
Abstract
Background
Congenital tricuspid valve malformations are known to occur, but tricuspid valve malformations associated with twins are rarely reported. We report this case from the point of view of a medical history, an auxiliary examination and a genetic pathogenesis to provide a reference for our peers.
Case presentation
We report a rare case of congenital heart disease in monozygotic twins of Hui nationality in Yunnan-Guizhou Plateau, they are normal conception. Twin 1 had Ebstein’s anomaly, and received surgical treatment and recovered satisfactorily. Twin 2 had only partial tricuspid septal prolapse, and pulmonary hypertension occurred during follow-up.
Conclusions
It is necessary to carry out individualized diagnosis and treatment for twins and follow-up observation by echocardiography for a long time. Choosing the right time for cardiac surgery is of great significance to the treatment of the disease.
Similar content being viewed by others


Background
CHD is more common in twins than in singletons, but the overall survival prognosis of CHD in human MZ twins is similar to that in singletons [1]. EA is a congenital tricuspid malformation, usually with a tricuspid septal valve, posterior valve downward movement, and valvular abnormality. The anterior valve usually has a sail-shaped dilatation, and the specific embryological mechanism is not clear. The individual treatment of children with EA varies greatly, and the standard of surgical intervention is also controversial. Critically ill children with severe cyanosis and metabolic acidosis in infancy may need early surgical intervention, while patients with asymptomatic or mild symptoms may only need observation [2]. Patients with obvious tricuspid regurgitation with symptoms or heart failure should also receive surgical intervention to avoid the increase in morbidity and mortality caused by delayed intervention. Tricuspid valve repair is usually preferable to replacement where technically feasible. Radiofrequency ablation should be performed when severe arrhythmia is present [3].
Case introduction
Twin 1 was an 11-year-old male with a height of 145 cm and weight of 36 kg. The patient was born prematurely at 36 weeks of gestation. On the 40th day after birth, the patient underwent routine physical examination, had a heart murmur during auscultation, and was diagnosed with congenital moderate tricuspid regurgitation by ultrasound in the local hospital. The hospital advised the patient's family members to continue to observe the condition. The patient was often prone to colds in infancy. Recently, the patient developed asthma after exercise, and his physical activity was slightly limited, so he went to Fuwai Yunnan Cardiovascular Hospital. A physical examination showed that a soft systolic murmur of grade 2 at beat 6 could be heard in the fourth intercostal space of the left margin of the sternum. The echocardiographic report was as follows: EA, type A according to the Carpentier classification, with right atrium enlargement, a TAPSE of 20 mm, tricuspid annulus enlargement, and an internal diameter up to 38 mm. The anterior tricuspid leaflet was long, the septal lobe moved downward approximately 12 mm, the abnormal chordae tendineae were attached to the right ventricular wall, the valvular lobe was poorly closed, and there was a large volume of tricuspid regurgitation (Fig. 1a). An electrocardiogram showed arrhythmias and complete right bundle branch block (Fig. 2a). The chest X-ray showed enlargement of the right atrium (Fig. 2b). CT showed tricuspid enlargement (Fig. 2c). Surgical intervention was performed under general anaesthesia and cardiopulmonary bypass. During the operation, the diameter ratio of the aorta to the pulmonary artery was approximately 1:1. A two-needle 5–0 double needle belt gasket was used to contract the tricuspid valve with a DeVega ring and to fold part of the atrialized right ventricle to correct three kinds of valve downward deformities. The patient’s chest was closed successfully and he was sent to the intensive care unit. The time of cardiopulmonary bypass (62 min) was 17 h after the operation in the intensive care unit, and 7 days after treatment, he was discharged safely. The patient was followed up for 2 years after the operation, and the prognosis was good.

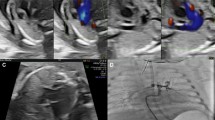
a The oblique four-chamber section of transthoracic echocardiography of Twin 1 shows that the septal lobe of the tricuspid valve moves downward to the apical part of the heart by transthoracic echocardiography, and the right ventricle inflow tract section by transthoracic echocardiography shows a large volume of tricuspid regurgitation by colour Doppler. b The oblique four-chamber section by transthoracic echocardiography of Twin 2 shows that the septal lobe of the tricuspid valve protrudes slightly into the right atrium during systole; the colour Doppler shows tricuspid regurgitation in the inflow section of the right ventricle by transthoracic echocardiography

