
Abstract
Background
The c-Jun N-terminal kinase (JNK) pathway is an evolutionarily conserved regulator of cell death, which is essential for coordinating tissue homeostasis. In this study, we have characterized the Drosophila Ste20-like kinase Slik as a novel modulator of JNK pathway-mediated apoptotic cell death.
Results
First, ectopic JNK signaling-triggered cell death is enhanced by slik depletion whereas suppressed by Slik overexpression. Second, loss of slik activates JNK signaling, which results in enhanced apoptosis and impaired tissue homeostasis. In addition, genetic epistasis analysis suggests that Slik acts upstream of or in parallel to Hep to regulate JNK-mediated apoptotic cell death. Moreover, Slik is necessary and sufficient for preventing physiologic JNK signaling-mediated cell death in development. Furthermore, introduction of STK10, the human ortholog of Slik, into Drosophila restores slik depletion-induced cell death and compromised tissue homeostasis. Lastly, knockdown of STK10 in human cancer cells also leads to JNK activation, which is cancelled by expression of Slik.
Conclusions
This study has uncovered an evolutionarily conserved role of Slik/STK10 in blocking JNK signaling, which is required for cell death inhibition and tissue homeostasis maintenance in development.
Similar content being viewed by others
Background
Tissue homeostasis depends on highly coordinated cellular processes, including cell regeneration, proliferation, differentiation, and apoptosis [1,2,3,4,5]. The evolutionarily conserved c-Jun N-terminal kinase (JNK) signaling is one of the key pathways that govern a wide range of biological processes including stress response, cell proliferation and apoptosis, which facilitates the maintenance of tissue homeostasis in development [6,7,8,9]. JNK signaling is initiated by various intrinsic and extrinsic signals, and is mediated by a mitogen-activated protein kinase (MAPK) cascade [10, 11]. In Drosophila, JNK signaling is activated by the tumor necrosis factor (TNF) ortholog Eiger (Egr), which binds to its receptor Wengen (Wgn) or Grindelwald (Grnd) that recruits the TNF receptor-associated factor 2 (dTRAF2) [12,13,14] to trigger the conserved MAP kinase cascade including dTAK1 (JNKK kinase), Hemipterous (Hep, JNK kinase) and Basket (Bsk, Drosophila JNK) [15,16,17]. Phosphorylated Bsk translocates into the nucleus to phosphorylate and activate the transcription factor complex AP-1, which upregulates the transcription of target genes [11]. The puckered (puc) gene, which encodes a JNK phosphatase, is a transcriptional target of JNK signaling, and thereby regulates JNK activity in a negative feedback manner [18, 19].
The Sterile-20 (Ste20) kinases have been implicated in numerous physiological processes including cell proliferation and survival [20], cytoskeletal dynamics [21, 22], and ion transport [23]. The Ste20-kinase family consists of a heterogeneous group of Ser/Thr kinases including two large families (PAK- and GCK-like) and ten divergent subfamilies [21, 24]. Drosophila Slik and its mammalian ortholog STK10 belong to the GCK-V subfamily [25], which has emerged as an evolutionarily conserved modulator of ezrin/radixin/moesin (ERM) family proteins [26, 27]. In Drosophila, Moesin and Merlin, two FERM domain proteins, are the best known substrates of Slik [28]. While Slik-mediated phosphorylation activates Moesin to stabilize microtubule organization and epithelial integrity [29, 30], phosphorylation by Slik inactivates Merlin to adjust cell proliferation [31, 32]. Most of the physiological activities are governed by signaling pathways, though Slik is known to participate in a variety of cellular processes, the connection between Slik and potential signaling pathway remains poorly understood.
Drosophila is one of the best model organisms to study genetics, with many components of signal pathways being identified in fruit flies. We have previously performed a genetic screen with ectopic JNK-triggered cell death, and have characterized multiple components and regulators of the JNK pathway in Drosophila [33, 34]. In the current study, we report that Slik is required for maintaining tissue homeostasis by preventing JNK-mediated apoptotic cell death in development. Loss of slik leads to JNK activation and triggers JNK-mediated cell death. Genetic epistasis analysis suggests that Slik acts upstream of or in parallel to Hep to suppress JNK pathway. Finally, the human ortholog STK10 has retained Slik functions to modulate JNK activity when introduced into Drosophila or in human cells. Taken together, this study unravels a crucial role of Slik in maintaining tissue homeostasis and organ development by preventing JNK-mediated apoptotic cell death.
Results
Slik inhibits ectopic JNK-induced cell death in development
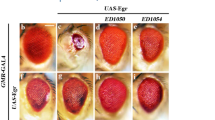
Given that JNK signaling is of vital importance in regulating cell death, to identify new factors that regulate JNK-mediated cell death, we performed a genetic screen for dominant modifiers of GMR > Egr-induced cell death in the Drosophila eyes using the Bloomington Drosophila Stock Center Deficiency kit [5, 35]. We found that three overlapping deficiencies, Df(2R)BSC603, Df(2R)ED4065 and Df(2R)ED4071, enhanced the small eye phenotype caused by GMR > Egr, indicating a potential enhancer is located within the common region (Additional file 1: Fig. S1A–E). This was confirmed by subsequent experiments showing that either mutation or knockdown of slik, one of the genes in this region, significantly enhanced the GMR > Egr-induced small eye phenotype (Additional file 11 Fig. S1F, G). Furthermore, ectopic expression of a constitutive active form of Hep (HepCA) triggered strong cell death posterior to the morphogenetic furrow (MF) in the eye imaginal discs (Fig. 1F, G), indicated by Acridine Orange (AO) staining that detects dying cells [36], and produced a small eye phenotype in the adults (Fig. 1A, B). We found that GMR > HepCA-induced cell death and small eye phenotype were enhanced in heterozygous slik mutants, but were significantly suppressed by overexpression of Slik (Fig. 1C–E, H–J).

