
Abstract
Background
A growing number of non-coding regulatory mutations are being identified in congenital disease. Very recently also some exons of protein coding genes have been identified to act as tissue specific enhancer elements and were therefore termed exonic enhancers or “eExons”.
Methods
We screened a cohort of 134 unrelated families with split-hand/split-foot malformation (SHFM) with high resolution array CGH for CNVs with regulatory potential.
Results
In three families with an autosomal dominant non-syndromic SHFM phenotype we detected microdeletions encompassing the exonic enhancer (eExons) 15 and 17 of DYNC1I1. In a fourth family, who had hearing loss in addition to SHFM, we found a larger deletion of 510 kb including the eExons of DYNC1I1 and, in addition, the human brain enhancer hs1642. Exons 15 and 17 of DYNC1I1 are known to act as tissue specific limb enhancers of DLX5/6, two genes that have been shown to be associated with SHFM in mice. In our cohort of 134 unrelated families with SHFM, deletions of the eExons of DYNC1I1 account for approximately 3% of the cases, while 17p13.3 duplications were identified in 13% of the families, 10q24 duplications in 12%, and TP63 mutations were detected in 4%.
Conclusions
We reduce the minimal critical region for SHFM1 to 78 kb. Hearing loss, however, appears to be associated with deletions of a more telomeric region encompassing the brain enhancer element hs1642. Thus, SHFM1 as well as hearing loss at the same locus are caused by deletion of regulatory elements. Deletions of the exons with regulatory potential of DYNC1I1 are an example of the emerging role of exonic enhancer elements and their implications in congenital malformation syndromes.
Similar content being viewed by others

Background
Split-hand/split-foot malformation (SHFM; OMIM 183600), also referred to as ectrodactyly, is a congenital limb malformation, characterized by a claw-like appearance of the distal portion of the upper and lower limbs with profound median clefts of hands and feet and missing or malformed central fingers [1]. This disorder is clinically heterogeneous, comprising both isolated as well as syndromic forms [2]. SHFM is typically inherited as an autosomal dominant trait with incomplete penetrance, variable expressivity and segregation distortion [3]. However, autosomal recessive inheritance has been reported [4]-[6]. The pathogenesis of SHFM has been studied in various mouse models. A failure to maintain signalling from the median apical ectodermal ridge (AER) has been shown to be a main pathogenic mechanism [7]-[9]. Without this signalling, cells of the underlying progress zone stop proliferation and differentiation which in turn results in defects of the central rays. The time point and extent of this signalling deficit determines the phenotype. Early and more extensive defects will result in more severe deficiencies and vice versa.
At least 5 different loci have been associated with non-syndromic SHFM. The most common form, which may also feature long bone (tibial hemimelia) deficiency (SHFLD) is caused by duplications involving the gene BHLHA9[10] (SHFLD3; MIM 612576). SHFM3 (OMIM 600095) is associated with the duplication of chromosome 10q24-q25, which encompasses the Dactylin gene (FBXW4) [11]. SHFM4 (OMIM 605289) is caused by mutations in TP63 on 3q27 [12]. The gene appears to be essential for ectodermal development and a deficiency affects function and maintenance of the AER. SHFM5 (OMIM 606708) maps to 2q31. Although the disease gene has not been identified for this locus yet, a contribution of HOXD gene cluster has been suggested for SHFM5 [13]. SHFM1 (OMIM 183600) maps to 7q21.2-q21.3 including the genes DLX5, DLX6 and SHFM1.
SHFM1 is an autosomal dominant trait with reduced penetrance and variable expression ranging from mild defects of a single limb to severe abnormalities of all four extremities [14]. SHFM1 is associated with mental retardation in 33% of patients, craniofacial malformations in more than 35% of patients, and deafness in 35% of patients (SHFM1D; OMIM 220600) [15]. Physical mapping of the SHFM1 locus on chromosome 7 defined a critical interval of about 1.5 Mb that encompasses SHFM1 (deleted in the split-hand/split-foot 1 region), Dlx5 and Dlx6 (distal less-related homeobox gene 5 and 6), of which only Dlx5/6 have been shown to play a role in early limb development [16],[17]. The Dlx5 and Dlx6 are expressed in the AER of the embryonic limb buds, craniofacial prominence, otic vesicle, and in the brain. Dlx5 deficient mice do not show any limb defects [18]. However, disruption of both Dlx5 and Dlx6 in mice leads to ectrodactyly [7]. So far a single consanguineous family with severe SHFM1 phenotype and hearing impairment was demonstrated to carry a homozygous missense mutation in DLX5[19]. Furthermore, Wang et al. identified a novel heterozygous missense mutation in exon 3 of DLX5 in a Chinese family with autosomal dominant SHFM1 [20].
