
Abstract
Background
Niemann-Pick disease Type C (NP-C) is a rare, progressive neurodegenerative disorder characterized by progressive neurodegeneration and premature death. We report data at closure of the NPC Registry that describes the natural history, disease course and treatment experience of NP-C patients in a real-world setting.
Methods
The NPC Registry was a prospective observational cohort study that ran between September 2009 and October 2017. Patients with a confirmed diagnosis of NP-C were enrolled regardless of treatment status. All patients underwent clinical assessments and medical care as determined by their physicians; data were collected through a secure internet-based portal.
Results
At closure on October 19, 2017, 472 patients from 22 countries were enrolled in the NPC Registry. Mean (standard deviation) age at enrollment was 21.2 (15.0) years, and 51.9% of patients were male. First neurological symptom onset occurred during the early-infantile (< 2 years), late-infantile (2 to < 6 years), juvenile (6 to < 15 years), or adolescent/adult (≥ 15 years) periods in 13.5, 25.6, 31.8, and 29.1% of cases, respectively. The most frequent neurological manifestations prior to enrollment included ataxia (67.9%), vertical supranuclear gaze palsy (67.4%), dysarthria (64.7%), cognitive impairment (62.7%), dysphagia (49.1%), and dystonia (40.2%). During infancy, splenomegaly and hepatomegaly were frequent (n = 199/398 [50%] and n = 147/397 [37.0%], respectively) and persisted in most affected patients. Of the 472 enrolled patients, 241 were continuously treated with miglustat during the NPC Registry observation period, of whom 172 of these 241 patients were treated continuously for ≥12 months. A composite disability score that assesses impairment of ambulation, manipulation, language, and swallowing was highest in the early-infantile population and lowest in the adolescent/adult population. Among the continuous miglustat therapy population, 70.5% of patients had improved or had stable disease (at least 3 of the 4 domains having a decreased or unchanged score between enrollment and last follow-up). The NPC Registry did not identify any new safety signals associated with miglustat therapy.
Conclusions
The profiles of clinical manifestations in the final NPC Registry dataset agreed with previous clinical descriptions. Miglustat therapy was associated with a stabilization of neurological manifestations in most patients. The safety and tolerability of miglustat therapy was consistent with previous reports.
Similar content being viewed by others


Background
Niemann-Pick disease Type C (NP-C) is a rare, progressive neurodegenerative disorder characterized by intracellular accumulation of cholesterol and complex lipids, such as sphingolipids and phospholipids, within the endosomal/lysosomal system [1, 2].
NP-C is caused by autosomal recessive mutations in either the NPC1 or NPC2 gene [3,4,5] and has an estimated incidence of between 1:100,000 and 1:120,000 live births, although this may be an underestimate [2, 6]. Patients with NP-C present at all ages with a heterogeneous spectrum of signs and symptoms across visceral, neurologic, and psychiatric domains, with characteristic symptomatology depending on the age of onset [2, 4, 7]. Disease onset in early infancy is characterized by visceral signs, such as liver and respiratory dysfunction, which in some cases can be rapidly progressive and fatal. Onset in childhood is characterized by a spectrum of visceral signs and neurological deficits, and onset in adults is characterized by a range of nonspecific neurological and psychiatric signs [1, 8,9,10,11,12,13,14,15,16]. The age at presentation of the first neurological manifestation is a predictor of disease progression and prognosis, with early-onset forms progressing more rapidly than late-onset forms [2, 4].
NP-C is invariably progressive, but reducing or halting progression of symptoms is key to optimal disease management [4, 7, 17]. Miglustat (Zavesca®, Actelion Pharmaceuticals Ltd.) is the only disease-specific therapy approved for NP-C.Footnote 1 It has been shown to delay disease progression and to stabilize neurological symptoms in several randomized controlled clinical trials, observational studies, and long-term extension studies [7, 16, 18,19,20]. The NPC Registry was initiated in May 2009 as a post-approval commitment to the European Medicines Agency (EMA) following approval of a new indication for miglustat for the treatment of progressive neurological deterioration in adults and children with NP-C [1, 12]. The NPC Registry describes the natural history, disease course, clinical outcomes, and treatment experience in real-world clinical settings, and the data collected by the NPC Registry has proven invaluable to describe the natural history of the disease and treatment experience of patients [1, 12, 21].