Slik inhibits ectopic Hep-induced cell death. Light micrographs of Drosophila adult eyes A–D and fluorescence micrographs of third instar larval eye discs F–I are shown. Compared with the GMR-Gal4 controls (A, F, GMR > HepCA-induced small eye phenotype B and cell death in eye discs G were partially inhibited by overexpression of Slik C, H, whereas enhanced in heterozygous slik mutants D, I. Statistical analysis of the adult eye sizes (E, n = 10 for each genotype) and AO-positive cell numbers in eye discs (J, n = 10 for each genotype) are shown. Light micrographs of Drosophila adult wings K–N and fluorescence micrographs of third instar larval wing discs P–S are shown. Compared with the ptc-Gal4 controls K, P, ectopic expression of Hep driven by ptc-Gal4 generated a loss-of-ACV phenotype in adults L and induced cell death in larval wing discs (Q), which were strongly blocked by expressing Slik (N, S), but not LacZ (M, R). The lower panels show high magnification view of the boxed areas in upper panels K–N). Quantification of the presence of ACV in adult wings (O, n = 15 for each genotype) and cell death in wing discs (T, n = 10 for each genotype) are shown. One-way ANOVA was used to compute P-values, ****p < 0.0001, ***p < 0.001, **p < 0.01, *P < 0.05, n.s indicates not significant. See the supplementary material for detailed genotypes. Scale bar:100 μm in A–D, F–I, K–N (upper panels) and P–S, 50 μm in K–N (lower panels)
To investigate whether Slik inhibits JNK-mediated cell death in other tissues, we turned to the developing wing. Compared with the controls (Fig. 1K, P), expression of Hep along the anterior/posterior (A/P) compartment border driven by patched (ptc)-Gal4 [37] resulted in increased cell death in the wing imaginal discs (Fig. 1Q), and generated a loss of anterior cross vein (ACV) phenotype in the adult wings (Fig. 1L). Both phenotypes were considerably suppressed by ectopic expression of Slik, but remained unaffected by that of LacZ (Fig. 1M–O, R–T). Collectively, these results indicate that Slik negatively regulates ectopic JNK-induced cell death during eye and wing development.
Depletion of slik triggers apoptotic cell death in development
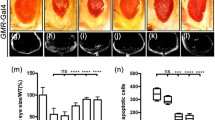
Given that Slik is an inhibitor of ectopic JNK-mediated cell death, we wonder whether slik is physiologically required for preventing cell death in development. To test this, we depleted slik with three independent RNA interference (RNAi), whose knockdown efficiencies were confirmed by the quantitative reverse transcription polymerase chain reaction (RT-qPCR) assay (Fig. 2P). Compared with the GMR-Gal4 or ptc-Gal4 controls, cell death was significantly increased upon slik knockdown in the corresponding areas of 3rd instar larval eye (Fig. 2A–E) or wing (Fig. 2F–J) discs, suggesting that endogenous Slik inhibits cell death in normal development.

Depletion of slik triggers apoptotic cell death. Fluorescence micrographs of third instar larval eye discs A–D are shown. Compared with the GMR-Gal4 controls (A), downregulation of slik with three independent RNA interference (RNAi) triggered extensive cell death posterior to the MF (B–D). Fluorescence micrographs of third instar larval wing discs F–I, K–N are shown. Compared with the ptc-Gal4 controls (F, K), depletion of slik induced massive cell death (G–I) and apoptosis L–N in wing discs. Quantification of cell death in eye discs (E, n = 12; n = 9; n = 10; n = 9) and statistical analysis of AO positive cell number (J, n = 10 for each genotype) and cleaved Dcp-1 (cDcp-1) activity (O, n = 10 for each genotype) in wing discs are shown. P The knock-down efficacies of slik RNAi lines. Expression of three independent slik RNAi significantly reduces the level of slik mRNA, as measured by quantitative RT-PCR. One-way ANOVA with Bonferroni multiple-comparison test was used to compute P-values, ****P < 0.0001, ***P < 0.001. See the supplementary material for detailed genotypes. Scale bar: 50 μm
Apoptosis is the major form of cell death in Drosophila, which is mediated by the cleavage and activation of caspases [38, 39]. To check whether Slik regulates apoptotic cell death, we utilized an anti-cleaved Dcp-1 antibody that specifically recognizes the activated form of the effector caspase Dcp-1 [40]. We found that knockdown of slik induced strong apoptosis in the wing discs (Fig. 2K–O). Consistently, slik depletion-induced cell death was efficiently blocked by expressing P35, a viral caspase inhibitor [41], but not a lacZ RNAi (Additional file 1: Fig. S2). Taken together, these data suggest that slik is physiologically required for preventing apoptotic cell death in normal development.
Depletion of slik promotes JNK pathway activation
As JNK signaling plays significant roles in regulating apoptotic cell death [42,43,44,45], slik depletion-triggered developmental apoptosis may depend on JNK pathway activation. In agreement with this assumption, knockdown of slik, but not lacZ, by ptc-Gal4 resulted in upregulated expression of TRE-RFP (Fig. 3A–D, A’-D’), a general reporter of JNK signaling [46, 47]. Moreover, JNK phosphorylation, detected by an antibody specific to the phosphorylated JNK (p-JNK) [5, 48], was considerably elevated upon slik depletion (Fig. 3E–H, E’–H’). Together, these data suggest that loss of slik potentiates JNK signaling in development.

Loss of slik promotes JNK pathway activation in vivo. Merged fluorescence micrographs of Drosophila third instar larval wing discs are shown A–H. The red channels detected only RFP A’–D’ or pJNK signal E’–H’. Compared with the controls A, E, expressing three independent slik RNAi notably elevated the expression of TRE-RFP B–D and JNK phosphorylation (F-H) along the A/P compartment boundary. See the supplementary material for detailed genotype. Scale bar: 50 μm
JNK signaling is required for loss of slik-induced apoptotic cell death
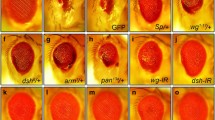
Given that slik depletion activates JNK and apoptosis, we hypothesized that loss of slik might promotes cell death through activating JNK signaling. In agreement with this speculation, we found that ptc-Gal4 (Fig. 4A) driven slik depletion-caused cell death (Fig. 4B) was soundly suppressed by knocking down bsk, which encodes the Drosophila JNK, or expressing a dominant negative form of Bsk (BskDN) or Puc, an inhibitor of JNK [18, 19] (Fig. 4H–K). Consistently, loss of slik-induced apoptosis, visualized by anti-cleaved Dcp-1 (cDcp-1) antibody staining, was remarkably inhibited by BskDN (Fig. 4L, M, P, Q). In conclusion, these results suggest that slik depletion triggers JNK-dependent apoptotic cell death.