Recently, exons 15 and 17 of DYNC1I1 (dynein cytoplasmic 1 intermediate chain 1) were identified as “coding” limb enhancers of Dlx5/6 in mice and zebrafish [21]. The two coding exons act as “exonic enhancers” or “eExons” and were shown to physically interact with the promoters of Dlx5/6 in mice using chromosome conformation capture analysis [21]. Dync1i1 itself is not expressed during limb development. In addition, an enhancer element within the intronic region of SLC25A13 (solute carrier family 25 member 13) was shown to regulate tissue specific Dlx5/6 expression in the otic vesicle, forebrain, branchial arch and limb bud mesenchyme. The deletion of Slc25a13 in mice is not associated with a SHFM phenotype [22]-[24].
Here we report on three unrelated families with overlapping deletions of the cis-regulatory eExons 15 and 17 of DYNC1I1 located over 900 kb centromeric to their target genes DLX5/6 on chromosome 7q21.3. In a fourth family presenting with SHFM and hearing loss we detected a 510 kb deletion including the eExons 15 and 17 of DYNC1I1 but also SLC25A13, C7orf76, and several brain enhancers. In our cohort of 134 unrelated families with SHFM and SHFLD deletions of the eExons of DYNC1I1 on chromosome 7q21.3 account for approximately 3% of the cases. These data have a direct impact on SHFM1 diagnostics and further highlight the role of regulatory mutations and structural variations in congenital disease.
Material and methods
Patients
In a cohort of 134 unrelated families with SHFM we excluded the known causes of SHFM and SHFLD i.e. 17p13.3 duplications with ectrodactyly and tibia hemimelia [25], 10q24 duplications and mutations in TP63 with the clinical manifestation of ectrodactyly without tibial involvement by quantitative real-time PCR (qPCR) and Sanger sequencing, respectively [25]. Balanced rearrangements were not excluded. The remaining families were used to screen for CNVs using high resolution (1 M Agilent) array CGH. Written informed consent was obtained from all individuals studied, or their parents or guardians in the case of children under 16, to participate in the study and for publication of this manuscript and any accompanying images. The study was approved by the Charité Universitätsmedizin Berlin ethics committee as well as by the Institutional Review Board of Poznan University of Medical Sciences.
Microarray-based comparative genomic hybridization (array CGH)
All experiments were done with genomic DNA extracted from blood samples. Array CGH for family 1, 3, and 4 was carried out using a whole genome 1 M oligonucleotide array (Agilent, Santa Clara, CA). 1 M arrays were analyzed by Feature Extraction v9.5.3.1 and CGH Analytics v3.4.40 software or Cytogenomics v2.5.8.11, respectively (Agilent, Santa Clara, CA). Family 2 was tested on Roche NimbleGen platform with use of 1.4 M oligo arrays according to standard protocols provided by the manufacturer. Analysis settings: aberration algorithm: ADM-2; threshold: 6.0; window size: 0.2 Mb; filter: 5probes, log2ratio = 0.29. The genomic profile was visualized by the SignalMap software (SignalMap v1.9.0.03, NimbleGen Systems Inc.). Data were submitted to the DECIPHER database (http://decipher.sanger.ac.uk); accession numbers: BER284939, BER284938, BER284937, and BER285016.
Quantitative real-time PCR (qPCR)
qPCR was performed as described previously [26],[27] using genomic DNA of the index patients and further family members to confirm the deletions and to show segregation with the phenotype. Primer sequences are given in Additional file 1: Table S1.
Breakpoint analysis
The exact determination of the deletion size was done by breakpoint spanning PCR following the qPCR analyses and sequencing of the junction fragment (primer sequences and positions in Additional file 1: Table S1 and Additional file 1: Figure S1).
PCR was performed in a total volume of 20 μl with 40 ng genomic DNA as template, 2 μl 10× PCR buffer, 0.6 μl dNTP mix (10 mM), 0.5 μl primer (10 pMol/μl), 0.6 μl MgCl2 (50 mM), 0.2 μl Taq polymerase (Rapidozym, Germany). PCR conditions are available upon request. The PCR products were purified by enzymatic treatment (Exonuclease I, NEB; Shrimp Alkaline Phosphatase, Roche Diagnostics). For the sequencing of the PCR products the BigDye v3.1 (Applied Biosystems) sequencing kit was used. PCR products were analyzed by capillary automat ABI3730 (Applied Biosystems). The sequencing results were processed by DNA-STAR software (DNA-Star).
Results
Phenotypic analysis
We investigated a total of 16 affected from 4 unrelated families.