Here we describe the characteristics of the patient population enrolled in the NPC Registry at closure in October 2017 and report the treatment experience of patients with NP-C who had received continuous miglustat therapy for more than 1 year during the observation period in the NPC Registry.
Methods
Study design and patients
The NPC Registry was an international, multicenter, prospective, observational cohort study in patients diagnosed with NP-C (EUPAS4622). All patients with a diagnosis of NP-C were eligible for inclusion in the NPC Registry regardless of their treatment. The methodology of the NPC Registry has previously been published [1, 12]. Data were collected via a secure internet-based portal, and written informed consent was obtained from all patients and/or their legal guardians before any clinical visit data were entered. Data entered to the NPC Registry included information routinely collected during clinical investigations for NP-C management as determined as appropriate by the treating physician.
These analyses include all patients in the NPC Registry from the commencement of enrollment in September 2009 up to database closure on 19th October 2017. The analysis to describe the patient treatment experience included all patients who received continuous miglustat therapy between the enrollment visit and their last follow-up visit. Continuous miglustat therapy was defined as the patient receiving miglustat for ≥90% of the observation period with no single period without receiving miglustat lasting > 28 days. A miglustat switcher was defined as a patient who had been treated with miglustat for < 90% of the observation time or had at least one period of > 28 days without miglustat treatment; switching does not imply that patients have switched from miglustat to other therapies. Patients were stratified based on the previously published age at neurological onset categories into early-infantile (< 2 years), late-infantile (2 to < 6 years), juvenile (6 to < 15 years), and adolescent/adult (≥ 15 years) populations [2, 4].
Due to small sample sizes, patients who switched to other therapies and those who were not treated with miglustat were not included in analyses of treatment evaluation.
Assessments of disease status and progression
An assessment of disability status was performed using a previously described modified NP-C disability scale [7, 11]; composite disability scores were calculated as the average of the scores from each of the 4 individual domains (ambulation, manipulation, language, swallowing) of the disability scale. Scores for each domain ranged from 0 with no disability, to 1 being the most affected (Table 1). The extent of change in each of the 4 domains of the disability scale was evaluated from enrollment to the last follow-up visit as improved (decrease in score), stable (no change in score), or progressed (increase in score). Overall neurological progression was considered improved or stable if at least 3 of the 4 individual domain scores were improved or stable during the observation period. The annual progression rate of the composite disability score was computed as the change in disability scale score from enrollment to the last follow-up visit divided by the time from enrollment to the last follow-up visit.
Data related to biomarkers of disease progression were not available from the NPC Registry.
Safety-relevant information, that included adverse drug reactions and adverse events, was collected as part of the NPC Registry.
Statistical analyses
Descriptive analyses were performed on the whole enrolled NPC Registry population and the continuous miglustat therapy population. Clinical disability assessments as a reflection of clinical outcomes were further described in the continuous miglustat therapy population and in patients continuously treated with miglustat for ≥ 12 months. Safety-relevant data are descriptively summarized for the continuous miglustat therapy population only.
Analyzes of data at enrollment are purely descriptive in nature. Continuous variables are summarized using descriptive statistics including mean, standard deviation (SD), median, range and 95% confidence interval (CI) of the mean. Categorical variables are summarized using counts and percentages. As this study is a registry, this analysis is of observational data with all summary statistics and percentages calculated relative to number of patients with available data. Denominators for analysis were the numbers of patients with the corresponding data available; different parameters may have different denominators.
Results
Demographics and patient characteristics
Enrolled population
At database closure, 472 patients from 22 countries were enrolled in the NPC Registry (see Additional file 1), with a similar proportion of males (n = 245; 51.9%) and females (n = 227; 48.1%). The mean (standard deviation [SD]) age at enrollment was 21.2 (15.0) years, with the majority (n = 291/470; 61.9%) aged between 10 and 40 years; 47.7% of enrolled patients were aged < 18 years. Patients with known age at onset of neurological symptoms (n = 422) were categorized as early-infantile (n = 57; 13.5%), late-infantile (n = 108; 25.6%), juvenile, (n = 134; 31.8%), or adolescent/adult (n = 123; 29.1%) onset (Table 2). A diagnostic delay was apparent in each of these age at neurological onset categories, with a mean delay between the appearance of first neurological symptoms and a confirmed diagnosis of NP-C of 2.5 years in early-infantile, 4.3 years in late-infantile, 6.2 years in juvenile, and 6.3 years in adolescent/adult patients (Table 2).