Slik acts upstream of or in parallel to Hep to adjust JNK-dependent apoptotic cell death. Fluorescence micrographs of third instar larval wing discs are shown (A–J). Compared with the control (A), RNAi-mediated down-regulation of slik along the A/P compartment boundary by ptc-Gal4 induced obvious cell death (B), which was suppressed by depletion of hep (G) or bsk (H), or expressing a dominant-negative form of Bsk (I) or Puc (J), but remained unaffected by expressing RNAi of lacZ (C), egr (D), dTRAF2 (E) or dTAK1 (F). Merged fluorescence micrographs of third instar larval wing discs are shown (L–P). Compared with the control (L), loss of slik-induced apoptosis M was blocked by expressing hep RNAi O or BskDN (P), but not by egr RNAi (N). Statistical analysis of the AO positive cell numbers (K, n = 10 for each genotype) and cleaved Dcp-1 (cDcp-1) activity (Q, n = 10 for each genotype) in wing discs are shown. One-way ANOVA with Bonferroni multiple-comparison test was used to compute P-values, ****P < 0.0001, n.s indicates not significant. See the supplementary material for detailed genotypes. Scale bar: 50 μm
Slik functions upstream of or in parallel to Hep
To understand how Slik regulates JNK signaling-mediated cell death, we performed genetic epistasis analysis between Slik and core components of the Egr-JNK pathway. We found that slik knockdown-induced cell death was significantly suppressed by depletion of hep, but not by that of egr, dTRAF2, dTAK1 or lacZ serving as a negative control (Fig. 4A–G, K). Consistently, loss of slik-induced Dcp-1 activation was abolished by expression of hep-IR or BskDN, but remained unaffected upon depletion of egr (Fig. 4L–Q). Taken together, these results suggest that Slik acts upstream of or in parallel to Hep to regulate JNK-mediated apoptotic cell death.
Slik modulates JNK-dependent tissue homeostasis in normal development
Strict regulation of cell death, which contributes to the adjustment of cell number and elimination of unwanted cells, is vital for tissue homeostasis and organism fitness [49]. To characterize the physiological function of slik in tissue homeostasis, we generated RNAi-mediated knockdown clones in the wing discs by the FLP/FRT-mediated recombination. Compared with the control clones, slik depletion resulted in decreased clone (GFP+) sizes and numbers, which were appreciably restored by expressing BskDN (Fig. 5A–D, Additional file 1: Fig. S3A). Furthermore, slik knockdown either in the posterior compartment by en-Gal4 or in the wing pouch by nub-Gal4 resulted in diminished sizes of corresponding areas, which were largely recovered by blocking Bsk activity (Fig. 5E–L). Moreover, we also generated slik mutant clones by FLP/FRT-mediated genetic mosaic technique [50], and observed that slik mutant clones (GFP−) were significantly smaller than their simultaneously generated wild-type twin clones (2×GFP), while the control clones were almost the same size as their counterparts (Additional file 1: Fig. S3B–D). Together, these data suggest that slik is required to maintain JNK-dependent tissue homeostasis in normal development.

slik is physiologically required for proper tissue homeostasis in development. Merged fluorescence micrographs of Drosophila third instar larval wing discs are shown (A–C, E–G, I–K). From the Flp-out clones (marked by GFP), compared with the control (A), slik knockdown clones (GFP+) displayed decreased clonal size (B), which was rescued by expressing BskDN (C). Statistical analysis of clone size/total size ratio in A–C are shown (D, n = 10 for each group). Besides, compared with the en-Gal4 (E) and nub-Gal4 controls (I), slik depletion-evoked decreased GFP area in the posterior compartment F or the whole wing pouch J was restored by BskDN (G, K). Statistical analysis of GFP+ area/GFP− area ratio in E–G (H, n = 10 for each group) and I-K (L, n = 11; n = 8; n = 7) are shown. One-way ANOVA with Bonferroni multiple-comparison test was used to compute P-values, ****P < 0.0001, ***p < 0.001, *P < 0.05, n.s indicates not significant. See the supplementary material for detailed genotypes. Scale bar: 100 μm
Human STK10 rescues slik depletion-induced developmental defects in Drosophila
Slik encodes a member of the Sterile-20 kinase family, sharing 46% similarity and 32% identity with its human ortholog STK10. To check whether STK10 has retained the developmental functions of Slik in Drosophila, we produced UAS-STK10 transgenic flies and checked whether expression of STK10 could rescue slik loss-caused developmental defects. We noted that ectopic expression of Slik or STK10, verified by RT-qPCR (Additional file 1: Fig. S4), could largely rescue ptc > slik-IR-induced L3-L4 area reduction in adult wings (Fig. 6A–E) and robust cell death along A/P compartment boundary in larval wing discs (Fig. 6F–J). In addition, area reduction in the posterior compartment of en > slik-IR wing discs was efficiently rescued by expressing Slik or STK10 (Fig. 6K–O). Furthermore, MARCM analyses showed that the augmented apoptosis and diminished size of slik mutant clones (marked by GFP) were effectively mitigated upon the expression of BskDN, Slik, or STK10 (Additional file 1: Fig. S5). Thus, these data demonstrate that the developmental functions of Slik are evolutionarily conserved by STK10.

Human STK10 reliefs slik depletion-caused cell death. Light micrographs of adult wings (A–D) and fluorescence micrographs of third instar larval wing discs F–I, K–N are shown. Compared with the controls (A, F), loss of slik driven by ptc-Gal4 resulted in reduced L3-L4 area in adult wings B and intense AO staining in wing discs (G), which were effectively rescued by expressing Slik (C, H) or STK10 (D, I). Statistical analysis of the adult wing L3-L4 size/total size ratio (E, n = 10 for each genotype) and cell death number (J, n = 10 for each genotype) in wing discs are shown. Comparable with the en-Gal4 control (K), slik knockdown-induced GFP area reduction in the posterior compartment L was restored by expressing Slik M or STK10 N. Statistical analysis of GFP+ area/GFP− area ratio in K-N is shown (O, n = 10 for each genotype). One-way ANOVA with Bonferroni multiple-comparison test was used to compute P-values, ****P < 0.0001, n.s indicates not significant. See the supplementary material for detailed genotypes. Scale bar: 100 μm
Slik function in the JNK pathway is retained by STK10
As the JNK pathway is evolutionarily conserved from fly to human, and STK10 is able to rescue slik loss-induced developmental defects, it is plausible that STK10 has retained the regulatory role of Slik in the JNK pathway. Consistently, GMR > Egr-triggered cell death in the larval eye discs and the small eye phenotype in the adults were effectively restored by expressing STK10, while hep depletion was included as a positive control (Additional file 1: Fig. S6). Since Egr triggers both JNK-dependent and JNK-independent cell death in development [12, 35, 51], to determine whether STK10 modulates JNK-mediated cell death, we activated JNK signaling by overexpressing the JNK kinase Hep. We found that expression of STK10 markedly inhibited GMR > HepCA-caused small eye in adults (Fig. 7A–D) and increased AO staining in larval eye discs (Fig. 7E–H), and ptc > Hep-induced loss-of-ACV phenotype in adult wings (Fig. 7I–L) and cell death in larval wing discs (Fig. 7M–P). Hence, we conclude that STK10 could functionally substitute for Slik to regulate JNK-dependent cell death in Drosophila.