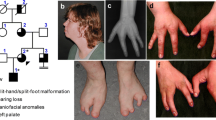
Family 1 is a non-consanguineous family from Poland with three affected individuals (grandfather, father and son), all showing the classical SHFM phenotype in all four extremities. Figure 1 (a-d & f-i) shows the hand and feet of the affected one-year-old son. Neither long bone involvement nor craniofacial abnormalities were noted. There was no history of hearing impairment in the family. Neurological and motor development was normal.

Limb phenotypes and pedigrees of families 1 and 2. Family 1 is a non-consanguineous family from Poland with three affected individuals. All affected individuals showed split hands and feet in all four extremities. Hands (a-d) and feet (f-i) of the affected one-year-old son. No long bone involvement or other bone phenotype was reported. There was no history of hearing impairment in the family. Neurological and motor development was normal. Family 2 is a non-consanguineous German family with five affected individuals. All affected individuals showed SHFM in both hands and feet and severe hearing loss. The hands (e) and feet (j) of a 69-year-old affected female. No long bone involvement or other bone phenotype was reported. Neurological and motor development was normal in all family members.
Family 2 is a non-consanguineous German family with five affected individuals (a grandfather, two affected sisters and their two daughters). Four affected individuals showed SHFM in both hands and feet and, in addition, severe hearing loss. One female showed SHFM in both hands but only one foot and hearing loss. Figure 1 (e and j) shows the hands and feet of a 69-year-old affected female. Neither long bone involvement nor craniofacial abnormalities were noted. Neurological and motor development was normal in all family members.
Family 3 is a non-consanguineous family of Polish origin with six individuals from three generations all affected by SHFM. All patients presented with variable limb malformations, ranging from a severe cleft in all limbs (see index in Figure 2a, b) to milder defects, such as absent or hypoplastic central digit (aunt of the index in Figure 2c). None of the patients from family 3 manifested long bone deficiency or hypoplasia. Psychomotor development and hearing of all affected family members was normal.

Limb phenotypes and pedigree of family 3. Family 3 is a non-consanguineous family from Poland with six affected individuals (grandmother, two affected sisters and their brother, and two male offspring of the brother and one of the sisters). All patients showed SHFM in all for limbs with no long bone involvement. Hearing loss was not reported in this family. The proband shows bilateral SHFM of the hands (a) and feet (b). The father of the proband shows central ray deficiency of the hands (c) and right foot (d). The aunt of the proband presents milder phenotype on the left hand (e) and feet (f).
Family 4 is a non-consanguineous German with two affected sisters (monozygotic twins) showing a SHFM phenotype in the hands only. Neither long bone involvement nor craniofacial abnormalities were noted. There was no history of hearing impairment in the family. Neurological and motor development was normal.
In our cohort of patients with deletions of the eExons of DYNC1I1 the penetrance was high. However, due to the small sample size future studies are needed to determine the penetrance of DYNC1I1 deletions.
Deletions of the eExons 15 and 17 of DYNC1I1 are associated with SHFM1
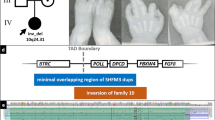
We identified three unrelated families with overlapping deletions of the cis-regulatory eExons of DYNC1I1 over 900 kb centromeric to DLX5/6 with a split-hand/split foot phenotype (Figure 3). In families 1, 3, and 4 we detected overlapping microdeletions of 167 kb, 205 kb and 167 kb, respectively. The deletions encompass the eExons 15 and 17 of DYNC1I1 as well as the last three exons of SLC25A13 including the enhancer elements eDlx#23 within its intronic region (Figure 3). The latter has been shown to drive expression in the otic vesicle, forebrain, branchial arch and limb bud mesenchyme. The DYNC1I1 exons 15 and 17 have previously been shown to act as tissue-specific enhancers of Dlx5/6 in mouse and zebrafish [21]. The deletions are therefore likely to result in tissue-specific loss of function of DLX5/6 in the developing hand or foot causing SHFM.

Schematic representation of the microdeletions on 7q21.3 associated with SHFM. In three families (Family 1, 3 and 4) with an autosomal dominant non-syndromic SHFM phenotype overlapping microdeletions were detected. The deletions encompass the eExons 15 and 17 of DYNC1I1 (red) as well as the last three exons of SLC25A13 including the enhancer elements eDlx#23 (blue) within its intronic region driving expression in otic vesicle. The DYNC1I1 exons 15 and 17 have previously been shown to act as tissue-specific enhancers of Dlx5/6 in mouse and zebrafish [21]. In family 2 presenting with SHFM and hearing loss a 510 kb deletion was identified including the eExons of DYNC1I1 but also SLC25A13, C7orf76, the brain enhancer element hs1642 and two branchial arch enhancers (green) [28]. The deletion of hs1642 might be responsible for hearing loss in family 2. The SHFM family reported by Kouwenhoven et al. developed hearing loss during adolescents [29]. P1- P4 indicated the qPCR amplicons used in this study. Breakpoint sequences are shown in Additional file 1: Figure S1.