The most frequent neurological manifestations reported in the medical history prior to enrollment include ataxia (n = 304/448; 67.9%), vertical supranuclear gaze palsy (n = 302/448; 67.4%), dysarthria (n = 290/448; 64.7%), cognitive impairment (n = 281/448; 62.7%), dysphagia (n = 220/448; 49.1%) and dystonia (n = 180/448; 40.2%) (Table 3). A number of other neurological manifestations were less frequently present within the population (Table 3). During infancy, splenomegaly and hepatomegaly were frequent (n = 199/398; 50.0%, and n = 147/397; 37.0%, respectively; Table 3) and persisted in most of the afflicted patients. Most patients with splenomegaly during infancy also presented with hepatomegaly (n = 139/199; 69.8%).
Continuous miglustat therapy population
Of the 472 enrolled patients, 241 were continuously treated with miglustat during the NPC Registry observation period, of whom 172 were continuously treated with miglustat for ≥ 12 months (Fig. 1). A further 47 patients were not treated with miglustat, and 113 were miglustat switchers; no data on miglustat treatment were available for 10 patients. Of the 241 patients who had continuous miglustat therapy, the majority (n = 216) had received miglustat prior to enrollment with a mean (SD; median [range]) duration of treatment of 2.06 (2.07; 1.39 [0.00–9.97]) years prior to enrollment. Mean (SD; median [range]) duration of treatment during the observation period of the NPC Registry was 3.27 (1.95; 3.29 [0.11–7.62]) years, which does not include treatment exposure prior to the observation period. Within the continuous miglustat therapy population, the mean age (SD) at neurological onset was 11.2 (10.2) years, at diagnosis was 15.0 (11.9) years (Table 4), and at enrollment was 20.0 (12.4) years.

Patient flow. * Patients who had periods of treatment and nontreatment and had been treated with miglustat for < 90% of the observation time or had one period without miglustat lasting more than 28 days. † Among patients continuously treated with miglustat, 216/241 had received miglustat prior to enrollment for a varying period
During infancy, 54.5% (n = 109/200) and 36.0% (n = 73/203) of patients continuously treated with miglustat presented with splenomegaly and hepatomegaly, respectively (Table 5), and most patients with splenomegaly also presented with hepatomegaly (n = 70/109; 64.2%). Of 227 patients with available data, 71.4% presented with ataxia (n = 162), 70.5% with vertical supranuclear gaze palsy (n = 160), 68.7% with dysarthria (n = 156), 59.9% with cognitive impairment (n = 136), 48.9% with dysphagia (n = 111), and 42.7% with dystonia (n = 97) prior to enrollment (Table 5). Of patients continuously treated with miglustat with data at the time of enrollment, 92/113 (81.4%) had neurological abnormalities, 46/191 (24.1%) had psychiatric manifestations, 50/113 (44.2%) had behavioral signs, and 17/197 (8.6) had respiratory tract abnormalities (Table 5).
Disease progression in the continuous miglustat therapy population
At enrollment, the mean (SD) composite disability score for the continuous miglustat therapy population (n = 221) was 0.38 (0.26). It was highest in the early-infantile group (0.59 [0.35]) and lowest in the adolescent/adult group (0.32 [0.16]). At last follow-up the mean (SD) composite disability score was 0.48 (0.29). It was highest in the early-infantile population (0.70 [0.34]), and lowest in the adult/adolescent population (0.39 [0.23]) (Table 4).
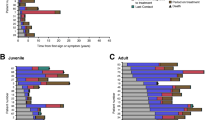
Overall, 70.5% (n = 153/217) of the continuous miglustat therapy population had improved or stable disease, with at least 3 of the 4 domains having a decreased score or remaining unchanged between enrollment and last follow-up. Decreased scores, indicative of lessened severity of symptoms, were observed in a small proportion of patients across all domains. Stable scores were observed in the majority of patients in all domains (Fig. 2). Stable or decreased scores were observed for all domains: ambulation (n = 156/230; 67.8%), manipulation (n = 155/224; 69.2%), language (n = 170/230; 73.9%), and swallowing (n = 164/230; 71.3%) (Fig. 2).