Human STK10 inhibits JNK signaling in Drosophila. Light micrographs of Drosophila adult eyes (A–C) and wings (I–K), and fluorescence micrographs of third instar larval eye (E–G) and wing discs (M–O) are shown. Compared with the controls (A, E), GMR > HepCA-evoked small eye phenotype (B) and cell death in eye discs (F) were inhibited by expressing STK10 (C, G). Statistical analysis of the adult eye sizes (D, n = 10 for each genotype) and AO-positive cell numbers in eye discs (H, n = 10 for each genotype) are shown. Consistently, compared with the ptc-Gal4 controls (I, M), ectopic Hep induced a loss-of-ACV phenotype in adult wings (J) and robust cell death in wing discs (N), both of which were suppressed by expressing STK10 (K, O). The lower panels show high magnification view of the boxed areas in upper panels (I–K). The presence of ACV in adult wings (L, n = 15 for each genotype) and AO positive cell numbers in wing discs (P, n = 10 for each genotype) were quantified. One-way ANOVA with Bonferroni multiple-comparison test was used to compute P-values, ****p < 0.0001, ***p < 0.001. See the supplementary material for detailed genotypes. Scale bar: 50 μm in A–C, E–G, I–K (lower panels) and M-O, 100 μm in I–K (upper panels)
Slik and STK10 modulates physiological JNK-mediated cell death
The above results suggest that Slik negatively regulates ectopic JNK-mediated cell death, triggered by overexpression of Egr or Hep, yet it remains unknown whether Slik modulates the physiological JNK signaling. It has been reported that disruption of cell polarity promotes JNK-mediated cell death in development [52, 53]. Consistently, knockdown of the cell polarity gene scrib along the A/P compartment boundary by ptc-Gal4 induced severe cell death in wing discs (Fig. 8F, G) and ACV loss in adult wings (Fig. 8A, B). These phenotypes, caused by the activation of endogenous JNK signaling [5], were effectively inhibited by expressing Slik or STK10 (Fig. 8C–E, H–J), while expression of BskDN and Puc served as the positive controls (Additional file 1: Fig. S7). Importantly, ptc > scrib-IR-increased cell death was potently aggravated in heterozygous slik mutants (Fig. 8K–M). Together, these results indicated that Slik modulates physiological JNK signaling-induced cell death in development.

Slik/STK10 prevents physiological JNK-mediated cell death. Light micrographs of Drosophila adult wings (A–D) and fluorescence micrographs of third instar larval wing discs (F–I, K–L) are shown. RNAi-mediated down-regulation of scrib along the A/P compartment boundary by ptc-Gal4 triggered the loss-of-ACV phenotype in adult wings (B), which was resulted from cell death in larval wing discs (G). Both phenotypes were inhibited by expressing Slik or STK10 (C–D, H–I). The bottom panels show high magnification views of the boxed areas in upper panels (A–D). Importantly, ptc > scrib-IR-induced cell death (K) was enhanced in heterozygous slik mutants (L). Statistical analysis of the presence of ACV in adult wings (E, n = 15 for each genotype) and cell death number in wing discs (J and M, n = 10 for each genotype) were shown. (N) Immunoblot analysis of p-JNK and total JNK protein level in SW480 cells treated by nonspecific siRNA (siCtrl) or two independent siRNA targeting STK10 (referred to as siSTK10-1 and siSTK10-2). p-JNK protein level was increased upon STK10 knockdown, while total JNK protein level remained unchanged. One-way ANOVA with Bonferroni multiple-comparison test and unpaired two tailed t-test were used to compute P-values, ****p < 0.0001, ***p < 0.001, **p < 0.01, *P < 0.05. See the supplementary material for detailed genotypes. Scale bar: 100 μm in A–D (upper panels), F–I and K–L, 50 μm in A–D (lower panels)
Finally, to examine whether STK10 regulates JNK activity in human cells, we knockdown STK10 in SW480 human colon cancer cells by two independent siRNA, whose efficacies were verified by RT-qPCR (Additional file 1: Fig. S8). We found that p-JNK level was up-regulated whereas total JNK protein level remained unaffected upon STK10 knockdown (Fig. 8N). Intriguingly, STK10 depletion-augmented p-JNK level was effectively mitigated by the expression of Slik, but not its kinase-deficient variant (Additional file 1: Fig. S9). These observations provide further evidence supporting the notion that Slik/STK10 play a conserved role in regulating the JNK pathway across both Drosophila and human cells, and the kinase activity of Slik/STK10 likely being crucial for this function.
Discussion
Control of tissue growth requires coordinate actions of cell proliferation, growth and death [54, 55]. Cell death modulates a great deal of physiological processes, including regulation of cell number, sculpting of tissue structures, and elimination of abnormal or aged cells, which plays a fundamental role in tissue homeostasis and organ development [56, 57]. JNK pathway has been shown to play a critical role in tissue homeostasis by maintaining the proper balance of cell death and proliferation [58, 59]. In this study, we took advantage of the fly genetics and identified Slik, the Drosophila ortholog of mammalian STK10, as an essential regulator of JNK-mediated apoptosis. Consistent with our data, slik exhibits moderately high expression in 3rd instar larval imaginal discs [60]. In particular, loss of slik activates JNK signaling and promotes JNK-dependent apoptotic cell death, while overexpression of Slik or its human ortholog STK10 significantly suppresses ectopic or physiologic JNK-induced cell death in development. Furthermore, our genetic epistasis analysis established that Slik acts upstream of or in parallel to Hep, to regulate the JNK signaling in cell death (Additional file 1: Fig. S10). Notably, knockdown of STK10 in human cells also results in JNK activation, suggest the inhibitory role of Slik/STK10 on JNK signaling has been evolutionarily conserved from fly to human.