In family 2 presenting with SHFM and hearing loss we detected a 510 kb deletion encompassing the eExons of DYNC1I1 but also SLC25A13, C7orf76 and the human brain enhancer element hs1642 (Figure 3).
The array CGH results were confirmed by qPCR in the index patients. Analysis of further affected and unaffected family members of all 4 families showed that the microdeletions segregate with the phenotype, i.e. unaffected family members had a normal copy number of the region. The qPCR data showed only one copy of the analyzed amplicons, i.e. a deletion, in all affected individuals. Similar deletions were not observed in 200 control DNA samples and are were not present in the Database of Genomic Variants (DGV) (http://dgv.tcag.ca/dgv/app/home).
Breakpoint analysis
In order to determine the exact breakpoints of the microdeletions and to exclude translocations or inversions, junction fragments that included the telomeric and centromeric breakpoints were PCR amplified and subsequently sequenced in all affected individuals. Deletion breakpoints were detectable only in the affected, but not in the unaffected individuals. In family 1 we detected a 167 kb deletion on 7q21.3 (genomic position chr7:95,615,187-95,783,313). In family 2 we detected a 510 kb deletion on 7q21.3 (genomic position chr7:95,624,825-96,135,521). In family 3 a 205 kb deletion on 7q21.3 (genomic position chr7:95,667,046-95,872,044) was detected (Table 1). Breakpoint spanning sequences are shown in Additional file 1: Figure S1. In family 4 we detected a 169 kb deletion on 7q21.3 (maximum genomic position chr7:95,693,341-95,862,369). For family 4, it was not possible to obtain additional DNA to performer breakpoints sequencing. All positions are given in hg19.
Deletions of eExons of DYNC1I1 account for approximately 3% of SHFM/SHFLD cases
In our cohort of 134 unrelated families with SHFM and SHFLD the following molecular analyses were performed: qPCR for 17p13.3 duplications, qPCR for 10q24 duplications, qPCR for deletions of the eExons 15 and 17 of DYNC1I1 on 7q21.3, and mutational analysis for TP63. WNT10B mutation analysis was only performed in seven consanguineous families. Balanced rearrangements were not excluded.
In our cohort, 17p13.3 duplications were identified in 13% (18/134) of the families, 10q24 duplications in 12% (16/134), TP63 mutations were detected in 4% (6/134), and deletions of the eExons of DYNC1I1 in 3% (3/134) (Figure 4). Thus, in 68% of the families with SHFM and SHFLD no molecular diagnosis was possible.

Deletions of eExons of DYNC1I1 account for approximately 3% of SHFM/SHFLD cases: In our cohort of SHFM/SHFLD cases, 17p13.3 duplications were identified in 13% (18/134) of the families, 10q24 duplications in 12% (16/134), TP63 mutations were detected in 4% (6/134), and deletions of the eExons of DYNC1I1 in 3% (3/134). WNT10B mutation analysis was negative in all seven consanguineous families investigated. In the majority of families with SHFM and SHFLD (68%) no molecular diagnosis could be made.
Conclusions
Congenital malformations represent a major medical problem with a prevalence of 2-6% in humans [30]. Elucidation of the genetic basis of this heterogeneous group of disorders has been tremendously successful over the last decades by the interpretation of disease related variations in the 1.5% of protein coding DNA and the identification of variants that result in the disruption of specific gene function [31]. However, the growing number of regulatory mutations mainly located in the non-coding DNA affecting cis-regulatory elements highlights the clinical importance of long range gene regulation in congenital disease [32],[33]. Very recently also some exons of protein coding genes have been identified to act as tissue specific enhancer elements and were therefore termed exonic enhancers or “eExons” [34].