Change in disability scale scores from enrollment to the last follow-up visit in patients continuously treated with miglustat
The mean annual progression rate of the composite disability score in the entire continuous miglustat therapy population (n = 194/210) was 0.034 (95% confidence interval [CI] 0.025–0.042) after a mean (SD) observation period of 3.67 (1.77) years. Of the individual domains, swallowing had the highest mean annual progression rate (0.043; 95% CI 0.028–0.058); annual progression of the other domains was slower (ambulation [0.032; 95% CI 0.020–0.043]; manipulation [0.031; 95% CI 0.017–0.045]; language [0.028; 95% CI 0.018–0.038]) (Fig. 3). In patients who received continuous miglustat therapy for ≥ 12 months (n = 160/172), the mean annual progression rate of the composite disability score was similar to that of the entire continuous miglustat therapy population (0.036; 95% CI 0.025–0.047), as were the scores for the individual domains (ambulation [0.035; 95% CI 0.021–0.049], manipulation [0.033; 95% CI 0.017–0.048], language [0.028; 95% CI 0.016–0.040], swallowing [0.049; 95% CI 0.029–0.070]).

Annual progression rate of disability scale scores from enrollment to the last follow-up visit in patients continuously treated with miglustat. CI, confidence interval
Safety in the continuous miglustat therapy population
In this final output from the NPC Registry, no new safety concerns were identified; the safety-relevant information obtained from the NPC Registry was consistent with the known safety profile of miglustat in NP-C (Table 6).
Known safety/tolerability considerations associated with miglustat, including chronic diarrhea, thrombocytopenia, and seizures, were frequently reported in the overall population. Chronic diarrhea (i.e., diarrhea lasting > 3 months) occurred in 11.2% (n = 27/241) of patients during the observation period and in 4.6% (n = 11/241) of patients before treatment. Seizures were present in a total of 33.6% (n = 81/241) of patients before miglustat therapy, and 46.6% of patients with available data (n = 48/103) had a new occurrence or worsened seizures during follow-up. Thrombocytopenia was recorded in 20.3% (n = 49/241) of patients at enrollment and in 51.7% (n = 109/211) of patients during follow-up; almost all cases of thrombocytopenia were mild or moderate. Amongst patients with thrombocytopenia it is likely related to splenomegaly, which was present in 71.1% of evaluable patients at enrollment; 72.7% of evaluable patients during follow-up. Tremor was present before miglustat initiation in 35.7% (n = 86/241) of patients, and newly occurring or worsened tremor was reported for 37/241 (15.4%) patients during follow-up. Neuropathy was present in 7.5% (n = 18/240) of patients before miglustat initiation; newly occurring or worsened neuropathy was reported for 19/241 (7.9%) patients during follow-up.
The most common reasons for discontinuation of miglustat therapy were death and progression of NP-C disease (Supplementary Table 2).
Discussion
Here, we present the characteristics of the 472 patients enrolled in the NPC Registry at database closure, expanding on previous reports of the NPC Registry dataset [1, 12]. The NPC Registry is the largest repository of data for patients with this ultra-rare disease and allows reporting of the natural history, disease course, clinical outcomes, and treatment experience of patients with NP-C in real-world clinical settings. The description of NP-C from these data is consistent with previous reports of the natural history and disease course [1, 15, 16].
Since the most recent previous description of the entire enrolled population of the NPC Registry [1], the number of patients has greatly increased from 163 in the 2013 report to 472 in the present report, providing a greater wealth of data for all age at neurological onset categories. The proportion of patients within each of the age at neurological onset categories has remained relatively consistent (early-infantile, [2013] 11% vs [present] 13.5%; late-infantile, 31% vs 25.6%; juvenile, 31% vs 31.8%; adolescent/adult, 27% vs 29.1%) [1]. The present data, reporting the age at onset of neurological symptoms, the age at diagnosis by age of neurological onset, and the percentages of patients in which the individual cardinal symptoms of NP-C manifestations occur, are also consistent between the two reports [1]. The proportions of patients who presented at enrollment with the characteristically wide range of presenting visceral, neurological, and psychiatric symptoms are consistent with current understanding of the disease [1, 2, 8, 10, 15, 16]. Neurological signs such as ataxia, vertical supranuclear gaze palsy, dysarthria, and cognitive impairment were the most common, being identified in around two-thirds of patients. Splenomegaly is relatively common and is usually accompanied by hepatomegaly in this population. It should be noted that the frequency of these findings may be an underestimate, as some signs, such as the vertical supranuclear gaze palsy, are often not recognized by less-experienced clinicians. Similarly, organomegaly may be missed in those patients who undergo only a physical examination and can often be detected only through ultrasound or other imaging modalities.
