
Abstract
Background
We investigated the underlying molecular mechanisms of bone overgrowth after femoral fracture by using high-throughput bioinformatics approaches.
Methods
The gene expression profile of GSE3298 (accession number) was obtained from the Gene Expression Omnibus database. Sixteen femoral growth plate samples, including nine samples without fracture and seven fracture samples for seven time points, were used for analysis. The Limma package was applied to identify differentially expressed genes (DEGs) between fractured and intact samples. The DAVID online tool was used for Gene ontology functional and pathway enrichment analysis. A protein-protein interaction (PPI) network established by String software was used to identify interactions between significant DEGs, and network modules were detected using plug-in MCODE. Additionally, a transcription regulatory network was constructed based on the ENCODE Project and PPI network.
Results
A total of 680 DEGs were screened in fractured femoral growth plate samples compared with controls, including 238 up- and 442 down-regulated genes. These DEGs were significantly involved in the calcium signaling pathway and cancer pathway. A PPI network was constructed with 167 nodes and 233 edges, and module analysis demonstrated that CCL2, CSF2, NOS2, and DLC1 may stimulate bone overgrowth after femoral fracture via anti-apoptosis-related functions. A transcription regulatory network was constructed with 387 interacting pairs, and overlapping nodes were significantly enriched in intracellular signaling cascade and regulation of cell proliferation, among others.
Conclusions
Bone overgrowth was associated with changes in the expression of identified DEGs such as CCL2, NOS2, CSF2, and DLC1 in the femoral head. They may be important in regulating bone overgrowth via the anti-apoptosis of osteoblasts.
Similar content being viewed by others
Background
Femoral fracture, which is one of the most commonly occurring fractures during childhood, always results from casual falls, motor vehicle accidents, or sporting accidents [1]. Treatment of femoral fracture typically includes open-reduction, traction, and internal fixation. However, pediatric femoral fracture often results in the stimulation of bone overgrowth, particularly in children younger than 12 years [2, 3]. Overgrowth is described as a universal phenomenon in patients with femoral shaft fractures and can elongate the lower limb by nearly 9 mm [4] or 11 mm [5]. It is crucial to explore the underlying molecular mechanism of bone overgrowth associated with femoral fracture.
In recent years, numerous studies have investigated the molecular mechanism of bone overgrowth after femoral fracture [6–8]. Bone homeostasis is thought to be maintained by a balance between bone formation by osteoblasts and bone resorption by osteoclasts in the growth plate. Various proteins such as β-catenin and triggering receptor expressed by myeloid cells-2 interact with each other by controlling the rate of osteoclastogenesis and further regulating bone homeostasis [9]. Several other factors were also found to be involved in this process. For instance, lipoprotein receptor-related protein 4 (LRP4) was found to be associated with the inhibitory function of sclerostin which is secreted by osteocytes and inhibits bone formation [6]. Additionally, the Wnt1/β-catenin signaling pathway is crucial for embryonic and bone homeostasis [10–12], and LRP4 may increase sclerostin secretion through Wnt1/β-catenin signaling [7]. Moreover, fibroblast growth factor receptor (FGFR) is also a critical gene in bone overgrowth and participates in FGFR3 signaling, further affecting chondrocyte proliferation [8]. Another report showed that osteocrin is highly expressed in osteoblasts and interacts with C-type natriuretic peptide receptors to modulate the action of the natriuretic system during bone elongation [13]. Therefore, expression changes of such related genes in cells after femoral fracture may provide insight into the physiological mechanisms of bone overgrowth.
In the past few years, DNA microarray technology has been increasingly utilized to comprehensively test for changes in the messenger RNA (mRNA) expression of genes and search for evidence of overgrowth after mid-femoral fracture [3, 14]. However, the potential molecular mechanism of bone overgrowth after mid-shaft femur fracture also remains unclear. The aim of this study was to explore potentially important genes associated with bone overgrowth after femoral fracture and clarify this phenomenon using high-throughput bioinformatics methods.
Methods
Data source
The gene expression profile of GSE3298 [3, 14], which describes mRNA expression in the rat proximal femoral growth plate after mid-shaft fracture, was derived from the Gene Expression Omnibus (GEO, http://ncbi.nlm.nih.gov/geo/) database based on the GPL1355 Affy metrix Rat Genome 230 2.0 Array platform (Santa Clara, CA, USA). A total of 16 femoral growth plate samples were used for analysis, including nine samples without fracture and seven fracture samples for seven time points: 1 day, 3 days, 1 week, 2 weeks, 3 weeks, 4 weeks, and 6 weeks, after mid-shaft fracture.