In this study, we report on four unrelated families with overlapping deletions of the cis-regulatory eExons 15 and 17 of DYNC1I1 that act as tissue specific limb enhancers of DLX5/6 which is located over 900 kb telomeric on chromosome 7q21.3. In three families with an autosomal dominant non-syndromic SHFM phenotype affecting all four limbs we detected overlapping microdeletions of 167 kb, 169 kb and 205 kb. The deletions encompass the two limb specific eExons 15 and 17 of DYNC1I1 as well as the last three exons of the SLC25A13 gene. In family 2 presenting with SHFM and hearing loss we detected a 510 kb deletion including the eExons 15 and 17 of DYNC1I1 but also SLC25A13, C7orf76, two branchial arch enhancers and the human brain enhancer element hs1642. All four deletions encompass the eExons of DYNC1I1. Interestingly, the enhancer element eDlx#23 that was shown to drive reporter expression in otic vesicle, forebrain, branchial arch and limb bud mesenchyme was also deleted in all three families, but only family 2 manifested hearing loss (Figure 3) [21]. Lango Allen et al. reported on a family with SHFM but without hearing loss carrying a similar 106 kb deletion also including the eExons of DYNC1I1 and the otic vesicle enhancer elements eDlx#23 [35]. These data suggest that the loss of otic vesicle enhancer eDlx#23 is not pathogenic in these patients. Rather, it is more likely that hearing loss in family 2 results from the deletion of the telomeric 264 kb region encompassing two branchial arch enhancers, the human enhancer element hs1642, and C7orf76 (Figure 3) [21],[28]. The human enhancer element hs1642 shows a tissue specific reporter expression in the forebrain, the hindbrain (rhombencephalon), and the neural tube (Figure 3) [28]. This region also includes the auditory forebrain and might therefore be a potential candidate for hearing loss [36].
Another large deletion of over 900 kb was described by Kouwenhoven et al. in an individual that was affected with SHFM and later also developed hearing loss. In this study, they showed that p63 binds to an enhancer element that is located between SHFM and DLX5/6[29]. In another recent study Restelli et al. report that the fibroblast growth factor 8 (FGF8) locus is a downstream target of DLX5. Furthermore they show that DLX5, p63, Pin1 and FGF8 are essential for AER stratification and normal patterning and skeletal morphogenesis of the limb buds [37]. Several other translocations and inversions at the SHFM1 locus have been described that separate DLX5/6 from their regulators located between DYNC1I1 and DLX5/6[21]. Depending on the location of the breakpoint a genotype-phenotype correlation seems now feasible. Our results confirm that deletions of the cis-regulatory eExons 15 and 17 of DYNC1I1 are associated with split-hand/split foot and we reduce the SHFM1 minimal critical region to 78 kb (Figure 3). Hearing loss, however, seems to be associated with deletions of a more telomeric 264 kb region encompassing the brain enhancer element hs1642 (Figure 3). Craniofacial defects seem to result from breakpoints located in the region closest to DLX5/6[38].
In order to determine the clinical relevance and prevalence of deletions of the eExons of DYNC1I1 we screened our cohort of SHFM and SHFLD cases. In our cohort of 134 unrelated families with SHFM and SHFLD, deletions of the eExons of DYNC1I1 on chromosome 7q21.3 account for approximately 3% of the cases. 17p13.3 duplications mainly associate with ectrodactyly and tibia hemimelia [25], while mutations in TP63 are mainly associated with ectrodactyly without tibial involvement. 10q24 duplications show the highest phenotypical variability ranging from single digit shortening to absence of hands and feet. The introduction of whole exome sequencing into clinical diagnostics will reveal the role of mutations in DLX5 and WNT10B in SHFM. Our data have a direct impact on diagnostics for SHFM since they further highlight the importance of array-CGH as a diagnostic tool for SHFM/SHFLD and to detect regulatory mutations [39]. Furthermore the deletions of the eExons of DYNC1I1 are an example of the emerging role of exonic enhancer elements and their role in congenital malformations.
Additional file
References
Sifakis S, Basel D, Ianakiev P, Kilpatrick M, Tsipouras P: Distal limb malformations: underlying mechanisms and clinical associations. Clin Genet. 2001, 60: 165-172. 10.1034/j.1399-0004.2001.600301.x.
Basel D, Kilpatrick MW, Tsipouras P: The expanding panorama of split hand foot malformation. Am J Med Genet A. 2006, 140: 1359-1365. 10.1002/ajmg.a.31304.
Scherer SW, Poorkaj P, Massa H, Soder S, Allen T, Nunes M, Geshuri D, Wong E, Belloni E, Little S, Zhou L, Becker D, Kere J, Ignatius J, Nllkawa J, Fukushlma Y, Hasegawa T, Weissenbach J, Boncinelli E, Trask B, Tsui L, Evans JP: Physical mapping of the split hand/split foot locus on chromosome 7 and implication in syndromic ectrodactyly. Hum Mol Genet. 1994, 3: 1345-1354. 10.1093/hmg/3.8.1345.
Freire-Maia A: A recessive form of ectrodactyly, and its implications in genetic counseling. J Hered. 1971, 62: 53-
Verma IC, Joseph R, Bhargava S, Mehta S: Split-hand and split-foot deformity inherited as an autosomal recessive trait. Clin Genet. 1976, 9: 8-14. 10.1111/j.1399-0004.1976.tb01543.x.
