
Abstract
Introduction
Patellar tendon overuse injuries are common in athletes. Imaging may show a change in tissue structure with tendon thickening and disruption of the intratendinous substance. We wish to test the hypothesis that both autologous bone marrow expanded mesenchymal stem cells and autologous leukocyte-poor platelet-rich plasma (LP-PRP) implanted into the area of the disrupted tendinopathic patellar tendon will restore function, but tendon regeneration tissue will only be observed in the subjects treated with autologous bone marrow expanded mesenchymal stem cells.
Methods and analysis
This is a single-centre, pilot phase I/II, double-blinded clinical trial with randomisation with active control. Twenty patients with a diagnosis of patellar tendinopathy with imaging changes (tendon thickening and disruption of the intratendinous substance at the proximal portion of the patellar tendon) will be randomised in a 1:1 ratio to receive a local injection of either bone-marrow autologous mesenchymal stem cells (MSC), isolated and cultured under GMP at The Institute of Biology and Molecular Genetics (IBGM) (Spain) or P-PRP. The study will have two aims: first, to ascertain whether a clinically relevant improvement after 3, 6 and 12 months according to the visual analogue scale (VAS), Victorian Institute of Sport Assessment for patellar tendons (VISA-P) and dynamometry scales (DYN) will be achieved; and second, to ascertain whether the proposed intervention will restore tendon structure as determined by ultrasonography (US), Doppler ultrasonography (DUS), and innovative MRI and ultrasound techniques: Magnetic Resonance T2 FAT SAT (UTE, Ultrashort Echo TE) sequence and Ultrasound Tissue Characterization (UTC). Patients who are randomised to the P-PRP treatment group but do not achieve a satisfactory primary endpoint after 6 months will be offered treatment with MSC.
Trial registration
Similar content being viewed by others

Background
Overload patellar tendon injuries are common in elite athletes (7 to 45%) and activities where repetitive jumping is required [1,2,3,4]. Pain may become recurrent and refractory to any conservative management, leading to a sports-related or occupational disability [5]. The pathogenesis of patellar tendinopathy is still elusive, but a failed healing response remains at its basis [6, 7]. At the time of surgery, typically no inflammatory cells or inflammatory mediators are detected in the tendon tissue, suggesting that tendinopathy is not the result of typical inflammation [8, 9]. Poor vascularization is often reported as a factor that contributes to tendinopathy, but tendinopathy is often associated with neovascularization and increased blood-flow through the tendon, which might be a restorative and beneficial response [10]. Although the source of pain has not been defined [11], in some studies small substance P-positive nerve fibres are closely related to the neovascularization process and may transmit pain perception [6, 12,13,14].
Other pathological changes related to patellar tendinopathy include (a) intra-tendinous calcification; (b)accumulation of adipose cells; (c) disruption and disorganisation of collagen fibres; (d) conversion of type I collagen to type III collagen; (e) a reduction in number of fibroblasts; (f) changes in tenocyte morphology; and (g) an increase in proteoglycan and glycosaminoglycan content [6].
Although the accepted standard treatment for patellar tendinopathy is a graded rehabilitation programme, not all patients respond to exercise therapy [15]. Medical or surgical management may provide improvement in symptoms but does not result in regeneration of normal tendon tissue [16]. Recently, MSC have been shown to promote tendon healing in an animal model of acute tendon injury [17, 18]. In addition, these cells may control inflammation by influencing differentiation, migration or apoptosis in different immune cells [19,20,21,22,23,24,25].
MSC have the following distinctive features: (a) ability to differentiate into osteoblasts, adipocytes and chondrocytes in vitro; (b) ability to adhere to plastic during culture; (c) more than 90% of the cells show a positive immunophenotype for CD73, CD105, CD90 and CD166 antigens; less than 10% of the cells show a positive immunophenotype for CD45, CD14, CD34, CD31 and HLA-DR antigens [26]. MSCs are found in several organs and tissues, where they may be harvested. Nevertheless, bone marrow is the optimal and most easily accessible source of MSC. Once obtained, these cells can be isolated and grown ex vivo under pharmaceutical standards and used in human therapies known as an ‘advanced therapy medicine’ [27]. Our research group has used MSC successfully in several osteoarticular injuries [28,29,30,31,32,33,34,35,36]. Given the immunomodulatory and anti-inflammatory abilities of MSC, harvesting and grafting these cells into an area affected by patellar tendinopathy are feasible options that may provide satisfactory results.
The use of autologous leukocyte-poor platelet-rich plasma (LP-PRP) in tendon injuries is well established [37], including patellar tendon injuries, at times showing good clinical response [38, 39]. Our research group has also successfully used this approach in various musculoskeletal conditions in several areas of the human body [40, 41]. A recent systematic review and meta-analysis [42] demonstrated improved outcomes with the use of leukocyte-rich-PRP (LR-PRP) compared to LP-PRP.
We wish to test the hypothesis that an analgesic effect will be achieved using either treatment, but significant regeneration of tendon tissue will only be achieved in the cell therapy group. We will assess clinical results by clinical examination, visual analogue scale (VAS), the validated Spanish version of VISA-P43 and manual dynamometry. We will evaluate tissue regeneration using several imaging techniques. The use of MRI with a UTE sequence in the sagittal plane makes it possible to determine tendon tissue features at very low T2 values in all regions, including the ‘gap’ and healthy tendon tissue. In addition, UTE sequences allow to visualize and delineate defects that are not detected by conventional imaging.