We also present longitudinal analysis of functional disability in patients who were treated continuously with miglustat for at least 1 year. Functional disability, measured as a composite of deficits in ambulation, manipulation, language, and swallowing, was higher for patients in whom neurological symptoms manifest at a younger age than in patients who are older when neurological signs first appear. This is consistent with our understanding of NP-C as a disease, which has more severe presentation and rapid progression of symptoms in patients whose neurological manifestations appear at a younger age [2]. Functional disability was stable or improved in a majority (70.5%) of patients who had received continuous miglustat therapy and is consistent with previously reported findings using this same disability assessment method [7]. These findings are also consistent with earlier analyses of the NPC Registry, which reported a mean annual composite disability score increase of 0.038 (95% CI 0.018, 0.059), compared with 0.036 (95% CI 0.025,0.047) [12]. More broadly, these functional disability findings complement the wealth of data that support the use of miglustat as an effective therapy for the stabilization of the neurological manifestations of NP-C [16, 18,19,20, 22, 23].
A majority of the patients were continuously treated with miglustat for at least 12 months, which is sufficient to observe treatment benefits [4, 24]. Most of these patients had been receiving miglustat therapy prior to their enrollment to the NPC Registry; some had been receiving miglustat for several years. In line with the product label for miglustat, most subjects who have progressive neurological manifestations will have been enrolled into the miglustat treated cohort, apart from a very small number of subjects who did not or could not commence miglustat treatment. The remainder of the untreated cohort are those subjects in whom NP-C has not presented as the progressive neurological form. Consequently, the disease characteristics of the two cohorts are not easily comparable, confounding any side-by-side comparisons of treatment efficacy. A challenge also remains to account for confounding by indication, as reasons for initiation of miglustat treatment were not collected, as well as other unmeasured confounders. Further limiting these comparisons is the imbalance in numbers between cohorts, e.g. only 47 subjects for the not-treated cohort. For these reasons, it is not possible to compare the disease course and prognosis between those patients who were treated or untreated with miglustat.
No new safety findings were observed since the start of the NPC Registry. The tolerability of miglustat treatment within the NPC Registry dataset was consistent with observations from previous clinical and observational studies [18,19,20] and with those reported in previous descriptions of the NPC Registry [1, 12]. Diarrhea was relatively common but is known to be associated with miglustat treatment; simple dietary modifications and up-titration of drug dose can alleviate these symptoms in many patients [4, 25, 26]. Although the incidence of both tremor and seizures increased during the follow-up period, this is likely reflective of the worsening of neurological manifestations as part of the natural disease course. No increase in neuropathy was reported during the observation period.
These observations from the final NPC Registry population of 472 patients represent the largest database of patients with NP-C reported to date. As with any disease registry data, caution must be taken with their interpretation, as the integrity of any disease registry database relies on the accurate entry of patient information by the treating physicians and the staff at each participating center; this may explain some outlier data, for which age at neurological onset may have been recalled or entered incorrectly. However, due to the wealth of data available in this dataset on such a large number of patients with NP-C, these findings provide a valuable contribution to our existing knowledge of the disease and patient characteristics.
Conclusions
In addition to its important role in post-approval monitoring, the NPC Registry has provided an unparalleled wealth of data on NP-C. This final report of the NPC Registry database provides the largest ever NP-C-specific clinical dataset, which confirms and strengthens our understanding of the natural history and disease course of NP-C and supports previous findings of the effectiveness of miglustat as a disease-modifying therapy that can stabilize progression of the disease.