Previous studies have revealed Moesin and Merlin as the main substrates of Slik. In addition, STRIPAK is shown to act as an upstream regulator that dephosphorylates Slik, which facilitates its association with the cortex [30]. In this work, we characterized Slik as a negative regulator of JNK pathway, yet the direct target of Slik in JNK signaling remains to be identified. It is worth noting that a previous study has documented the involvement of Moesin in the regulation of JNK-dependent cell death via the Rho1-Slpr-dTAK1 axis [61]. Notably, the inhibition of cell death induced by Moesin was observed upon the knockdown of dTAK1 [61], but no such effect was observed in the case of slik loss-triggered cell death (Fig. 4F, K). Therefore, it can be inferred that Slik may regulate JNK-mediated cell death through a distinct mechanism that is independent of Moesin.
As Slik encodes a kinase, Slik-mediated phosphorylation may suppress JNK activity via two possible mechanisms, activating a negative regulator or inactivating a positive regulator. It is well known that phosphorylation can either activate or inactivate a substrate protein. Intriguingly, Slik-mediated phosphorylation promotes the function of Moesin, but inhibits that of Merlin, which results in a coordinate regulation of proliferation and epithelial integrity [27, 31]. If Slik acts upstream of Hep, a possible mechanism is that Slik phosphorylates and activates an inhibitor of Hep, thus inhibits Hep-mediated JNK activation. The protein phosphatase PP2A has been shown to inactivate JNK signaling by dephosphorylating MKK7, the ortholog of Hep, in hepatocellular carcinoma (HCC) cells [61]. Interestingly, Cka, a regulatory subunit of PP2A, acts upstream of Hep to inhibit JNK signaling during Drosophila spermatogenesis [62]. Furthermore, a Cka-Slik physical interaction has been reported, which contributes to PP2A-mediated Slik dephosphorylation [30]. Thus, it is likely that Slik, in a feed-back manner, phosphorylates and activates Cka, which dephosphorylates Hep and inhibits JNK signaling. If Slik acts in parallel to Hep, a possible target is Puc, a serine/threonine protein phosphatase for Bsk. Kinase-mediated phosphorylation has been reported to promote phosphatases activities. For instance, the extracellular signal-regulated kinase 2 (ERK2) activates MAP kinase phosphatase-3 (MKP-3) [63], and protein kinase A (PKA) activates protein phosphatase 2 A (PP2A) by phosphorylation [64]. Of particular interest, mTORC2-induced phosphorylation on DUSP10, the human ortholog of Puc, blocks its proteasome-mediated degradation [65]. Hence, it is plausible that Slik/STK10 may inhibit JNK signaling via phosphorylation and activation of Puc/DUSP10. Apart from this, while Slik regulates Moesin and Merlin through its kinase function, a kinase independent genetic interaction of Slik with Raf has also been described [32, 66].
Conclusions
Loss of slik triggers JNK-mediated apoptosis and impaired tissue homeostasis. Slik is necessary and sufficient for preventing physiological JNK-mediated apoptosis. Human STK10 rescues slik loss-induced cell death and impaired tissue homeostasis. Knockdown of STK10 in human cancer cells also leads to JNK activation, which is cancelled by expressing Slik. Thus, the inhibitory role of Slik/STK10 on JNK signaling has been evolutionarily conserved from fly to human.
Materials and methods
Drosophila strains
Flies were reared on a standard cornmeal and brown sugar medium at 25 °C unless otherwise indicated. The following strains were used in this work: ptc-Gal4, en-Gal4, nub-Gal4, GMR-Gal4, UAS-GFP [67], TRE-RFP [68], UAS-Egr, UAS-Hep [53], UAS-Puc, UAS-BskDN [69], UAS-scrib-IR, UAS-P35 [34], yw hs-Flp; act > CD2 > Gal4 UAS-GFP [70], UAS-dTRAF2-IR, UAS-dTAK1-IR [14] and UAS-bsk-IR [71] were previously described. UAS-slik-IR (55,626), UAS-hep-IR (28,710), UAS-HepCA (6406), Df(2R)BSC603 (25,436), Df(2R)ED4065 (9069), Df(2R)ED4071 (24,117), UAS-LacZ (3956) and hs-Gal4 (1799) were obtained from the Bloomington stock center, UAS-egr-IR (45,253) was acquired from the Vienna Drosophila RNAi Center, UAS-slik-IR (4527R-3) was received from Japanese National Institute of Genetics (NIG), FRT42D slikKG04837 (114,386) was obtained from Kyoto Stock Center, UAS-slik-IR (02634) was got from Tsing Hua Fly Center. yw hs-Flp; FRT42D ubi-GFP was provided by professor Haiyun Song, FRT42D was provided by professor Xianjue Ma, hs-Flp; FRT42D tub-Gal80; tub-Gal4 UAS-GFP was provided by professor Chenhui Wang. The human STK10 and Drosophila Slik expression plasmid, generated by PCR and subcloned into PUAST vector, was further used to produce the transgenic flies.
Acridine Orange (AO) staining
Eye and wing discs were dissected from third-instar larvae in 0.1% PBST (phosphate-buffered saline + 0.1% Tween-20) and incubated in 1×10-5 M AO for 5 min at room temperature prior to imaging.
Immunostaining
Antibody staining was performed by standard procedures for imaginal discs. Dissected discs were fixed in 4% formaldehyde for 20 min. After several washes with 0.3% (v/v) PBST, discs were stained overnight with primary antibodies at 4 ℃, then washed with PBST and incubated with the secondary antibody for 2 h at room temperature. The following primary antibodies were used for immunostaining: rabbit anti-Cleaved Dcp-1 (Cell Signaling Technology, 9578, 1:100), rabbit anti-phospho-JNK (Calbiochem, 559,309, 1:200), secondary antibody is goat anti-rabbit CY3 (Life technologies, A10520, 1:1000).
Heat shock induction of clones
Fluorescently labelled clones were produced in larval imaginal discs using the yw hs-Flp; act > CD2 > Gal4 UAS-GFP strain. Clones were induced by heat shock at 37 °C for 30 min, late third-instar larvae were dissected after recovering for 4 days (Fig. 5A–C).
Fluorescently labelled slik mutant MARCM clones were generated by hs-Flp; FRT42D tub-Gal80; tub-Gal4 UAS-GFP strain and clones were created by heat shock at 37 °C for 30 min, late third-instar larvae were dissected after recovering for 4 days (Additional file 1: Fig. S5). Twinspot clones in larval imaginal discs using yw hs-Flp; FRT42D ubi-GFP were induced by heat shock at 37 °C for 30 min, recovering for 3 days (Additional file 1: Fig. S3).
RT-qPCR
For RNAi-knockdown efficiency experiments, hs-Gal4 driver was used. Animals were raised at 25 °C, heat-shocked at 37 °C for 30 min, and recovered at 29 °C for 2 h before dissection.