UTC is an innovative imaging technique for assessing tendon injuries. Using a conventional ultrasound device, 600 axial sections of the tendon are performed at 0.2 mm intervals and compounded to form a 3D image. Using this technique, collagen fibre alignment may be quantified into four different types: (a) normal aligned fibrillar structure; (b) slightly misaligned fibrillar structure; (c) disorganised fibrillar structure; (d) complete disintegration of tendon tissue [43]. Ultrasonography is the most accessible imaging technique used to confirm the clinical diagnosis and determine the exact location of the lesion. Ultrasonography also helps calculate tendon size, defines eco-texture, allows measurement of any tendon gaps in several planes and assesses the degree of tendon vascularization. In addition, ultrasonography allows to dynamically monitor tendon function [44].
Should the results be better for the cell therapy group, the patients in the active control group (who were injected with LP-PRP) who have not achieved similar benefits to those of patients in the MSC group after 6 months will be offered cell therapy. We believe that this will increase the feasibility of the clinical trial.
Characteristics of the MSC intervention
MSCs exhibit the typical stem cell features defined by the International Society of Cellular Therapy [23]. MSC are an advanced therapy drug registered with the Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) with IND number 10–134. They are bone marrow MSC, processed according to the GMP practices at the IBGM and re-suspended in an isotonic medium composed of Ringer lactate solution, 0.2% human serum albumin and 5mM glucose. The product is supplied by the laboratory after a 23-day processing period. Cell viability is ≥ 90%, and negative microbiological results are obtained for Gram and Mycoplasma microorganisms. Endotoxin levels are below 0.5 IU/mL. The product is stable for 8 h at 4–12 °C. The product is supplied in two 5 mL syringes, one containing 10 × 106 MSC suspended in a 2 mL solution, and the other containing 10 × 106 MSC suspended in a 4 mL solution. The total dose of cells supplied is 20 × 106 ± 2 × 106 MSC.
General characteristics of P-PRP intervention
LP-PRP is obtained under strict aseptic conditions. Blood components are fractioned by centrifugation. In a surgical environment, peripheral venous blood is collected into four 9 mL citrate tubes. The anticoagulated blood is centrifuged at 1,200 rpm for 8 min (460g). Six milliliters of LP-PRP are obtained from a 36-mL sample of autologous peripheral venous blood. The LP-PRP suspension contains a two-fold increase in the number of platelets compared to peripheral blood and a very low number of leukocytes (average basal peripheral blood 228 × 103 platelets/mm3, range 165–329 × 103; LP-PRP: 563 × 103 platelets/mm3; range 407–801 × 103). Before application, plasma coagulation is activated by adding 5% CaCl2 [38, 45].
Methods/design
Main objectives
-
1.
To assess the clinical efficacy of MSC injection and compare the results with those obtained in the LP-PRP group. Outcomes will be assessed using subjective patient reports, VAS and VISA-P scales, and strength determination of the extensor muscle group using manual dynamometry (DYN).
-
2.
To confirm changes in the architecture for regeneration of the patellar tendon gap following peritendinous and intratendinous injection of MSC and compare these results with those obtained in the LP-PRP group. Assessment will be performed using UTE-MRI, UTC and ultrasonography.
Secondary objectives
To assess the viability and safety of the use of medical devices MSC and LP-PRP when injected into the patellar tendon; prove that all procedures described in the present protocol are feasible; and register potential adverse events related to either treatment, as well as any other adverse events that occur during the clinical trial, whether related to the trial or not.
Trial design
Study population
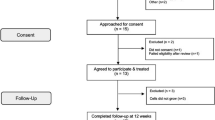
Twenty male recreational athletes with a patellar tendinopathy who meet the inclusion and exclusion criteria will be recruited and will be followed for 1 year (Fig. 1).

Study flow chart
Inclusion criteria:
-
1.
Male participants aged between 18 and 48
-
2.
Pain and tenderness at the area of attachment of the patellar tendon on the lower pole of the patella [46] for more than 4 months, with no clinically relevant response after conservative treatments
-
3.
US confirming disruption ≥ 3 mm of the fibrillar structure in the proximal portion of the patellar tendon, tendon thickening and a hypoechoic injury MRI of the patellar tendon in T2 FAT SAT sequence (fat saturation) showing an increase of ≥ 3 mm in longitudinal diameter at the proximal insertion
-
4.
Informed signed written consent by the participant
-
5.
The participant is able to understand the nature of the study.
Exclusion criteria:
-
1.
Participant younger than 18 (or legally dependent) or over 48 years of age
-
2.
MRI with a grade III–IV osteochondral lesion in any compartment of the knee
-
3.
Injury to the anterior or posterior cruciate ligament
-
4.
Local injection with corticosteroid during the previous 12 months
-
5.
Local injection with PRP during the previous 6 months
-
6.
Evidence of local or systemic infection
-
7.
Patients presenting with positive serology to HIV 1 or 2, hepatitis B (HBsAg, HBcAc), hepatitis C (anti-HCV-Ab) and syphilis
-
8.
Congenital or acquired conditions involving malformation and/or significant deformation of the knee that hampers the administration of the planned intervention and evaluation of the results
-
9.
Body mass index (BMI) greater than 30. 5 (obesity grade II)
-
10.
Active neoplastic disease
-
11.
Immunosuppression
-
12.
Simultaneous participation in another clinical trial or treatment with another research product during the 30 days prior to inclusion in the study
-
13.