Faiyaz ul Haque M, Uhlhaas S, Knapp M, Schuler H, Friedl W, Ahmad M, Propping P: Mapping of the gene for X-chromosomal split-hand/split-foot anomaly to Xq26-q26.1. Hum Genet. 1993, 91: 17-19. 10.1007/BF00230215.
Robledo RF, Rajan L, Li X, Lufkin T: The Dlx5 and Dlx6 homeobox genes are essential for craniofacial, axial, and appendicular skeletal development. Genes Dev. 2002, 16: 1089-1101. 10.1101/gad.988402.
Capdevila J, Izpisua Belmonte JC: Patterning mechanisms controlling vertebrate limb development. Annu Rev Cell Dev Biol. 2001, 17: 87-132. 10.1146/annurev.cellbio.17.1.87.
Duijf PH, van Bokhoven H, Brunner HG, et al: Pathogenesis of split-hand/split-foot malformation.Hum Mol Genet 2003 :R51–R60. 12 Spec No 1.,
Armour CM, Bulman DE, Jarinova O, Rogers RC, Clarkson KB, DuPont BR, Dwivedi A, Bartel FO, McDonell L, Schwartz CE, Boycott KM, Everman DB, Graham GE: 17p13.3 microduplications are associated with split-hand/foot malformation and long-bone deficiency (SHFLD). Eur J Hum Genet: EJHG. 2011, 19: 1144-1151. 10.1038/ejhg.2011.97.
Gurrieri F, Prinos P, Tackels D, Kilpatrick MW, Allanson J, Genuardi M, Vuckov A, Nanni L, Sangiorgi E, Garofalo G, Nunes ME, Neri G, Schwartz C, Tsipouras P: A split hand-split foot (SHFM3) gene is located at 10q24– > 25. Am J Med Genet. 1996, 62: 427-436. 10.1002/(SICI)1096-8628(19960424)62:4<427::AID-AJMG16>3.0.CO;2-Q.
van Bokhoven H, Hamel BC, Bamshad M, Sangiorgi E, Gurrieri F, Duijf PH, Vanmolkot KR, van Beusekom E, van Beersum SE, Celli J, Merkx GF, Tenconi R, Fryns JP, Verloes A, Newbury-Ecob RA, Raas-Rotschild A, Majewski F, Beemer FA, Janecke A, Chitayat D, Crisponi G, Kayserili H, Yates JR, Neri G, Brunner HG: p63 Gene mutations in eec syndrome, limb-mammary syndrome, and isolated split hand-split foot malformation suggest a genotype-phenotype correlation. Am J Hum Genet. 2001, 69: 481-492. 10.1086/323123.
Goodman FR, Majewski F, Collins AL, Scambler PJ: A 117-kb microdeletion removing HOXD9-HOXD13 and EVX2 causes synpolydactyly. Am J Hum Genet. 2002, 70: 547-555. 10.1086/338921.
Sowinska-Seidler A, Socha M, Jamsheer A: Split-hand/foot malformation - molecular cause and implications in genetic counseling. J Appl Genet. 2014, 55: 105-115. 10.1007/s13353-013-0178-5.
Elliott AM, Evans JA: Genotype-phenotype correlations in mapped split hand foot malformation (SHFM) patients. Am J Med Genet A. 2006, 140: 1419-1427. 10.1002/ajmg.a.31244.
Scherer SW, Poorkaj P, Allen T, Kim J, Geshuri D, Nunes M, Soder S, Stephens K, Pagon RA, Patton MA, Berg MA, Donlon T, Rivera H, Pfeiffer RA, Naritomi K, Hughes H, Genuardi M, Gurrieri F, Neri G, Lovrein E, Magenis E, Tsui L, Evans JP: Fine mapping of the autosomal dominant split hand/split foot locus on chromosome 7, band q21.3-q22.1. Am J Hum Genet. 1994, 55: 12-20.
van Silfhout AT, van den Akker PC, Dijkhuizen T, Verheij JB, Olderode-Berends MJ, Kok K, Sikkema-Raddatz B, van Ravenswaaij-Arts CM: Split hand/foot malformation due to chromosome 7q aberrations(SHFM1): additional support for functional haploinsufficiency as the causative mechanism. Eur J Hum Genet. 2009, 17: 1432-1438. 10.1038/ejhg.2009.72.
Acampora D, Merlo GR, Paleari L, Zerega B, Postiglione MP, Mantero S, Bober E, Barbieri O, Simeone A, Levi G: Craniofacial, vestibular and bone defects in mice lacking the Distal-less-related gene Dlx5. Development. 1999, 126: 3795-3809.