Availability of data and materials
The data sharing policy of the Sponsor is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
Notes
Miglustat is indicated for the treatment of progressive neurologic manifestations in adult and pediatric patients with NP-C. Miglustat is approved for NP-C under the brand name Zavesca® in Argentina, Australia, Brazil, Canada, Chile, China, Colombia, Ecuador, European Union/European Economic Area, Iceland, Liechtenstein, Iran, Israel, Mexico, New Zealand, Norway, Palestine, Panama, Peru, Russia, South Korea, Thailand, Turkey, and Venezuela. In Japan, miglustat is approved for NP-C under the brand name Brazaves®. Miglustat is not approved for the treatment of NP-C in the US and Taiwan.
Abbreviations
- CI:
-
Confidence interval
- EMA:
-
European Medicines Agency
- NP-C:
-
Niemann-Pick disease type C
- SD:
-
Standard deviation
References
Patterson MC, Mengel E, Wijburg FA, Muller A, Schwierin B, Drevon H, et al. Disease and patient characteristics in NP-C patients: findings from an international disease registry. Orphanet J Rare Dis. 2013;8:12.
Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis. 2010;5:16.
Geberhiwot T, Moro A, Dardis A, Ramaswami U, Sirrs S, Marfa MP, et al. Consensus clinical management guidelines for Niemann-Pick disease type C. Orphanet J Rare Dis. 2018;13:50.
Patterson MC, Hendriksz CJ, Walterfang M, Sedel F, Vanier MT, Wijburg F, et al. Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol Genet Metab. 2012;106:330–44.
Vanier MT, Millat G. Niemann-Pick disease type C. Clin Genet. 2003;64:269–81.
Wassif CA, Cross JL, Iben J, Sanchez-Pulido L, Cougnoux A, Platt FM, et al. High incidence of unrecognized visceral/neurological late-onset Niemann-Pick disease, type C1, predicted by analysis of massively parallel sequencing data sets. Genet Med. 2016;18:41–8.
Wraith JE, Baumgartner MR, Bembi B, Covanis A, Levade T, Mengel E, et al. Recommendations on the diagnosis and management of Niemann-Pick disease type C. Mol Genet Metab. 2009;98:152–65.
Garver WS, Francis GA, Jelinek D, Shepherd G, Flynn J, Castro G, et al. The National Niemann-Pick C1 disease database: report of clinical features and health problems. Am J Med Genet A. 2007;143A:1204–11.
Imrie J, Heptinstall L, Knight S, Strong K. Observational cohort study of the natural history of Niemann-Pick disease type C in the UK: a 5-year update from the UK clinical database. BMC Neurol. 2015;15:257.
Kelly DA, Portmann B, Mowat AP, Sherlock S, Lake BD. Niemann-Pick disease type C: diagnosis and outcome in children, with particular reference to liver disease. J Pediatr. 1993;123:242–7.
Iturriaga C, Pineda M, Fernandez-Valero EM, Vanier MT, Coll MJ. Niemann-Pick C disease in Spain: clinical spectrum and development of a disability scale. J Neurol Sci. 2006;249:1–6.
Patterson MC, Mengel E, Vanier MT, Schwierin B, Muller A, Cornelisse P, et al. Stable or improved neurological manifestations during miglustat therapy in patients from the international disease registry for Niemann-Pick disease type C: an observational cohort study. Orphanet J Rare Dis. 2015;10:65.
Jahnova H, Dvorakova L, Vlaskova H, Hulkova H, Poupetova H, Hrebicek M, et al. Observational, retrospective study of a large cohort of patients with Niemann-Pick disease type C in the Czech Republic: a surprisingly stable diagnostic rate spanning almost 40 years. Orphanet J Rare Dis. 2014;9:140.
Wraith JE, Guffon N, Rohrbach M, Hwu WL, Korenke GC, Bembi B, et al. Natural history of Niemann-Pick disease type C in a multicentre observational retrospective cohort study. Mol Genet Metab. 2009;98:250–4.
Wraith JE, Sedel F, Pineda M, Wijburg FA, Hendriksz CJ, Fahey M, et al. Niemann-Pick type C Suspicion Index tool: analyses by age and association of manifestations. J Inherit Metab Dis. 2014;37:93–101.
Pineda M, Jurickova K, Karimzadeh P, Kolnikova M, Malinova V, Insua JL, et al. Disease characteristics, prognosis and miglustat treatment effects on disease progression in patients with Niemann-Pick disease Type C: an international, multicenter, retrospective chart review. Orphanet J Rare Dis. 2019;14:32.