Other pathologies or circumstances that may compromise participation in the study according to medical criteria.
Patients will be randomized by the Clinical Research Organization (CRO) (Adknoma) using SAS statistical software (version 9. 4) into either the MSC group or PRP group in a 1: 1 ratio. After randomization, either MSC or LP-PRP will be injected intralesionally and around the affected area of the patellar tendon. To keep patients and evaluating physicians blind to treatment, sham procedures will be performed in both groups (i.e. the MSC group will have blood extracted and will have their tendon injected with normal saline at the first stage of treatment, and the LP-PRP group will undergo a sham bone marrow harvesting at the first stage of treatment). The treating physicians will not be blind to the treatment administered.
Experimental phase A
MSC treatment group
Ten patients will be treated with saline solution (sham) and with 20 × 10E6 MSC after 23 days.
Stage one
-
1.
Under sterile conditions, a total of 4 citrate tubes (9 mL per tube; total volume 36 mL) of venous blood are withdrawn from the antecubital vein of the non-dominant arm as a sham procedure of P-PRP preparation.
-
2.
With the patient prone, and after sedation induced by midazolam and propofol, local anaesthetic injections (lidocaine 1%; 10 mL) are administered in both the posterior superior iliac crests and are penetrated using an 11 gauge trocar. Using sequential aspirations of 1–2 mL each, 100 mL of bone marrow is collected in a heparinized container to be sent to IBGM for isolation and culture of MSC.
-
3.
The patient is then positioned supine. Under ultrasonography guidance, 1 mL of normosaline solution is injected using a 22 gauge needle in the tendinous and 1 mL in the paratendinous medial and lateral areas of the patellar tendon. The participant is then transferred to the postoperative recovery room, where they remain for 1 h before discharge.
Stage two (after 23 days)
-
1.
Under sterile conditions, a total of 4 citrate tubes (9 mL per tube; total volume 36 mL) of venous blood are withdrawn from the antecubital vein of the non-dominant arm as a sham procedure of P-PRP preparation.
-
2.
With the patient supine under sedation with midazolam and propofol, under ultrasonography guidance, a 2-mL solution containing 10 × 106 MSV is injected into the tendon; a 2-mL solution containing 5 × 106 MSC is injected into the medial peritendinous area, and a 2-mL solution containing 5 × 106 MSC is injected into the lateral peritendinous area.
LP-PRP control group
Ten patients will be treated with two doses of LP-PRP, each containing 6 mL of solution-PRP. The second dose will be injected 23 days after the first one.
Stage one
-
1.
Under sterile conditions, a total of 4 citrate tubes (9 mL per tube; total volume 36 mL) of venous blood are withdrawn from the antecubital vein of the non-dominant arm for P-PRP preparation. One to two supplementary tubes of peripheral venous blood are withdrawn for preservation in a serum bank for security reasons. LP-PRP is prepared according to ITRT’s PRP protocol.
-
2.
With the patient prone, and after sedation induced by midazolam and propofol, 10 mL of lidocaine 1% is administered in the posterior superior iliac crests, and an 11-gauge trocar is inserted in the subcutaneous tissue as part of the sham procedure for obtaining MSC.
-
3.
With the patient supine, and sedation induced by midazolam and propofol, using ultrasound guidance, 2 mL of LP-PRP are injected intratendinously, 2 mLof LP-PRP are injected into the medial peritendinous area, and 2 mL of LP-PRP are injected in the lateral peritendinous zone adjacent to the area of tendinopathy.
Second stage (after 23 days)
-
1.
The procedure for obtaining blood and LP-PRP preparation is performed again under sterile conditions.
-
2.
With the patient supine, and sedation induced by midazolam and propofol, using ultrasound guidance, 2 mL of LP-PRP are injected intratendinously, 2 mL of LP-PRP are injected into the medial peritendinous area, and 2 mL of LP-PRP are injected in the lateral peritendinous zone adjacent to the area of tendinopathy.
Experimental phase B
Results will be evaluated 6 months after the treatment. If the results support the hypothesis that a higher efficacy is obtained in the MSC group, the cell therapy trial will be offered to subjects from the LP-PRP group who show no evidence of clinically relevant improvement: if they wish, this group will be treated with MSC. This will be ‘experimental phase B’, which will be evaluated with the same clinical and imaging assessment protocol as described above.
Post-treatment procedures (rehabilitation program)
The participants will be discharged 1 h after injection of the cell suspension or LP-PRP solution and advised to rest for 48 h. Analgesia (ibuprofen 600 mg, every 8 h, and omeprazole 20 mg once daily) are prescribed for the participant to self-administer if needed.
After treatment, a specific rehabilitation protocol will be followed, applying appropriate progressive load to the affected limb [6]. Table 1 shows the rehabilitation program over 3 months. For greater details, please refer to Malliaras et al 2015 [15] and Mascaro et al. 2018 [47].
After the 3 months, and after evaluating each patient by physical examination, US and MRI, the athletes will be progressively allowed to gradually return to their sport.
The follow-up consists of clinical examination visits and image monitoring by ultrasonography and by UTC, MRI-UTE, as specified in the protocol schedule.
Concomitant medications
Any concomitant medications administered from the day of the initial treatment until the end of the trial will be recorded. No fluoroquinolone-based antibiotic drugs will be administered during the entire study period [48, 49]. If they must be administered, the subject will be excluded from the clinical trial and will continue therapy using standard treatments. Corticosteroids or anti-inflammatory therapy should not be administered. If they must be administered, the patient will not be excluded from the trial, but the administration will be recorded in the clinical records. If necessary, treatment with ibuprofen 600 mg will be allowed.