Data preprocessing and differentially expressed genes (DEGs) screening
Each sample in the obtained dataset had a probe ID, which was converted into the corresponding gene name. Multiple probe IDs targeting the same gene were averaged as the gene expression value. After expression values were log2 transformed, quantile normalization was carried out [15]. The Limma package (http://www.bioconductor.org/packages/release/bioc/html/limma.html) [16] in R language was used to screen DEGs between intact and fractured samples. Gene expression differences were assessed using Student’s t test, and expression changes were considered to be significant when by P < 0.05.
Functional annotation and pathway enrichment of DEGs
Functional annotation of genes was carried out using the Database for Annotation, Visualization, and Integrated Discovery (DAVID, http://david.abcc.ncifcrf.gov/) [17]. Gene ontology (GO, http://geneontology.org/) annotation and pathway enrichment analysis were performed to derive all associated functions with their enrichment scores and P values. Fisher’s exact test was used to evaluate the differences between the intact and fractured femora. Only results showing enrichment scores of more than 2 and P values <0.05 were considered to be statistically significant.
Construction of protein-protein interaction (PPI) network
The PPI is thought to be important for understanding the potential functions of a certain protein. The up- and down-regulated genes identified as described above were respectively mapped to the Search Tool for the Retrieval of Interacting Genes (STRING, http://string-db.org/) software which is commonly used to predict PPI pairs [18]. The PPI network was constructed with interesting PPI pairs and visualized by Cytoscape 2.8 (http://cytoscape.org/) [19].
Module detection
Molecular complex detection (MCODE) [20] is a clustering algorithm used to identify molecular complexes in the PPI network. Degree ≥2 and k-score ≥2 were selected as cutoff criteria. Next, Bingo [21] was applied to perform GO functional enrichment analysis with a threshold of adjusted P value <0.05, with multiple test adjustment conducted as described by Benjamini-Hochberg.
Transcription regulatory network
The ENCODE (ENCyclopedia of DNA Elements) Project is designed to identify all functional components in the human genome sequence, including protein-coding genes, non-protein-coding genes, sequences that mediate chromosome structure and dynamics, and transcriptional regulatory elements [22]. Based on transcription factor information determined using ENCODE and the PPI network, a transcription regulatory network was constructed with interacting pairs using Cytoscape. Additionally, overlapping nodes in the network with node degrees of >2 were further analyzed for functional enrichment using DAVID. Only the results showing P values <0.05 were considered to be statistically significant.
Results
Identification of DEGs
After data preprocessing and quantile normalization, the gene expression profile of GSE3298 was used to screen for DEGs in the proximal femoral growth plate between intact and fractured samples. A total of 680 DEGs were screened out with P < 0.05, including 238 up- and 442 down-regulated genes (Additional file 1: Table S1).
Functional analysis and pathway enrichment for DEGs
To evaluate DEG functions, GO and pathway analysis were performed for up-regulated and down-regulated genes, respectively. As shown in Table 1, up-regulated genes were mainly enriched in seven GO terms including cell fraction, response to organic substance, and response to wounding, among others, while down-regulated genes mainly function in the extracellular region, vesicles, cytoplasmic vesicles, and membrane-bound vesicles, among others. Additionally, up-regulated genes were found to be mainly enriched in six pathways such as the calcium signaling pathway (P = 0.001) and neuroactive ligand-receptor interactions (P = 0.001), while the 442 down-regulated DEGs were significantly enriched in four pathways, including pathways in cancer (P = 0.008), calcium signaling pathway (P = 0.016), hedgehog signaling pathway (P = 0.025), and MAPK signaling pathway (P = 0.028) (Table 2). These results suggest that perturbations in genes involved in these functions and/or pathways are associated with bone overgrowth following femoral fracture.