Eastep Super (Shanghai Promega) was used to isolate total RNA from third instar larvae of indicated genotypes, and RT-qPCR was performed using SYBR Green PCR Premix Kit (TaKaRa). Primers used were as follows:
rp49 FP: CCACCAGTCGGATCGATATGC.
rp49 RP: CTCTTGAGAACGCAGGCGACC.
slik FP: TAGGACAGCAGCAATGAGCTGG.
slik RP: TTCACGTAGCTCCTGCTTACGG.
STK10 FP: ATCCTGCGCCTGTCTACCTT.
STK10 RP: GCCTTGTAAACCTTGCCGAA.
Cell culture and transfection
Human cells were cultured at 37 °C in DMEM (Gibco) containing 10% FBS, 100 U/mL penicillin, and 100 µg/mL streptomycin in a humidified incubator with 5% CO2. Lipofectamine RNAiMAX (Invitrogen, 13,778,150) was used for small-interfering RNA (siRNA) transfection as instructed. For STK10 knockdown, two different siRNAs were used. The sequences are as follows:
siRNA-STK10-1: 5′-AGGAGGAGCUGGAGGACUATT-3′ (sense);
siRNA-STK10-1: 5′-UAGUCCUCCAGCUCCUCCUTT-3′ (antisense);
siRNA-STK10-2: 5′-CGACAGCAGCGGAAGGAAATT-3′ (sense);
siRNA-STK10-2: 5′-UUUCCUUCCGCUGCUGUCGTT-3′ (antisense);
siRNA-Control: 5′-UUCUCCGAACGUGUCACGUTT-3′ (sense);
siRNA-Control: 5′-ACGUGACACGUUCGGAGAATT-3′ (antisense).
Immunoblotting
Cells were harvested and washed in ice-cold PBS, then lysed with RIPA lysis buffer (WELLBIO, WB0101, China) supplemented with protease inhibitor cocktails (Yeasen, 20124ES03, China) on ice for 30 min. Cell lysates were then centrifuged at 15,000 rpm for 10 min at 4 °C. Proteins were separated by SDS-PAGE following standard procedures. The primary antibodies were used as follows: Rabbit anti-β actin (CST, 8457, 1:2000), Rabbit anti-SAPK/JNK (CST, 9252, 1:2000) and Rabbit anti-phospho SAPK/JNK (CST, 9251, 1:2000).
Image of fly wings and eyes
Three-day-old flies were collected and frozen at − 80 °C. When taking pictures, flies were unfrozen at room temperature and placed on 1% agarose plate. Light images of eyes were taken by OLYMPUS stereo microscope SZX16 (Olympus Corporation, Shinjuku, Tokyo, Japan). Light images of wings, dissected and placed on slide with alcohol/glycerol (1:1) medium, were taken by OLYMPUS BX51 microscope.
Statistical analysis
Statistical analysis was performed with GraphPad Prism 8.0.2 software. The data were analyzed by One-way ANOVA with Bonferroni’s multiple comparison test and two-tailed unpaired t-test to calculate statistical significance (*P < 0.05; **P < 0.01; ***P < 0.001, ****P < 0.0001; ns, no significant difference). Error bar indicates standard deviation. The experiments were repeated at least three times.
Availability of data and materials
The datasets generated during the current study are available from the corresponding author on reasonable request.
References
Zhou J, Edgar BA, Boutros M. ATF3 acts as a rheostat to control JNK signalling during intestinal regeneration. Nat Commun. 2017;8:14289.
Wu M, Pastor-Pareja JC, Xu T. Interaction between ras(V12) and scribbled clones induces tumour growth and invasion. Nature. 2010;463(7280):545–8.
Zhou J, Valentini E, Boutros M. Microenvironmental innate immune signaling and cell mechanical responses promote tumor growth. Dev Cell. 2021;56(13):1884–99. e5.
Eisenhoffer GT, et al. Crowding induces live cell extrusion to maintain homeostatic cell numbers in epithelia. Nature. 2012;484(7395):546–9.
Wu C, et al. Snail modulates JNK-mediated cell death in Drosophila. Cell Death Dis. 2019;10(12):893.
Mundorf J, et al. Ets21c governs tissue Renewal, stress tolerance, and aging in the Drosophila intestine. Cell Rep. 2019;27(10):3019–3033e5.
Demay Y, et al. The PERK pathway independently triggers apoptosis and a Rac1/Slpr/JNK/Dilp8 signaling favoring tissue homeostasis in a chronic ER stress Drosophila model. Cell Death Dis. 2014;5:e1452.
Takino K, Ohsawa S, Igaki T. Loss of Rab5 drives non-autonomous cell proliferation through TNF and ras signaling in Drosophila. Dev Biol. 2014;395(1):19–28.
Li Z, et al. Toll signaling promotes JNK-dependent apoptosis in Drosophila. Cell Div. 2020;15:7.
Igaki T, Miura M. The Drosophila TNF ortholog Eiger: emerging physiological roles and evolution of the TNF system. Semin Immunol. 2014;26(3):267–74.
Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol. 2007;19(2):142–9.
Igaki T, et al. Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J. 2002;21(12):3009–18.
Kanda H, et al. Wengen, a member of the Drosophila tumor necrosis factor receptor superfamily, is required for Eiger signaling. J Biol Chem. 2002;277(32):28372–5.
Xue L, et al. Tumor suppressor CYLD regulates JNK-Induced cell death in Drosophila. Dev Cell. 2007;13(3):446–54.
Takatsu Y, et al. TAK1 participates in c-Jun N-terminal kinase signaling during Drosophila development. Mol Cell Biol. 2000;20(9):3015–26.
Glise B, Bourbon H, Noselli S. Hemipterous encodes a Novel Drosophila map kinase kinase, required for epithelial-cell sheet Movement. Cell. 1995;83(3):451–61.
Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103(2):239–52.
McEwen DG, Peifer M. Puckered, a Drosophila MAPK phosphatase, ensures cell viability by antagonizing JNK-induced apoptosis. Development. 2005;132(17):3935–46.
Martin-Blanco E, et al. Puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Develop. 1998;12(4):557–70.
Avruch J, et al. Protein kinases of the Hippo pathway: regulation and substrates. Semin Cell Dev Biol. 2012;23(7):770–84.
Dan I, Watanabe NM, Kusumi A. The Ste20 group kinases as regulators of MAP kinase cascades. Trends Cell Biol. 2001;11(5):220–30.
Rane CK, Minden A. P21 activated kinase signaling in cancer. Sem Cancer Biol. 2019;54:40–9.