Patterson MC, Clayton P, Gissen P, Anheim M, Bauer P, Bonnot O, et al. Recommendations for the detection and diagnosis of Niemann-Pick disease type C: an update. Neurol Clin Pract. 2017;7:499–511.
Patterson MC, Vecchio D, Jacklin E, Abel L, Chadha-Boreham H, Luzy C, et al. Long-term miglustat therapy in children with Niemann-Pick disease type C. J Child Neurol. 2010;25:300–5.
Patterson MC, Vecchio D, Prady H, Abel L, Wraith JE. Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. Lancet Neurol. 2007;6:765–72.
Wraith JE, Vecchio D, Jacklin E, Abel L, Chadha-Boreham H, Luzy C, et al. Miglustat in adult and juvenile patients with Niemann-Pick disease type C: long-term data from a clinical trial. Mol Genet Metab. 2010;99:351–7.
Bonnot O, Gama CS, Mengel E, Pineda M, Vanier MT, Watson L, et al. Psychiatric and neurological symptoms in patients with Niemann-Pick disease type C (NP-C): findings from the international NPC Registry. World J Biol Psychiatry. 2019;20:310–9.
Nadjar Y, Hütter-Moncada AL, Latour P, Ayrignac X, Kaphan E, Tranchant C, et al. Adult Niemann-Pick disease type C in France: clinical phenotypes and long-term miglustat treatment effect. Orphanet J Rare Dis. 2018;13:175.
Pineda M, Walterfang M, Patterson MC. Miglustat in Niemann-Pick disease type C patients: a review. Orphanet J Rare Dis. 2018;13:140.
Sedel F, Chabrol B, Audoin B, Kaphan E, Tranchant C, Burzykowski T, et al. Normalisation of brain spectroscopy findings in Niemann-Pick disease type C patients treated with miglustat. J Neurol. 2016;263:927–36.
Champion H, Ramaswami U, Imrie J, Lachmann RH, Gallagher J, Cox TM, et al. Dietary modifications in patients receiving miglustat. J Inherit Metab Dis. 2010;33(Suppl 3):S379–83.
Belmatoug N, Burlina A, Giraldo P, Hendriksz CJ, Kuter DJ, Mengel E, et al. Gastrointestinal disturbances and their management in miglustat-treated patients. J Inherit Metab Dis. 2011;34:991–1001.
Acknowledgements
The authors would like to thank all who contributed data to the NPC Registry. MCP would like to acknowledge the support of the Peggy Furth Fund at Mayo Clinic; MP would like to thank the Spanish Foundation of NP-C for all their help.
Funding
The NPC Registry was funded by Actelion Pharmaceuticals Ltd. Medical writing support was provided by Andrew Smith, PhD, and Gosia Carless, PhD, of Fishawack Communications, funded by Actelion Pharmaceuticals Ltd.
Author information
Authors and Affiliations
Contributions
All authors have reviewed and interpreted the data, reviewed each draft of the manuscript, and approved the final version for submission.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Before enrollment into the Registry, the patient and/or legal guardian must have signed a Patient Informed Consent Form, which authorized data collection.
Consent for publication
Not applicable.
Competing interests
MCP has been a member of Actelion Pharmaceuticals Ltd. Advisory Board, and has received honoraria for talks from Actelion Pharmaceuticals Ltd.
EM has been a member of Actelion Pharmaceuticals Ltd. Advisory Board, and has received honoraria for talks and/or research grants from Actelion Pharmaceuticals Ltd.
MTV has been a member of Actelion Pharmaceuticals Ltd. Advisory Board, and has received travel reimbursement and honoraria for talks from Actelion Pharmaceuticals Ltd.
PM and DR are employees of Actelion Pharmaceuticals Ltd.
MP has received honoraria for talks and travel reimbursement from Actelion Pharmaceuticals Ltd.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Supplementary Table 1.
Countries from which patients were enrolled to the NPC Registry up until database closure. Additional file 1: Supplementary Table 2. Reasons for discontinuation of miglustat treatment.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Patterson, M.C., Mengel, E., Vanier, M.T. et al. Treatment outcomes following continuous miglustat therapy in patients with Niemann-Pick disease Type C: a final report of the NPC Registry. Orphanet J Rare Dis 15, 104 (2020). https://doi.org/10.1186/s13023-020-01363-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-020-01363-2