Construction of PPI network
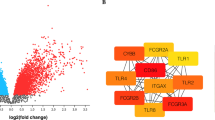
To build the PPI network, significant protein interactions were predicted; the results are displayed in Additional file 2: Table S2. Based on these interaction pairs, a PPI network was constructed with 167 nodes and 233 edges (Fig. 1). Among all nodes, three DEGs showed relatively higher degrees, including chemokine (C–C motif) ligand 2 (CCL2), nitric oxide synthase 2 (NOS2), and colony-stimulating factor 2 (CSF2). Moreover, GO analysis suggested that up-regulated CCL2 participated in the chemokine signaling pathway, and down-regulated NOS2 involved cancer and calcium signaling pathways. In addition, down-regulated CSF2 was mainly enriched in the hematopoietic cell lineage pathway.

Protein-protein interaction network for differentially expressed genes (DEGs). Red circles indicate protein products of significant DEGs; blue lines indicate interaction between different proteins
Significant module analysis
The PPI network was processed by MCODE and a total of 3 significant modules were obtained (Fig. 2). Module A contained one up-regulated DEG (CCL2) and four down-regulated DEGs (NOS2, CSF2, APOE, GAPDHS) . Five down-regulated DEGs made up module B, including potassium voltage-gated channel, shaker-related subfamily, member 6 (KCNA6), potassium voltage-gated channel, KQT-like subfamily, member 1 (KCNQ1), potassium voltage-gated channel, Shaw-related subfamily, member 3 (KCNC3), potassium voltage-gated channel, shaker-related subfamily, beta member 2 (KCNAB2), and potassium voltage-gated channel, Isk-related family, member 1 (KCNE1). Moreover, module C was constituted by one up-regulated DEGDEP domain containing seven (DEPDC7) and five down-regulated DEGs including Rho guanine nucleotide exchange factor 7 (ARHGEF7), Rho guanine nucleotide exchange factor 12 (ARHGEF12), deleted in liver cancer 1 (DLC1), kalirin, RhoGEF kinase (KALRN), and neogenin 1 (NEO1). According to functional analysis (Table 3), DEGs in module A were mainly associated with cGMP-mediated signaling (P = 1.47E−06) and anti-apoptosis-related functions (P = 7.95E−06); genes in module B were mainly related to ion transport-associated functions (P = 1.94E−05) while DEGs in module C were most significantly involved in the regulation of Rho and Ras protein signal transduction (P = 3.21E−08).

Three significant modules identified from PPI network. Red circles indicate protein products of differentially expressed genes; blue lines indicate interaction between different proteins
Transcription regulatory network
Based on information of transcription factors from ENCODE and the PPI network, 387 interacting pairs (Additional file 3: Table S3) were screened out for construction of the transcription regulatory network (Fig. 3). Analysis of functional enrichment showed that the overlapping nodes were significantly enriched in GO terms such as intracellular signaling cascade, regulation of cell proliferation, and regulation of apoptosis, among others.

Transcription regulatory network. Triangles represent transcription factors and circles represent targeted genes
Discussion
Currently, the incidence of fractures of proximal femur have increased as industrial societies become older [23]. Femur fracture is thought to be associated with bone overgrowth, which is a common phenomenon, particularly in children. However, the underlying mechanism remains unclear. In this study, we identified potential genes involved in the molecular mechanism of bone overgrowth after femoral fracture in juvenile rats by using high-throughput bioinformatics. Based on gene expression profiles, a total of 680 DEGs were screened out, including 238 up- and 442 down-regulated DEGs. The up-regulated DEGs were found to be significantly enriched in six pathways, while down-regulated DEGs were strikingly enriched in the cancer pathway and calcium signaling pathway. PPI network construction accompanied by module detection revealed key genes such as CCL2, CSF2, NOS2, and DLC1 were identified to be potentially related with femoral overgrowth.
CCL2, also known as monocyte chemoattractant protein-1 and small inducible cytokine A2, is a chemokine ligand and plays a crucial role in the recruitment and activation of macrophages/monocytes during inflammation after bone injury [24]. It is well known that activation of macrophages and monocytes can stimulate osteoclastic bone resorption or bone formation [25, 26]. However, the recruitment of macrophages and monocytes to the inflamed bone by CCL2 is regulated by rhTNF to regulate bone formation and further improve fracture healing, which only occurs in the fractured environment [27–29]. Additionally, the production of CCL2 is stimulated by the receptor-activator of nuclear factor (NF)-κB ligand, which is regarded as an essential regulator of bone remodeling [30]. In the present study, CCL2 was found to be up-regulated at the proximal femoral growth plate of mid-shaft fracture samples compared with that of no fracture samples and was predicted to participate in cell functions; CCL2 was a key node in the PPI network constructed of significant interaction pairs of DEGs. Thus, CCL2 may be involved in bone overgrowth after femora fracture via recruiting macrophages and monocytes to fractured bone to accelerate bone formation.