Gagnon KB, Delpire E. Molecular physiology of Spak and Osr1: two Ste20-Related protein kinases regulating Ion Transport. Physiol Rev. 2012;92(4):1577–617.
Delpire E. The mammalian family of sterile 20p-like protein kinases. Pflugers Archiv Eur J Physiol. 2009;458(5):953–67.
Panneton V, et al. Regulation of catalytic and non-catalytic functions of the Drosophila Ste20 kinase Slik by activation segment phosphorylation. J Biol Chem. 2015;290(34):20960–71.
Viswanatha R, et al. Local phosphocycling mediated by LOK/SLK restricts ezrin function to the apical aspect of epithelial cells. J Cell Biol. 2012;199(6):969–84.
Hipfner DR, Keller N, Cohen SM. Slik Sterile-20 kinase regulates moesin activity to promote epithelial integrity during tissue growth. Genes Develop. 2004;18(18):2243–8.
Katzemich A, et al. Slik phosphorylation of Talin T152 is crucial for proper Talin recruitment and maintenance of muscle attachment in Drosophila. Development. 2019;146(20):dev176339.
Carreno S, et al. Moesin and its activating kinase Slik are required for cortical stability and microtubule organization in mitotic cells. J Cell Biol. 2008;180(4):739–46.
De Jamblinne CV, et al. STRIPAK regulates Slik localization to control mitotic morphogenesis and epithelial integrity. J Cell Biol. 2020;219(11):e201911035.
Hughes SC, Fehon RG. Phosphorylation and activity of the tumor suppressor Merlin and the ERM protein moesin are coordinately regulated by the Slik kinase. J Cell Biol. 2006;175(2):305–13.
Yang Y, et al. The PP1 phosphatase flapwing regulates the activity of Merlin and Moesin in Drosophila. Dev Biol. 2012;361(2):412–26.
Ma X, et al. Bendless modulates JNK-mediated cell death and migration in Drosophila. Cell Death Differ. 2014;21(3):407–15.
Ma X, et al. Rho1-Wnd signaling regulates loss-of-cell polarity-induced cell invasion in Drosophila. Oncogene. 2016;35(7):846–55.
Ma XJ, et al. NOPO modulates egr-induced JNK-independent cell death in Drosophila. Cell Res. 2012;22(2):425–31.
Abrams JM, et al. Programmed cell-death during Drosophila Embryogenesis. Development. 1993;117(1):29–43.
Phillips RG, et al. The Drosophila segment polarity gene patched is involved in a position-signaling mechanism in Imaginal Disks. Development. 1990;110(1):105–.
Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281(5381):1312–6.
Steller H. Regulation of apoptosis in Drosophila. Cell Death Differ. 2008;15(7):1132–8.
Sarkissian T, et al. Detecting apoptosis in Drosophila tissues and cells. Methods. 2014;68(1):89–96.
Ohtsubo T, Kamada S, Tsujimoto Y. Inhibition of apoptosis by a baculovirus p35 gene]. Nihon Rinsho. 1996;54(7):1907–11.
Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene. 2008;27(48):6245–51.
Wang XC, Liu ZG, Jin LH. Drosophila jumu modulates apoptosis via a JNK-dependent pathway and is required for other processes in wing development. Apoptosis. 2019;24(5–6):465–77.
Pushpavalli SNCVL, et al. Drosophila MOF regulates DIAP1 and induces apoptosis in a JNK dependent pathway. Apoptosis. 2016;21(3):269–82.
Lam D, et al. Drosophila happyhour modulates JNK-dependent apoptosis. Cell Death Dis. 2010;1:e66.
Chatterjee N, Bohmann D. A versatile ΦC31 based reporter system for measuring AP-1 and Nrf2 signaling in Drosophila and in tissue culture. PLoS ONE. 2012;7(4):e34063.
Wu C, et al. CtBP modulates snail-mediated tumor invasion in Drosophila. Cell Death Discov. 2021;7(1):202.
Ma X, et al. Hippo signaling promotes JNK-dependent cell migration. Proc Natl Acad Sci U S A. 2017;114(8):1934–9.
Tare M, et al. Cullin-4 regulates Wingless and JNK signaling-mediated cell death in the Drosophila eye. Cell Death Dis. 2016;7(12):e2566.
Theodosiou NA, Xu T. Use of FLP/FRT system to study Drosophila development. Methods a Companion Methods Enzymol. 1998;14(4):355–65.
Moreno E, Yan MH, Basler K. Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr Biol. 2002;12(14):1263–8.
Zhang SP, et al. Wingless modulates activator protein-1-mediated tumor invasion. Oncogene. 2019;38(20):3871–85.
Sun Y, et al. Lic regulates JNK-mediated cell death in Drosophila. Cell Prolif. 2019;52(3):e12593.
Dong JX, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130(6):1120–33.
Zhang YF, Cui C, Lai ZC. The defender against apoptotic cell death 1 gene is required for tissue growth and efficient N-glycosylation in Drosophila melanogaster. Dev Biol. 2016;420(1):186–95.
Baehrecke EH. How death shapes life during development. Nat Rev Mol Cell Biol. 2002;3(10):779–87.
Conradt B. Genetic control of programmed cell death during Animal Development. Annu Rev Genet. 2009;43:493–523.
Cosolo A, et al. JNK-dependent cell cycle stalling in G2 promotes survival and senescence-like phenotypes in tissue stress. Elife. 2019;8:e41036.
Corrales TD, Losada-Perez M, Casas-Tinto S. JNK Pathway in CNS Pathologies. Int J Mol Sci. 2021;22(8):3883.
Brown JB, et al. Diversity and dynamics of the Drosophila transcriptome. Nature. 2014;512(7515):393–9.
Song IS, et al. Inhibition of MKK7-JNK by the TOR signaling pathway regulator-like protein contributes to resistance of HCC cells to TRAIL-induced apoptosis. Gastroenterology. 2012;143(5):1341–51.
La Marca JE, et al. Strip and Cka negatively regulate JNK signalling during Drosophila spermatogenesis. Development. 2019;146(13):dev174292.
Camps M, et al. Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science. 1998;280(5367):1262–5.
Ahn JH, et al. Protein kinase A activates protein phosphatase 2A by phosphorylation of the B56delta subunit. Proc Natl Acad Sci U S A. 2007;104(8):2979–84.
Benavides-Serrato A, et al. mTORC2 modulates feedback regulation of p38 MAPK activity via DUSP10/MKP5 to confer differential responses to PP242 in glioblastoma. Genes Cancer. 2014;5(11–12):393–406.