a The electrocardiogram of Twin 1 shows a complete right bundle branch block. b the Chest X-ray of Twin 1 shows an enlarged right atrium, a clear lung field, and a cardiothoracic ratio of 0.5. c Twin 1’s CT scan, with an arrow showing tricuspid annulus enlargement
Twin 2 was also examined. The history of Twin 2 was similar to that of Twin 1, but there were no clinical symptoms. The ultrasonic electrocardiogram of Twin 2 showed that the tricuspid septal lobe was partially prolapsed, the right atrium was enlarged, the tricuspid valve leaf was slightly thickened, the tricuspid septal valve tip adhered to the interventricular septum, and the activity of the tricuspid septum was slightly limited. Part of the valve body near the posterior part of the tricuspid septal lobe slightly protruded into the right atrium during systole, resulting in valve opening, poor closure and moderate tricuspid regurgitation (Fig. 1b). The patient did not meet the indication for surgical treatment, but we still instructed the patient to follow up regularly, and the patient has been followed up for 2 years. Unfortunately, the most recent echocardiogram estimated mild pulmonary hypertension, but the patient refused to undergo further examination by right cardiac catheterization. We asked the patient to limit exercise, take oral diuretics, reduce heart load, and continue close follow-up.
Discussion
EA is a rare CHD, accounting for less than 1% of all CHDs. It was first described by Wilhelm Ebstein in a report published in 1866 [4]. EA is even more rare in twins; a single fertilized egg divides into twins, who share a placenta, and there is a large amount of vascular communication between twins. When blood exchange is not equal, twin transfusion syndrome can cause changes in foetal cardiac load, but the severity of tricuspid regurgitation is still unclear [5]. MZ twins are thought to be genetically identical, but inconsistent phenotypes have been found in MZ twins. Differences in chromosome structure or gene mutations are one of the reasons for these inconsistent phenotypes. Other mechanisms leading to CHD differences in MZ twins include environmental factors, epigenetic changes, noncoding DNA mutations or oligogenic mechanisms [6]. In addition to epigenetics, the influence of the local placenta is also considered to be an important factor in the difference in CHD in MZ twins. The vascular connection between the placenta and the insertion site of the umbilical cord leads to blood flow imbalance, resulting in relatively insufficient perfusion in MZ twins [7].
Most patients with EA have normal physical development, and ultrasonic electrocardiogram can diagnose this kind of disease [8, 9]. In the past, patients with severe EA often underwent tricuspid valve replacement, but the clinical results were not satisfactory. Modified tricuspid valvuloplasty greatly reduced the mortality of patients with EA. A 20-year follow-up showed a survival rate of 82% [10]. The MZ twins were in stable condition for 12 years after birth until Twin 1 showed obvious symptoms of fatigue, arrhythmia and tricuspid annulus enlargement, so the surgical indications were clear, and annuloplasty was performed with a DeVega ring [11].
Conclusion
In this case of MZ twins, the changes in the tricuspid valve were different. We took effective treatment measures for the twins and followed up for 2 years. Twin 1 underwent surgical intervention with a good prognosis, and no adverse events have been reported thus far. Twin 2 still needs further observation. In the future, individualized treatment for such patients is desirable; of course, this kind of disease is relatively less common and physicians lack experience; it is hoped that more cases are reported, and evidence-based treatments are obtained so that patients have better benefits.
Availability of data and materials
Not applicable.
Abbreviations
- CHD:
-
Congenital heart disease
- MZ:
-
Monozygotic
- EA:
-
Ebstein’s anomaly
References
Herskind AM, Larsen LA, Pedersen DA, et al. Comparison of late mortality among twins versus singletons with congenital heart defects. Am J Cardiol. 2017;119(10):1680–6.
Sainathan S, da Fonseca da Silva L, da Silva JP. Ebstein’s anomaly: contemporary management strategies. J Thorac Dis. 2020;12(3):1161–73.
Possner M, Gensini FJ, Mauchley DC, et al. Ebstein’s anomaly of the tricuspid valve: an overview of pathology and management. Curr Cardiol Rep. 2020;22(12):157.
Attenhofer Jost CH, Connolly HM, Dearani JA, et al. Ebstein’s anomaly. Circulation. 2007;115(2):277–85.
Ishido H, Masutani S, Mikami Y, et al. Modified underlying cardiac disease severity in twin–twin transfusion syndrome. Ann Pediatr Cardiol. 2019;12(3):336–8.
Xu Y, Li T, Pu T, et al. Copy number variants and exome sequencing analysis in six pairs of Chinese monozygotic twins discordant for congenital heart disease. Twin Res Hum Genet. 2017;20(6):521–32.
Imany-Shakibai H, Yin O, Russell MR, et al. Discordant congenital heart defects in monochorionic twins: risk factors and proposed pathophysiology. PLoS ONE. 2021;16(5):e0251160.
Rydzewska K, Sylwestrzak O, Krekora M, et al. Ebstein’s anomaly: epidemiological analysis and presentation of different prenatal management. J Matern Fetal Neonatal Med. 2020. https://doi.org/10.1080/14767058.2020.1818207.
Kumar TKS. Ebstein’s anomaly in the neonate. Indian J Thorac Cardiovasc Surg. 2021;37(Suppl 1):17–25.
Yuan SM. Ebstein’s anomaly: genetics, clinical manifestations, and management. Pediatr Neonatol. 2017;58(3):211–5.
Knott-Craig CJ, Overholt ED, Ward KE, et al. Repair of Ebstein’s anomaly in the symptomatic neonate: an evolution of technique with 7-year follow-up. Ann Thorac Surg. 2002;73(6):1786–92 (discussion 1792–1783).
Acknowledgements
Not applicable.
Funding
(1) The present study was funded by the National Natural Science Foundation of China (Grant No. 81960072). (2) Yunnan Provincial Cardiovascular Disease Clinical Medical Center Project (No. FZX2019-06-01). (3) The Medical Reserve Talented Person of Yunnan Provincial Health and Family Planning Commission (H-2017015).
Author information
Authors and Affiliations
Contributions
PS and QX, guided by YS, performed the literature search and wrote the preliminary manuscript. QW performed the surgery, YS assisted in the surgery. YD did echocardiographic follow-up. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
A written informed consent to publish the report was obtained from the patient before preparing the manuscript.
Competing interests
Authors declare that they do not have any competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shen, P., Xie, Q., Ma, R. et al. Diagnosis and treatment of congenital tricuspid valve malformation in a case of monozygotic twins. J Cardiothorac Surg 17, 176 (2022). https://doi.org/10.1186/s13019-022-01911-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13019-022-01911-w