CSF2, also known as granulocyte macrophage colony-stimulating factor (GM-CSF), encodes a monomeric glycoprotein which is regarded as a hemopoietic growth factor. CSF2 is released by osteoblast lineage cells [31]. CSF2 is associated with the production, differentiation, and function of granulocytes and macrophages in vitro [32]. The fusion of monocytes/macrophages can form osteoclasts, which were demonstrated to function in degrading bone [31, 33]. Additionally, CSF2 was confirmed to be a target of NF-κB for inducing osteoclastogenesis and further promoting osteolytic bone metastasis [34]. CSF2 has also been reported to enhance osteoclast development which was mediated by tumor necrosis factor α [35]. In addition, CSF2 is an anti-apoptotic factor that can minimize the extent of cell death (such as osteoclasts) in tissues surrounding the injured areas [36]. In this study, CSF2 was identified to be down-regulated. As a result, reduced CSF2 may negatively regulate osteoclastogenesis, resulting in partial recovery of bone formation.
NOS2 is an isoenzyme of nitric oxide (NO) synthase and plays an important role in producing NO, a multifunctional signal molecule. Osteoclasts have been confirmed to express NOS2 and release NO in a regulated manner [37–40]. In addition, NO is involved in the mechanism of osteoclastic activity by releasing bone-resorbing inflammatory cytokines [41]. Endogenously produced NO exerts potent biphasic actions that may significantly affect the proliferation, recruitment, differentiation, and/or survival of osteoblasts and osteoclasts [42–44]. Low levels of NO may support osteoblast bone formation and osteoclast-mediated bone remodeling as well as protect osteoblasts against apoptosis, while high NO levels inhibit osteoclastogenesis and prevent bone loss [45–47]. Additionally, NO is involved in the control of Ca2+ dynamics and mediates Ca2+-inhibited bone resorption [39, 48]. Furthermore, epidermal growth factor receptor/signal transducers and activators of transcription 3 can interact with the NOS2 promoter and activate NOS2 expression [49]. In this study, NOS2 was predicted to participate in anti-apoptosis-related functions and was found to be significantly enriched in the calcium pathway and cancer pathway. Therefore, NOS2 may participate in bone overgrowth after femora fracture via suppressing osteoblast apoptosis through the cancer pathway and calcium pathway. However, the regulation of NOS2 in bone overgrowth after fracture requires further investigation.
DLC1 encodes a Rho GTPase-activating protein that regulates osteoclastogenesis via Rho protein signal transduction [50]. Moreover, Rho GTPases have been confirmed to play a critical role in regulating the actin cytoskeleton organization of osteoclasts [51]. In the present study, DLC1 was found to be down-regulated and enriched in the functions of regulation of Rho protein signal transduction in module C. Thus, this gene may also be important for reducing osteoclastogenesis and bone resorption after femur fracture.
Conclusions
The identified DEGs, particularly those in significant gene modules, including CCL2, CSF2, NOS2, and DLC1, may play a vital role in bone overgrowth after mid-shaft femur fracture. Experimental studies including samples of a larger size will be performed in the future. These data underscore the complexity of the regulation of bone overgrowth. Additionally, these findings form a basis for future studies focusing on the role of these key genes in the molecular mechanisms of bone growth disturbances with the longer-term goal of investigating proper treatment for children with fractured growing bones.
Abbreviations
- CCL2:
-
Chemokine (C-C motif) ligand 2
- CSF2:
-
Colony-stimulating factor 2
- DEGs:
-
Differentially expressed genes
- ENCyclopedia of DNA Elements:
-
The ENCODE
- FGFR:
-
Fibroblast growth factor receptor
- GM-CSF:
-
Granulocyte macrophage colony-stimulating factor
- GO:
-
Gene ontology
- LRP4:
-
Lipoprotein receptor-related protein 4
- MCODE:
-
Molecular complex detection
- NO:
-
Nitric oxide
- NOS2:
-
Nitric oxide synthase 2
- PPI:
-
Protein-protein interaction
References
Loder RT, O’Donnell PW, Feinberg JR. Epidemiology and mechanisms of femur fractures in children. J Pediatr Orthop. 2006;26:561–6. doi:10.1097/01.bpo.0000230335.19029.ab.