Ukken FP, et al. Slik and the receptor tyrosine kinase breathless mediate localized activation of Moesin in terminal tracheal cells. PLoS ONE. 2014;9(7):e103323.
Ma XJ, et al. POSH regulates Hippo signaling through ubiquitin-mediated expanded degradation. Proc Natl Acad Sci USA. 2018;115(9):2150–5.
Xu M, et al. dGLYAT modulates Gadd45-mediated JNK activation and cell invasion. Cell Div. 2022;17(1):4.
Wang XJ, et al. Pontin/Tip49 negatively regulates JNK-mediated cell death in Drosophila. Cell Death Discovery. 2018;4:8.
Zhang S, et al. The canonical wg signaling modulates Bsk-mediated cell death in Drosophila. Cell Death Dis. 2015;6:e1713.
Igaki T, Pagliarini RA, Xu T. Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr Biol. 2006;16(11):1139–46.
Acknowledgements
We thank Bloomington Drosophila center, VDRC Drosophila center, NIG-FLY center, Kyoto Stock Center and Tsing Hua Fly Center for fly stocks, and members of Xue Lab for discussion and critical comments.
Funding
This work is supported by the National Natural Science Foundation of China (31970536) and Shanghai Committee of Science and Technology (09DZ2260100).
Author information
Authors and Affiliations
Contributions
CL, QZ and LX conceived and designed the experiments. CL, XZ, YS and WL conducted the experiments. CL, XZ, XG, PC, YS, QZ and LX analyzed the data. CL and LX wrote the manuscript with input from YS and QZ. All authors approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors have read and agreed to the final version of the manuscript.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
A genetic screen for modifiers of GMR>Egr-induced eye-ablation phenotype. A A schematic depiction of the three deficiencies Df(2R)BSC603, Df(2R)ED4065, and Df(2R)ED4071. B–G Light micrographs of adult eyes. GMR>Egr-induced small eye phenotype (B) was obviously enhanced by deficiency Df(2R)BSC603 (C), Df(2R)ED4065 (D) or Df(2R)ED4071 (E), or by slikKG04837(F) or slik RNAi (G). Scale bar: 100 µm. Figure S2. Depletion of slik causes apoptotic cell death. Fluorescence micrographs of third instar larval wing discs are shown (A–D). Compared with the control (A), knockdown of slik driven by ptc-Gal4 led to massive cell death along the A/P compartment boundary (B), which was completely impeded by expression of P35 (D), but not that of lacZ-IR (C). Statistical analysis of AO positive cell number in wing discs (E, n=10 for each genotype) is shown. One-way ANOVA with Bonferroni multiple comparison test was used to compute P-values, ****P < 0.0001; ns, no significant difference. Scale bar: 40 µm. Figure S3. Slik is required for maintaining tissue homeostasis in development. A Statistical analysis of clone numbers in Fig. 5A-C are shown (n=10 for each group). B–C Fluorescence micrographs of third instar larval wing discs with twin clones (marked with 2ÍGFP or the absence of GFP) are shown. Compared with the control (B–B’), slik mutant clone (black) was obviously smaller than its wild-type twin clone (2ÍGFP) (C–C’). Statistical analysis of GFP-area/2ÍGFP area ratio (D, n=10 for each genotype) is shown. Unpaired two tailed t-test was used to compute P-values,****p < 0.0001. Scale bar: 25 µm. Figure S4. The efficacies of Slik and STK10 overexpression. mRNA level of slik (A) or STK10 (B) was measured by quantitative RT-PCR (n=2). Unpaired two tailed t-test was used to compute P-values, ****p < 0.0001, **p < 0.01. Figure S5. STK10 suppresses JNK-mediated apoptosis in slik mutant clones. A–E Fluorescence micrographs of third instar larval wing discs with MARCM clones (marked with GFP) are shown. Compared with the control (A-A’’), apoptosis was triggered in slik mutant clone (B-B’’), which was suppressed by the expression of Slik (C-C’’), STK10 (D-D’’) or BskDN (E-E’’). Scale bar: 25 µm. Figure S6. Human STK10 suppresses Egr-triggered cell death in eye development. Light micrographs of adult eyes (A–D) and fluorescence micrographs of third instar larval eye discs (F–I) are shown. Compared with the controls (A, F), GMR>Egr-induced small eye phenotype (B) and massive cell death in eye discs (G) were obviously suppressed by expressing STK10 (C, H), or hep-IR (D, I) serving as a positive control. Statistical analysis of adult eye size (E, n=10 for each genotype) and AO positive cell numbers in eye discs (J, n=10 for each genotype) are shown. One-way ANOVA with Bonferroni multiple-comparison test was used to compute P-values, ****p < 0.0001, ***p< 0.001, **p < 0.01. Scale bar: 100 μm. Figure S7. scrib depletion-induced cell death depends on JNK pathway. Light micrographs of adult wings (A–C) and fluorescence micrographs of third instar larval wing discs (E–G) are shown. ptc>scrib-IR-triggered loss-of-ACV phenotype in adult wings (A) and cell death in larval wing discs (E) were suppressed by expression of BskDN (B, F) or Puc (C, G). Statistical analysis of the presence of ACV in adult wings (D, n=15 for each genotype) and cell death number in wing discs (H, n≥10 for each genotype) are shown. One-way ANOVA with Bonferroni multiple-comparison test was used to compute P-values, ****p < 0.0001, **p < 0.01. Scale bar: 100 µm in A–C (upper panels), 50 µm in A–C (lower panels) and E–G.Figure S8. The knock-down efficacies of STK10 RNAi. Validation of two independent siRNA-STK10 used in current study. SW480 cells treated with siRNA for 72 hours were subjected to RT-qPCR (n=3). One-way ANOVA with Bonferroni multiple-comparison test was used to compute P-values, ****p < 0.0001. Figure S9. Kinase activity is necessary for Slik/STK10 to regulate JNK pathway. Immunoblot analysis of p-JNK and total JNK protein level in SW480 cells. Compared with the control (lane 1), STK10 knockdown increased p-JNK level (lane 2), which was effectively suppressed by expressing Slik (lane 3), but not a truncated Slik with kinase domain deletion (lane 4). One-way ANOVA with Bonferroni multiple-comparison test was used to compute P-values, ****p< 0.0001, ***p < 0.001. Figure S10. Schematic summary of Slik in regulating JNK pathway. Slik inhibits JNK-mediated cell death. Genetic epistasis analysis suggests that Slik acts upstream of or in parallel to Hep to impede JNK pathway.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, C., