stationary night blindness type-2 CACNA1F mutations F742C, G1007R, and R1049W. Neuroscience. 2007;150:335–45.
Yu J, Shi Y, Zhao K, Yang G, Yu L, Li Y, et al. Enhanced expression of β cell Ca(V)3.1 channels impairs insulin release and glucose homeostasis. Proc Natl Acad Sci U S A. 2020;117:448–53.
Coutelier M, Blesneac I, Monteil A, Monin M-L, Ando K, Mundwiller E, et al. A recurrent mutation in CACNA1G Alters Cav3.1 T-type calcium-channel conduction and causes autosomal-dominant cerebellar ataxia. Am J Hum Genet. 2015;97:726–37.
Morino H, Matsuda Y, Muguruma K, Miyamoto R, Ohsawa R, Ohtake T, et al. A mutation in the low voltage-gated calcium channel CACNA1G alters the physiological properties of the channel, causing spinocerebellar ataxia. Mol Brain. 2015;8:89.
Rzhepetskyy Y, Lazniewska J, Blesneac I, Pamphlett R, Weiss N. CACNA1H missense mutations associated with amyotrophic lateral sclerosis alter Cav3.2 T-type calcium channel activity and reticular thalamic neuron firing. Channels (Austin). 2016;10:466–77.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Key Research and Development Program of China [No. 2016YFC1306202], Hunan Key Research and Developement Program [N0. 2019SK2081] and the National Natural Science Foundation of China [No. 81771408].
Author information
Authors and Affiliations
Contributions
MK is the first author who designed study, reviewed the articles, drafted and wrote the manuscript. BC and FY assisted in literature search and prepared tables and figures. LY, JP and FY revised the manuscript and supervised each step involved in the preparation of the manuscript. All co-authors have read and agreed to the content of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Search strategies utilized.
Additional file 2: Table S1.
Specific calcium channel genes, their mutations, OMIM number, functional significance, protein/enzyme activity change, type of change, phenotype, electrophysiology results, and MRI results.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kessi, M., Chen, B., Peng, J. et al. Calcium channelopathies and intellectual disability: a systematic review. Orphanet J Rare Dis 16, 219 (2021). https://doi.org/10.1186/s13023-021-01850-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-021-01850-0