Response evaluation
Subsequent assessments following the treatment include AE review, VAS (pain), VISA-P Questionnaire, ultrasound imaging (conventional ultrasound, UTC and MRI), dynamometry manual test and clinical assessment (Table 2 for phase A, and Table 3 for phase B). These will take place at 1, 2, 3, 6, 8 and 10 weeks and 3, 6, 12 months post-treatment.
Adverse events
Possible adverse events occurring during the trial period (regardless of whether they are related to the procedure) will be recorded and tabulated. In the event of a serious adverse effect, a notification will be sent to the Spanish agency for Medicines and Healthcare Products (AEMPS) through the Clinical Research Organization (CRO), according to established procedures and terms.
Pain and function
Pain will be recorded on the VAS, and function will be evaluated by measuring extension forces in the knee using a manual dynamometer.
The pain and functional evaluation will be performed using the VISA-P score, which specifically evaluates pain and function in patellar tendinopathy. The scale ranges from 0 to 100, where a value of 100 indicates the absence of pain at rest, when walking, when climbing up or downstairs or during exercise [50]. VISA-P score values for patients with chronic patellar tendinopathy usually range from 30 to 50. Physical activity may be undertaken again if values are above 60. Patients with score values over 70 are considered fit for competition, although demands are higher for professional athletes. The minimum significant difference has been set at 13 points.
The following VISA-P criteria are used to assess improvement in this trial [51]:
-
Desired: ≥ 60/100 (after 3 months)
-
Very satisfactory: ≥ 70/100 (after 6 months)
-
Ideal: ≥ 80/100 (after 12 months)
Ultrasonography
Ultrasound scanning will be performed with an Aplio 500 (TUS-500 5.0 Platinum Series, manufactured by Canon Medical Systems Corporation in Nasu, Japan) using a high-frequency linear array probe (PLT 1005BT), 5.0–14 MHz frequency range. The most commonly used 2D frequency will be differential harmonic of 14 MHz (diff THI 14 MHz). The depth used will be 5 cm, with a single focus at 1.8 cm and a dynamic range of 65 dB
The patellar tendon will be examined longitudinally and transversely using greyscale and colour measures, exerting the least possible pressure with the probe. Images will be recorded in static and dynamic formats.
Therapeutic results will be regarded as positive if the following findings will be evidenced: improvement in tendon echotexture, gap reduction and reduced neovascularization.
A change in thickness will also be considered, but to a lesser extent, as the significance of thickness change in terms of success of any intervention programme is unknown [44].
The following will be determined:
-
1.
Lesion location (medial, central, lateral) and (anterior, central, posterior).
-
2.
Tendon thickness 5 mm distal to the inferior pole of the patella.
-
3.
Eco-texture (using a 0–3 scale).
-
4.
Longitudinal and axial “gap” measures; area value.
-
5.
Vascularization degree I–V
UTC
Tendon eco-structure will be evaluated according to four subtypes I, II, II and IV in cross-section of region of interest (ROI): percentage of each subtype will be calculated. The distance (mm) from the lower patellar pole in the anteroposterior section and cross-section will also be measured (based on percentage points).
Subtype I is regarded as ideal, corresponding to a perfectly aligned fibrillar structure; Subtype IV is regarded as the most sub-optimal, corresponding to the absence of fibrillar structure. To establish a healing standard, the 4 aforementioned subtypes will be assessed, and changes will be evaluated by calculating the relative increase in percentage points of subtypes I and II (indicative of organised tissue), along with the relative decrease in percentage points of subtypes III and IV (indicative of disorganised tissue).
MRI
Regarding MR, we shall use a 3D sequence using a Toshiba TITAN 3T MRI (Canon Medical Systems, Corporation in Nasu, Japan), with a phase 3D MPV Voxel mVox T2 weight, isotropic Voxel (0.6 mm, 90.6 mm and 90.6 mm).
With a T2 FAT SAT sequence (fat saturation) in the sagittal plane, the dimensions of the intra-tendinous changes will be measured using several signal characteristics: hypo-signal, homogeneous signal, heterogeneous signal, isointense signal, and normal signal. Proximal, medial and distal areas of altered signal will be measured in millimetres. Hoffa fat oedema and patellar bone oedema will also be evaluated. An FFE (Fast FE) 3D sequence will be used in the coronal plane to measure the gap and the gap in cross-section diameter in millimetres. The presence of calcification will also be assessed. UTE (Ultrashort Echo TE) sequences will be obtained in the sagittal plane, and T2 mapping will be acquired in regions of Interest (ROI), and will be established for the areas of altered signal, in the medial and lateral areas, and in healthy tendon.
The intra-articular appearance of the knee joint will also be assessed, with quantification of the degree of arthrosis (I–IV) of each knee compartment.
Criteria for non-response to treatment
A lack of response to treatment will be recorded if no clinically relevant improvement or tendon repair changes occur after a 6-month period. Clinical changes will be assessed using a combination of physical examination, VAS score, VISA-P questionnaire and DYN. Tendon repair will be assessed using ultrasonography, UTC and MRI. A partial response will be considered if there is evidence of either clinical improvement or tendon repair.
Patient and public involvement
Patients and the public were not directly involved with the development of the current protocol. The primary research question is the efficacy of the use of culture-expanded MSCs versus LP-PRP on the treatment of patellar tendinopathy. This will enable the development of a larger RCT that will involve oversight from the patient and public representatives. Patient recruitment is described above, and the results will be disseminated to the participants directly in the doctor's office at the end of the study.
Statistical analysis
Statistical analysis will be performed using version 21. 0 of the Statistical Package for Social Sciences (SPSS Inc, Chicago, IL). We will evaluate: (i) adverse events (AE) and serious adverse events (SAE) showing relationships, causality and severity will be provided; (ii) frequency and percentage of AE and SAE; (iii) laboratory results including hematologic and biochemistry values will be collated: leukocytes levels, absolute neutrophils count, platelet count, creatinine levels, AST (SGOT)/ALT (SGTP) levels, etc. A list of laboratory parameters will be obtained for each patient and visit and will include reference range values. Values will be summarized in a basal and post-basal visit analysis will include the following values: (i) valid ‘n’; (ii) mean; (iii) standard deviation; (iv) median; (v) minimum and maximum.
The frequency and percentage values of patients showing evidence of tendon regeneration 6 months after treatment with MSC or P-PRP will be provided.
The following methods will be used to describe and analyse the results obtained:
-
1.
Descriptive statistics in numerical variables: number of valid cases; number of non-available values; mean value; standard deviation; 95% confidence interval for the mean value; median, minimum and maximum values. In discrete and categorical variables, the following values will be provided: frequency; percentage value for the total number of valid cases; percentage value for the total number of non-available values.
-
2.
Inferential statistics tests for comparing groups and time points: ANOVA with repeated measures (or ANCOVA) if covariables with a significant interaction are detected with their corresponding contrast values (Scheffe test), or non-parametric tests if the assumption of normality is rejected (Mann-Whitney U test for comparison between groups and Wilcoxon test for comparison between measurements). The significance level for all statistical analyses will be 0. 05.
Efficiency sample (per protocol (PP) population) includes all patients whose tendon lesion has been treated with an MSC or LP-PRP suspension. These patients must have had no major violations of the protocol, and at least one assessment of efficacy after treatment.
‘Full analysis set’ (FAS) (intention to treat analysis) includes all patients in the study, whether they finish the study or comply with the protocol.
The safety population will be used in the analysis of the primary endpoint and of all other safety parameters defined.
A list of all demographic and baseline data will be provided for each patient. All lists will be provided for FAS and PP populations.
Discussion
Patellar tendinopathy mainly occurs in young adults who practise sports, either professionally or as amateurs, which involve repeated running, hopping and jumping [52], with typical clinical and imaging diagnostics features [53].
To minimise uncontrolled factors that could affect the results, this study only includes male patients, as the viscoelastic properties of skeletal muscle and tendon are driven by hormone expression [54]. Disorders of insulin and thyroid hormones metabolism are related to tendinopathy both in vitro and in vivo [55], and sex hormones may promote changes in the structure of tendons, thus influencing their response to physical exercise [56]. If we obtain positive results in this study, a new study will be performed on female subjects only.
Both the cellular and applied LP-PRP treatments are based on safety and efficacy criteria obtained in animal studies and in clinical trials in which these treatments were applied successfully to treat other musculoskeletal pathologies [17, 18, 22, 29, 31,32,33, 41]. Future studies should try to establish the optimal cellular dose or the appropriateness of multiple doses. The composition and volume of the cell suspension medium should also be noted, as these variables can influence results.
Applying the guidelines of the Helsinki declaration of the World Medical Association, we have avoided using a true placebo in the control group, and plan to use an active control LP-PRP group. We hypothesise that this control treatment is very unlikely to exert the significant regenerative effect that we would attribute to MSC. However, this hypothesis must be confirmed, as no previous clinical trials have analysed the therapeutic effect of expanded BM-MSCs in patellar tendinopathy. The present study has several trengths. This is the first study to assess the effect of injecting autologous mesenchymal cells into the pathological zone of refractory proximal patellar tendinopathy in athletes. The study will monitor clinical scores, strength and imaging findings including 2D US, UTE MRI and ultrasound tissue characterization (UTC). The addition of UTE MRI and UTC will enable more precise measurement of structural tendon changes than 2D US. The control group will be offered mesenchymal cell therapy if the results of the control group prove ineffective. We are fully aware of the limitations of the present study. For example, the control group will receive active LP-PRP treatment rather than a true placebo, limiting the value of a control group. However, it will be easier to recruit athletes in a study with a control treatment that is perceived to be effective in an athletic cohort. The sample size is small, limiting the power of this study. Finally, this study fails to establish an optimal cell dose, which should be examined in future studies.
It is important to plan a second open experimental phase, where patients treated with LP-PRP who have not improved will be offered cell treatment. This approach, in addition to providing subjects included in the control group with a potentially more effective treatment, should also help determine the scope of the therapeutic effect of the MSC on LP-PRP, establishing a comparative cost-effective index.
This clinical trial also aims to advance current knowledge about the pathology of interest, and the hypothetical regenerative effect of incorporating innovative diagnostic systems and evaluations of this type of lesions. Both UTE magnetic resonance (Ultrashort Echo TE) and UTC are innovative non-invasive techniques that allow us to quantify regenerative effects using imaging.
Trial Status
-
Protocol version: V2. 05-July 2017
-
Recruitment starts December 2017.
-
Total subjects recruited on 28-10-2018: 20
Availability of data and materials
Not applicable.
Abbreviations
- AE (AA):
-
Adverse event
- AEMPS:
-
Agencia Española de Medicamentos y Productos Sanitarios (Spanish Agency for Medicines and Healthcare Products)
- CEIM:
-
Ethics Committee for ClinicalResearch with Drugs
- CRO:
-
Clinical Research Organization
- DYN:
-
Dynamometry
- FAS:
-
Full analysis set
- FFE:
-
FAST FE Sequence (Ultrafast Gradient Echo Sequence)
- GMP:
-
Good Manufacturing Practices
- IBGM:
-
Instituto de Biología Genética Molecular (Institutefor Molecular Biology and Genetics)
- ITRT:
-
Institut de TerapiaRegenerativaTissular (Institute for Tissue Regeneration Therapy)
- LP-PRP:
-
Leukocyte-poor platelet-rich plasma
- MO:
-
Bone marrow
- MRI:
-
Magnetic resonance imaging
- MSC:
-
Mesenchymal stem cells
- MSV:
-
Mesenchymal stem cells from valladolid
- PEI (IMP):
-
Investigational medicine product
- PP:
-
Per protocol analysis
- PRP:
-
Platelet-rich plasma
- ROI:
-
Region of interest
- SAE (AAG):
-
Serious adverse event
- T2:
-
T2 sequence
- UTC:
-
Ultrasound tissue characterisation
- UTE:
-
Ultrashort echo time sequence
- VAS:
-
Visual analogue scale for pain
- VISA:
-
‘Victorian Institute of Sport Assessment’ scale
- VISA-P:
-
‘Victorian Institute of Sport Assessment (patellar)’ scale
References
Magnusson SP, Hansen M, Langberg H, Miller B, Haraldsson B, Westh EK, Koskinen S, Aagaard P, Kjaer M. The adaptability of tendon to loading differs in men and women. Int J Exp Pathol. 2007;88:237–40.
Kaux JF, Forthomme B, Goff CL, Crielaard JM, Croisier JL. Current opinions on tendinopathy. J Sports Sci Med. 2011;10:238–53.
Øystein B, Engebretsen L, Bahr R. Prevalence of jumper’s knee among elite athletes from different sports: a cross-sectional study. Am. J Sports Med. 2005;33:561–7.
Pruna R, Medina D, Rodas G, Artells R. Patellar tendinopathy. Therapeutic model in sport. Med Clin (Bar). 2013;141(3):119–24.
Kettunen JA, Kvist M, Alanen E, Kujala UM. Long-term prognosis for jumper’s knee in male athletes. A prospective follow-up study. Am J Sports Med. 2002;30:689–92.
Magnusson SP, Langberg H, Kjaer M. The pathogenesis of tendinopathy: balancing the response to loading. Nat. Rev. Rheumatol. 2010;6:262–8.
Del Buono A, Battery L, Denaro V, Maccauro G, Maffulli N. Tendinopathy and inflammation: some truths. Int J Immunopathol Pharmacol. 2011;24(1 Suppl 2):45–50.
D'Addona A, Maffulli N, Formisano S, Rosa D. Inflammation in tendinopathy. Surgeon. 2017;15(5):297–302.
Battery L, Maffulli N. Inflammation in overuse tendon injuries. Sports Med Arthrosc Rev. 2011;19(3):213–7.
Tol JL, Spiezia F, Maffulli N. Neovascularization in Achilles tendinopathy: have we been chasing a red herring? Knee Surg Sports Traumatol Arthrosc. 2012;20(10):1891–4.
Khan KM, Cook JL, Maffulli N, Kannus P. Where is the pain coming from in tendinopathy? It may be biochemical, not only structural, in origin. Br J Sports Med. 2000;34(2):81–3.
Cook JL, Khan KM, Purdam C. Achilles tendinopathy. Manual Therapy. 2002;7:121–30.
Rees JD, Wilson AM, Wolman RL. Current concepts in the management of tendon disorders. Rheumatology (Oxford). 2006;45:508–21.
Riley G. Tendinopathy from basic science to treatment. Nature Clinical Practice Rheumatology. 2008;4:82–9.
Malliaras P, Cook J, Purdam C, Rio E. Patellar tendinopathy: clinical diagnosis, load management, and advice for challenging case presentations. J Orthop Sports Phys Ther. 2015;45(11):887–98.
Maffulli N, Giai Via A, Oliva F. Revision surgery for failed patellar tendinopathy exploration. Sports Med Arthrosc Rev. 2017;25(1):36–4.
Godwin EE, Young NJ, Dudhia J, Beamish IC, Smith RKW. Implantation of bone marrow-derived mesenchymal stem cells demonstrates improved outcome in horses with overstrain injury of the superficial digital flexor tendon. Equine Vet J. 2012;44:25–32.
Romero A, Barrachina L, Ranera B, Remacha AR, Moreno B, de Blas I, Sanz A, Vázquez FJ, Vitoria A, Junquera C, Zaragoza P, Rodellar C. Comparison of autologous bone marrow and adipose tissue derived mesenchymal stem cells, and platelet rich plasma, for treating surgically induced lesions of the equine superficial digital flexor tendon. Vet J. 2017;224:76–84.
Le Blanc K, Ringden O. Immunomodulation by mesenchymal stem cells and clinical experience. J Intern Med. 2007;262:509–25.
Tse WT, Pendleton JD, Beyer WM, Egalka MC, Guinan EC. Suppression of allogeneic T-cell proliferation by human marrow stromal cells: implications in transplantation. Transplantation. 2003;75:389–97.
Frank MH, Sayegh MH. Immunomodulatory functions of mesenchymal stem cells. Lancet. 2004;363(9419):1411–2.
Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 2005;105:1815–22.
Rasmusson I. Immunemodulation by mesenchymal stem cells. Exp Cell Res. 2006;312(Issue 12, 15):2169–79.
Nauta AJ, Fibbe WE. Immunomodulatory properties of mesenchymal stromal cells. Blood. 2007;110:3499–506.
Ma S, Xie N, Li W, Yuan B, Shi Y, Wang Y. Review Immunobiology of mesenchymal stem cells. Cell Death Diff. 2014;21:216–25.
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, Deans R, Keating A, ProckopDj, Horwitz E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315–7.
Andia I, Maffulli N. New biotechnologies for musculoskeletal injuries. Surgeon. 2019;17(4):244-55. https://doi.org/10.1016/j.surge.2018.08.004.
Orozco L, Soler Rich R, Morera C, Alberca M, Sánchez A, García Sancho J. Intervertebral disc repair by autologous mesenchymal bone marrow cells: a pilot study. Transplant J. 2011;92(Issue 7):822–8.
Orozco L, Munar A, Soler R, Alberca M, Huguet MJ, Sánchez A, García-Sancho J. Treatment of knee osteoarthritis with autologous mesenchymal stem cells: a pilot study. Transplant J. 2013;95(12):1535–41.
Soler F, Soler R, Peirau X, Orozco L. Rapid Isolation of Human Stem Cells. Arthroscopy. 2012;28:895–6 (letter).
Soler R, Munar A, Soler Romagosa F, Peirau X, Huguet M, Alberca M, Sánchez A, García Sancho J, Orozco L. Treatment of Knee Osteoarthritis with Autologous Expanded Bone Marrow Mesenchymal Stem Cells: 50 Cases Clinical and MRI Results at One YearFollow-Up. J Stem Cell Res Ther. 2015;5:285.
Soler R, Ll O, Munar A, Huguet M, López R, Vives J, Coll R, Codinach M, García-López J. Final results of a phase I–II trial using ex vivo expanded autologous Mesenchymal Stromal Cells for the treatment of osteoarthritis of the knee confirming safety and suggesting cartilage regeneration. The Knee. 2016;23:647–54.
Vega A, Martín-Ferrero A, Del Canto F, Alberca M, García V, Munar A, Orozco L, SolerR FJJ, Huguet M, Sánchez A, García-Sancho J. Treatment of knee osteoarthritis with allogeneic bone marrow mesenchymal stem cells: a randomized controlled trial. Transplantation. 2015;99(8):1681–90.
Barrachina L, Remacha AR, Romero A, Vázquez FJ, Albareda J, Prades M, Ranera B, Zaragoza P, Martín-Burriel I, Rodellar C. Inflammation affects the viability and plasticity of equine mesenchymal stem cells: possible implications in intra-articular treatments. J Vet Sci. 2017;18(1):39–49.
Barrachina L, Remacha AR, Romero A, Vázquez FJ, Albareda J, Prades M, Gosálvez J, Roy R, Zaragoza P, Martín-Burriel I, Rodellar C. Priming Equine Bone Marrow-Derived Mesenchymal Stem Cells with Proinflammatory Cytokines: Implications in Immunomodulation-Immunogenicity Balance, Cell Viability, and Differentiation Potential. Stem Cells Dev. 2017;26(1):15–24.
Barrachina L, Remacha AR, Romero A, Zaragoza P, Vázquez FJ, Rodellar C. Differentiation of equine bone marrow derived mesenchymal stem cells increases the expression of immunogenic genes. Vet Immunol Immunopathol. 2018;200:1–6.
Andia I, Maffulli N. Muscle and tendon injuries: the role of biological interventions to promote and assist healing and recovery. Arthroscopy. 2015;31(5):999–1015.
Kon E, Filardo G, Delcogliano M, Kon E, Presti ML, Russo A, Bondi A, Di Martino A, Cenacchi A, Fornasari PM, Marcacci M. Platelet-rich plasma: New clinical application: A pilot study for treatment of jumper’s knee. Injury. 2009;40:598–603.
Andia I, Maffulli N. Use of platelet-rich plasma for patellar tendon and medial collateral ligament injuries: best current clinical practice. J Knee Surg. 2015;28(1):11–8.
Soler R, Soler F, Peirau X, Anglés F, Buló J, Rodas G, Orozco L. My experience with PRP. ICRS Newsletter., 2011. Summer Issue 13, p: 31-32.
Martinez-Zapata MJ, Orozco L, Balius R, Soler R, Bosch A, Rodas G, Til LL, Peirau X, UrrutiaG GI, Bonfill X on behalf of the PRP-RICE group. Efficacy of autologous Platelet rich plasma for treatment of muscle rupture with haematoma: a multicentre, randomized, double-blind, placebo-controlled clinical trial. Blood Transfusion. 2016;14(3):245–54.
Fitzpatrick J, Bulsara M, Zheng MH. The effectiveness of platelet-rich plasma in the treatment of tendinopathy: a meta-analysis of randomized controlled clinical trials. Am J Sports Med. 2017;45(1):226–33.
van Schie HT, de Vos RJ, de Jonge S, Bakker EM, Heijboer MP, Verhaar JA, Tol JL, Weinans H. Ultrasonographic tissue characterization of human Achilles tendons: quantification of tendon structure through a novel non-invasive approach. Br J Sports Med. 2010;44:1153–9.
Warden SJ, Kiss ZS, Malara FA, Ooi ABT, Cook JL, Crossley KM. Comparative Accuracy of Magnetic Resonance Imaging and Ultrasonography in Confirming, Clinically Diagnosed Patellar Tendinopathy. Am J Sports Med. 2007;35:427–36.
Anitua E, Andia I, Ardanza B, Nurden P, Nurden AT. Autologous platelets as a source of proteins for healing and tissue regeneration. Thromb Haemost. 2004;91(1):4–15.
Maffulli N, Oliva F, Loppini M, Aicale R, Spiezia F, King J. The Royal London Hospital test for the clinical diagnosis of patellar tendinopathy. Muscles Ligaments Tendons J. 2017;7(2):315–22.
Mascaró A, Cos MA, Morral A, Roig A, Purdam C, Cook J. Load management in tendinopathy: Clinical progression for Achilles and patellar tendinopathy. Apunts Med Sport. 2018;53(197):19–27.
Budny AM, Ley AN. Fluoroquinolone-mediated Achilles rupture: a case report and review of the literature. J Foot Ankle Surg. 2015;54(3):494–6.
Bisaccia DR, Aicale R, Tarantino D, Peretti GM, Maffulli N. Biological and chemical changes in fluoroquinolone-associated tendinopathies: a systematic review. Br Med Bull. 2019;130:39–49.
Hernandez-Sánchez S, Hidalgo MD, Gomez A. Responsiveness of the VISA-P scale for patellar tendinopathy in athletes. Br J Sports Med. 2014;48:453–7.
Hoksrud A, Ohberg L, Alfredson H, Bahr R. Ultrasound-guided sclerosis of neovessels in painful chronic patellar tendinopathy: a randomized controlled trial. Am J Sports Med. 2006;34(11):1738–46.
de Groot R, Malliaras P, Munteanu S, Payne C, Morrissey D, Maffulli N. Foot posture and patellar tendon pain among adult volleyball players. Clin J Sport Med. 2012;22(2):157–9.
Longo UG, Berton A, Stelitano G, Madaudo C, Perna M, Ciuffreda M, Guarnieri A, Papalia R, Maffulli N, Denaro V. 2017 Marathon of Rome: Anthropometry and Sport Profile in 350 Runners and Association With Achilles and Patellar Tendinopathy. Clin J Sport Med. 2018. https://doi.org/10.1097/JSM.0000000000000695.
Oliva F, Piccirilli E, Berardi AC, Frizziero A, Tarantino U, Maffulli N. Hormones and tendinopathies: the current evidence. Br Med Bull. 2016;117(1):39–58.
Oliva F, Piccirilli E, Berardi AC, Tarantino U, Maffulli N. Influence of Thyroid Hormones on Tendon Homeostasis. Adv Exp Med Biol. 2016;920:133–8.
Denaro V, Ruzzini L, Longo UG, Franceschi F, De Paola B, Cittadini A, Maffulli N, Sgambato A. Effect of dihydrotestosterone on cultured human tenocytes from intact supraspinatus tendon. Knee Surg Sports Traumatol Arthrosc. 2010;18(7):971–6.
Acknowledgements
Carme Barbero, for her assistance in study coordination tasks; Alejandra Jiménez member of the surgical nursing staff in charge of bone marrow sampling procedures and P-PRP preparation procedures. Silvia Ortega, Football Club Barcelona physical therapist, for her assistance in DYN and UTC evaluation.
Funding
This work was supported by Institutde Terapia Regenerativa Tissular. Centro Médico Teknon (ITRT). (Institute for Tissue Regeneration Therapy), Barcelona, Spain.
Author information
Authors and Affiliations
Contributions
GR envisioned the study, participated in its design and coordination, and acted as a blind evaluator for clinical aspects and for UTC. RS envisioned the study, participated in its design and coordination and applied the treatment under study. RB participated in the study design and performed and evaluated (blind evaluation) ultrasonography imaging techniques. XA participated in the study design and performed and evaluated (blind evaluation) MRI imaging techniques. XP participated in the design and applied the treatment under study. MA was involved in cell production. AS was involved in cell production, JGS was involved in cell production. CR was involved in the preliminary study in an equine model. AR was involved in the preliminary study in an equine model. LM was involved in the evaluation of the study and to help to write this manuscript. LO planned the study, participated in its design and applied the treatment under study. NM advised on the theoretical framework of the study, critically revising the procedures and the writing of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The protocol was approved by the Agencia Española de Medicamentos y Productos Sanitarios on 21 August 2017. It had previously been approved by the Comité Ético de Investigación Clínica con medicamentos (Ethical Committee for Clinical Research involving medicines) at the Quirón Hospital Group on 18 July 2017. The approval includes the ‘Patient Information Sheet’ and the Informed Consent Document.
We obtained informed consent from all participants in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Rodas, G., Soler, R., Balius, R. et al. Autologous bone marrow expanded mesenchymal stem cells in patellar tendinopathy: protocol for a phase I/II, single-centre, randomized with active control PRP, double-blinded clinical trial. J Orthop Surg Res 14, 441 (2019). https://doi.org/10.1186/s13018-019-1477-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13018-019-1477-2