Jung ST, Park H, Lee JH, Kim JR. Residual angulation of distal tibial diaphyseal fractures in children younger than ten years. J Orthop Surg Res. 2014;9:84. doi:10.1186/s13018-014-0084-5.
Ashraf N, Meyer MH, Frick S, Meyer Jr RA. Evidence for overgrowth after midfemoral fracture via increased RNA for mitosis. Clin Orthop Relat Res. 2007;454:214–22. doi:10.1097/01.blo.0000238783.21478.5d.
Shapiro F. Fractures of the femoral shaft in children. The overgrowth phenomenon. Acta Orthop Scand. 1981;52:649–55.
Hougaard K. Femoral shaft fractures in children: a prospective study of the overgrowth phenomenon. Injury. 1989;20:170–2.
Leupin O, Piters E, Halleux C, Hu S, Kramer I, Morvan F, Bouwmeester T, Schirle M, Bueno-Lozano M, Fuentes FJR. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem. 2011;286:19489–500.
Leupin O, Piters E, Halleux C, Hu S, Kramer I, Morvan F, Bouwmeester T, Schirle M, Bueno-Lozano M, Fuentes FJ, Itin PH, Boudin E, de Freitas F, Jennes K, Brannetti B, Charara N, Ebersbach H, Geisse S, Lu CX, Bauer A, Van Hul W, Kneissel M. Bone overgrowth-associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem. 2011;286:19489–500. doi:10.1074/jbc.M110.190330.
Karolak MR, Yang X, Elefteriou F. FGFR1 signaling in hypertrophic chondrocytes is attenuated by the Ras-GAP neurofibromin during endochondral bone formation. Human Mol Genet. 2015;24:2552–64.
Otero K, Shinohara M, Zhao H, Cella M, Gilfillan S, Colucci A, Faccio R, Ross FP, Teitelbaum SL, Takayanagi H. TREM2 and β-catenin regulate bone homeostasis by controlling the rate of osteoclastogenesis. J Immunol. 2012;188:2612–21.
Johnson ML, Kamel MA. The Wnt signaling pathway and bone metabolism. Curr Opin Rheumatol. 2007;19:376–82. doi:10.1097/BOR.0b013e32816e06f9.
Balemans W, Piters E, Cleiren E, Ai M, Van Wesenbeeck L, Warman ML, Van Hul W. The binding between sclerostin and LRP5 is altered by DKK1 and by high-bone mass LRP5 mutations. Calcif Tissue Int. 2008;82:445–53. doi:10.1007/s00223-008-9130-9.
Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang J, Harris SE, Wu D. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280:19883–7. doi:10.1074/jbc.M413274200.
Gethin P. Thomas PM. The role of osteocrin and its interaction with the natriuretic system in bone growth. Handbook of Growth and Growth Monitoring in Health and Disease. Springer New York; 2012. p. 2825–37. doi:10.1007/978-1-4419-1795-9_168.
Meyer Jr RA, Meyer MH, Ashraf N, Frick S. Changes in mRNA gene expression during growth in the femoral head of the young rat. Bone. 2007;40:1554–64. doi:10.1016/j.bone.2007.01.013.
Fujita A, Sato JR, Rodrigues Lde O, Ferreira CE, Sogayar MC. Evaluating different methods of microarray data normalization. BMC Bioinf. 2006;7:469. doi:10.1186/1471-2105-7-469.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi:10.1093/nar/gkv007.
da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi:10.1038/nprot.2008.211.
Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–D8.
Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–2. doi:10.1093/bioinformatics/btq675.
Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinf. 2003;4:2.
Maere S, Heymans K, Kuiper M. BiNGO: a cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005;21:3448–9. doi:10.1093/bioinformatics/bti551.
Qu H, Fang X. A brief review on the human encyclopedia of DNA elements (ENCODE) project. Genomics Proteomics Bioinformatics. 2013;11:135–41. doi:10.1016/j.gpb.2013.05.001.
Lenich A, Vester H, Nerlich M, Mayr E, Stöckle U, Füchtmeier B. Clinical comparison of the second and third generation of intramedullary devices for trochanteric fractures of the hip—blade vs screw. Injury. 2010;41:1292–6.