Shamseldin HE, Faden MA, Alashram W, Alkuraya FS: Identification of a novel DLX5 mutation in a family with autosomal recessive split hand and foot malformation. J Med Genet. 2012, 49: 16-20. 10.1136/jmedgenet-2011-100556.
Wang X, Xin Q, Li L, Li J, Zhang C, Qiu R, Qian C, Zhao H, Liu Y, Shan S, Dang J, Bian X, Shao C, Gong Y, Liu Q, et al: Exome sequencing reveals a heterozygous DLX5 mutation in a Chinese family with autosomal-dominant split-hand/foot malformation.Eur J Hum Genet 2014, doi: 10.1038/ejhg.2014.7. [Epub ahead of print].,
Birnbaum RY, Everman DB, Murphy KK, Gurrieri F, Schwartz CE, Ahituv N: Functional characterization of tissue-specific enhancers in the DLX5/6 locus. Hum Mol Genet. 2012, 21: 4930-4938. 10.1093/hmg/dds336.
Kikuchi A, Arai-Ichinoi N, Sakamoto O, Matsubara Y, Saheki T, Kobayashi K, Ohura T, Kure S: Simple and rapid genetic testing for citrin deficiency by screening 11 prevalent mutations in SLC25A13. Mol Genet Metab. 2012, 105: 553-558. 10.1016/j.ymgme.2011.12.024.
Kobayashi K, Sinasac DS, Iijima M, Boright AP, Begum L, Lee JR, Yasuda T, Ikeda S, Hirano R, Terazono H, Crackower MA, Kondo I, Tsui LC, Scherer SW, Saheki T: The gene mutated in adult-onset type II citrullinaemia encodes a putative mitochondrial carrier protein. Nat Genet. 1999, 22: 159-163. 10.1038/9667.
Sinasac DS, Moriyama M, Jalil MA, Begum L, Li MX, Iijima M, Horiuchi M, Robinson BH, Kobayashi K, Saheki T, Tsui LC: Slc25a13-knockout mice harbor metabolic deficits but fail to display hallmarks of adult-onset type II citrullinemia. Mol Cell Biol. 2004, 24: 527-536. 10.1128/MCB.24.2.527-536.2004.
Klopocki E, Lohan S, Doelken SC, Stricker S, Ockeloen CW, Soares Thiele de Aguiar R, Lezirovitz K, Mingroni Netto RC, Jamsheer A, Shah H, Kurth I, Habenicht R, Warman M, Devriendt K, Kordass U, Hempel M, Rajab A, Makitie O, Naveed M, Radhakrishna U, Antonarakis SE, Horn D, Mundlos S: Duplications of BHLHA9 are associated with ectrodactyly and tibia hemimelia inherited in non-Mendelian fashion. J Med Genet. 2012, 49: 119-125. 10.1136/jmedgenet-2011-100409.
Klopocki E, Ott CE, Benatar N, Ullmann R, Mundlos S, Lehmann K: A microduplication of the long range SHH limb regulator (ZRS) is associated with triphalangeal thumb-polysyndactyly syndrome. J Med Genet. 2008, 45: 370-375. 10.1136/jmg.2007.055699.
Klopocki E, Lohan S, Brancati F, Koll R, Brehm A, Seemann P, Dathe K, Stricker S, Hecht J, Bosse K, Betz RC, Garaci FG, Dallapiccola B, Jain M, Muenke M, Ng VC, Chan W, Chan D, Mundlos S: Copy-number variations involving the IHH locus are associated with syndactyly and craniosynostosis. Am J Hum Genet. 2011, 88: 70-75. 10.1016/j.ajhg.2010.11.006.
Visel A, Minovitsky S, Dubchak I, Pennacchio LA: VISTA Enhancer Browser–a database of tissue-specific human enhancers. Nucleic Acids Res. 2007, 35: D88-D92. 10.1093/nar/gkl822.
Kouwenhoven EN, van Heeringen SJ, Tena JJ, Oti M, Dutilh BE, Alonso ME, de la Calle-Mustienes E, Smeenk L, Rinne T, Parsaulian L, Bolat E, Jurgelenaite R, Huynen MA, Hoischen A, Veltman JA, Brunner HG, Roscioli T, Oates E, Wilson M, Manzanares M, Gomez-Skarmeta JL, Stunnenberg HG, Lohrum M, van Bokhoven H, Zhou H: Genome-wide profiling of p63 DNA-binding sites identifies an element that regulates gene expression during limb development in the 7q21 SHFM1 locus. PLoS Genet. 2010, 6: e1001065-10.1371/journal.pgen.1001065.
Martinez-Frias ML, Bermejo E, Frias JL: Pathogenetic classification of a series of 27,145 consecutive infants with congenital defects. Am J Med Genet. 2000, 90: 246-249. 10.1002/(SICI)1096-8628(20000131)90:3<246::AID-AJMG12>3.0.CO;2-Q.
Kornak U, Mundlos S: Genetic disorders of the skeleton: a developmental approach. Am J Hum Genet. 2003, 73: 447-474. 10.1086/377110.
Spielmann M, Mundlos S: Structural variations, the regulatory landscape of the genome and their alteration in human disease. Bioessays. 2013, 35: 533-543. 10.1002/bies.201200178.
Spielmann M, Klopocki E: CNVs of noncoding cis-regulatory elements in human disease. Curr Opin Genet Dev. 2013, 23: 249-256. 10.1016/j.gde.2013.02.013.
Birnbaum RY, Clowney EJ, Agamy O, Kim MJ, Zhao J, Yamanaka T, Pappalardo Z, Clarke SL, Wenger AM, Nguyen L, Gurrieri F, Everman DB, Schwartz CE, Birk OS, Bejerano G, Lomvardas S, Ahituv N: Coding exons function as tissue-specific enhancers of nearby genes. Genome Res. 2012, 22: 1059-1068. 10.1101/gr.133546.111.
Lango Allen H, Caswell R, Xie W, Xu X, Wragg C, Turnpenny PD, Turner CL, Weedon MN, Ellard S: Next generation sequencing of chromosomal rearrangements in patients with split-hand/split-foot malformation provides evidence for DYNC1I1 exonic enhancers of DLX5/6 expression in humans. J Med Genet. 2014, 51 (4): 264-267. 10.1136/jmedgenet-2013-102142. doi: 10.1136/jmedgenet-2013-102142. Epub 2014 Jan 23
De Groof G, Poirier C, George I, Hausberger M, Van der Linden A: Functional changes between seasons in the male songbird auditory forebrain. Front Behav Neurosci. 2013, 7: 196-10.3389/fnbeh.2013.00196.
Restelli M, Lopardo T, Lo Iacono N, Garaffo G, Conte D, Rustighi A, Napoli M, Del Sal G, Perez-Morga D, Costanzo A, Merlo GR, Guerrini L: DLX5, FGF8 and the Pin1 isomerase control DeltaNp63alpha protein stability during limb development: a regulatory loop at the basis of the SHFM and EEC congenital malformations. Hum Mol Genet. 2014, 23: 3830-3842. 10.1093/hmg/ddu096.
Brown KK, Reiss JA, Crow K, Ferguson HL, Kelly C, Fritzsch B, Morton CC: Deletion of an enhancer near DLX5 and DLX6 in a family with hearing loss, craniofacial defects, and an inv(7) (q21.3q35). Hum Genet. 2010, 127: 19-31. 10.1007/s00439-009-0736-4.
Spielmann M, Brancati F, Krawitz PM, Robinson PN, Ibrahim DM, Franke M, Hecht J, Lohan S, Dathe K, Nardone AM, Ferrari P, Landi A, Wittler L, Timmermann B, Chan D, Mennen U, Klopocki E, Mundlos S: Homeotic Arm-to-Leg Transformation Associated with Genomic Rearrangements at the PITX1 Locus. Am J Hum Genet. 2012, 91: 629-635. 10.1016/j.ajhg.2012.08.014.
Acknowledgements
We would like to thank the families for their collaboration and contribution to this project. We acknowledge F. Trotier and R. Koll for technical assistance. This project was supported by a grant from the Deutsche Forschungsgemeinschaft to SM and EK. This work was supported in part by a grant from the Polish National Science Centre (UMO-2011-03-D-NZ2-06136) to AJ. MS was supported by a fellowship of the Berlin-Brandenburg School for Regenerative Therapies (BSRT), Berlin, Germany.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare no competing interests.
Authors’ contributions
Patient recruitment and phenotyping: MS, AJ, ASS, RF, SCD, WH, RH, BOJ, SM, and EK. Array-CGH experiments and analysis: NT, AJ, RF, MS, SM and EK. All the authors contributed in writing and reviewing the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
13023_2014_108_MOESM1_ESM.doc
Additional file 1:Figure S1. Confirmation of microdeletions on 7q21.3 by breakpoint spanning PCRs in family 1-3. Table S1: qPCR primers and sequencing primers for breakpoint detection in family 1-3. (DOC 286 kb) (DOC 286 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Tayebi, N., Jamsheer, A., Flöttmann, R. et al. Deletions of exons with regulatory activity at the DYNC1I1 locus are associated with split-hand/split-foot malformation: array CGH screening of 134 unrelated families. Orphanet J Rare Dis 9, 108 (2014). https://doi.org/10.1186/s13023-014-0108-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-014-0108-6