Mantovani A, Allavena P, Sica A. Tumour-associated macrophages as a prototypic type II polarised phagocyte population: role in tumour progression. Eur J Cancer. 2004;40:1660–7. doi:10.1016/j.ejca.2004.03.016.
Nathan CF. Secretory products of macrophages. J Clin Invest. 1987;79:319–26. doi:10.1172/JCI112815.
Mundy GR, Boyce B, Hughes D, Wright K, Bonewald L, Dallas S, Harris S, Ghosh-Choudhury N, Chen D, Dunstan C, et al. The effects of cytokines and growth factors on osteoblastic cells. Bone. 1995;17:71S–5S.
Alexander KA, Chang MK, Maylin ER, Kohler T, Muller R, Wu AC, Van Rooijen N, Sweet MJ, Hume DA, Raggatt LJ, Pettit AR. Osteal macrophages promote in vivo intramembranous bone healing in a mouse tibial injury model. J Bone Miner Res. 2011;26:1517–32.
Rahimi P, Wang CY, Stashenko P, Lee SK, Lorenzo JA, Graves DT. Monocyte chemoattractant protein-1 expression and monocyte recruitment in osseous inflammation in the mouse. Endocrinology. 1995;136:2752–9. doi:10.1210/endo.136.6.7750500.
Volejnikova S, Laskari M, Marks Jr SC, Graves DT. Monocyte recruitment and expression of monocyte chemoattractant protein-1 are developmentally regulated in remodeling bone in the mouse. Am J Pathol. 1997;150:1711–21.
Meng YH, Li H, Chen X, Liu LB, Shao J, Chang KK, Du MR, Jin LP, Li MQ, Li DJ. RANKL promotes the growth of decidual stromal cells in an autocrine manner via CCL2/CCR2 interaction in human early pregnancy. Placenta. 2013;34:663–71. doi:10.1016/j.placenta.2013.04.020.
Shinohara M, Takayanagi H. Novel osteoclast signaling mechanisms. Curr Osteoporos Rep. 2007;5:67–72.
Burgess AW, Metcalf D. The nature and action of granulocyte-macrophage colony stimulating factors. Blood. 1980;56:947–58.
He X, Andersson G, Lindgren U, Li Y. Resveratrol prevents RANKL-induced osteoclast differentiation of murine osteoclast progenitor RAW 264.7 cells through inhibition of ROS production. Biochem Biophys Res Commun. 2010;401:356–62.
Park BK, Zhang H, Zeng Q, Dai J, Keller ET, Giordano T, Gu K, Shah V, Pei L, Zarbo RJ. NF-κB in breast cancer cells promotes Osteolytic bone metastasis by inducing osteoclastogenesis via GM-CSF. Nat Med. 2007;13:62–9.
Atanga E, Dolder S, Dauwalder T, Wetterwald A, Hofstetter W. TNFalpha inhibits the development of osteoclasts through osteoblast-derived GM-CSF. Bone. 2011;49:1090–100. doi:10.1016/j.bone.2011.08.003.
Rehman J, Traktuev D, Li J, Merfeld-Clauss S, Temm-Grove CJ, Bovenkerk JE, Pell CL, Johnstone BH, Considine RV, March KL. Secretion of angiogenic and antiapoptotic factors by human adipose stromal cells. Circulation. 2004;109:1292–8. doi:10.1161/01.CIR.0000121425.42966.F1.
Kasten TP, Collin-Osdoby P, Patel N, Osdoby P, Krukowski M, Misko TP, Settle SL, Currie MG, Nickols GA. Potentiation of osteoclast bone-resorption activity by inhibition of nitric oxide synthase. Proc Natl Acad Sci U S A. 1994;91:3569–73.
Brandi ML, Hukkanen M, Umeda T, Moradi-Bidhendi N, Bianchi S, Gross SS, Polak JM, MacIntyre I. Bidirectional regulation of osteoclast function by nitric oxide synthase isoforms. Proc Natl Acad Sci U S A. 1995;92:2954–8.
Sunyer T, Rothe L, Kirsch D, Jiang X, Anderson F, Osdoby P, Collin-Osdoby P. Ca2+ or Phorbol ester but not inflammatory stimuli elevate inducible nitric oxide synthase messenger ribonucleic acid and nitric oxide (NO) release in avian osteoclasts: autocrine NO mediates Ca2 + -inhibited bone resorption. Endocrinology. 1997;138:2148–62. doi:10.1210/endo.138.5.5144.
Sunyer T, Rothe L, Jiang X, Osdoby P, Collin-Osdoby P. Proinflammatory agents, IL-8 and IL-10, upregulate inducible nitric oxide synthase expression and nitric oxide production in avian osteoclast-like cells. J Cell Biochem. 1996;60:469–83. doi:10.1002/(SICI)1097-4644(19960315)60:4<469::AID-JCB4>3.0.CO;2-Q.
Löwik C, Nibbering P, Van de Ruit M, Papapoulos S. Inducible production of nitric oxide in osteoblast-like cells and in fetal mouse bone explants is associated with suppression of osteoclastic bone resorption. J Clin Investig. 1994;93:1465.
Holliday LS, Dean AD, Lin RH, Greenwald JE, Gluck SL. Low NO concentrations inhibit osteoclast formation in mouse marrow cultures by cGMP-dependent mechanism. Am J Physiol. 1997;272:F283–91.
Chae HJ, Park RK, Chung HT, Kang JS, Kim MS, Choi DY, Bang BG, Kim HR. Nitric oxide is a regulator of bone remodelling. J Pharm Pharmacol. 1997;49:897–902.
van’t Hof RJ, Ralston SH. Nitric oxide and bone. Immunology. 2001;103:255–61.
van’t Hof RJ, Armour KJ, Smith LM, Armour KE, Wei XQ, Liew FY, Ralston SH. Requirement of the inducible nitric oxide synthase pathway for IL-1-induced osteoclastic bone resorption. Proc Natl Acad Sci U S A. 2000;97:7993–8. doi:10.1073/pnas.130511497.
Wimalawansa SJ. Restoration of ovariectomy-induced osteopenia by nitroglycerin. Calcif Tissue Int. 2000;66:56–60.
Choi B-M, Pae H-O, Jang SI, Kim Y-M, Chung H-T. Nitric oxide as a pro-apoptotic as well as anti-apoptotic modulator. J Biochem Mol Biol. 2002;35:116–26.
Zheng H, Yu X, Collin-Osdoby P, Osdoby P. RANKL stimulates inducible nitric-oxide synthase expression and nitric oxide production in developing osteoclasts. An autocrine negative feedback mechanism triggered by RANKL-induced interferon-beta via NF-kappaB that restrains osteoclastogenesis and bone resorption. J Biol Chem. 2006;281:15809–20. doi:10.1074/jbc.M513225200.
Lo HW, Hsu SC, Ali-Seyed M, Gunduz M, Xia W, Wei Y, Bartholomeusz G, Shih JY, Hung MC. Nuclear interaction of EGFR and STAT3 in the activation of the iNOS/NO pathway. Cancer Cell. 2005;7:575–89. doi:10.1016/j.ccr.2005.05.007.
Wang Y, Lei R, Zhuang X, Zhang N, Pan H, Li G, Hu J, Pan X, Tao Q, Fu D, Xiao J, Chin YE, Kang Y, Yang Q, Hu G. DLC1-dependent parathyroid hormone-like hormone inhibition suppresses breast cancer bone metastasis. J Clin Invest. 2014;124:1646–59. doi:10.1172/JCI71812.
Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–79.
Acknowledgements
The study was supported by a special fund for medical service of the Jilin Finance Department, project number: SCZSY201507.
Funding
The study was supported by the special fund for medical service of Jilin Finance Department, project number: SCZSY201507.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Authors’ contributions
YL performed the statistical analysis. CL carried out the study, together with WZ, and collected important background information. XL conceived the study, participated in the study design, and helped to draft the manuscript. All the authors have read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
All the DEGs screened between intact and fractured samples. (XLS 96 kb)
Additional file 2:
Significant protein interactions in the PPI network.ᅟ(XLS 39 kb)
Additional file 3:
The interacting pairs used for construction of the transcription regulatory network.ᅟ(XLS 24 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liu, C., Liu, Y., Zhang, W. et al. Screening for potential genes associated with bone overgrowth after mid-shaft femur fracture in a rat model. J Orthop Surg Res 12, 8 (2017). https://doi.org/10.1186/s13018-017-0510-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13018-017-0510-6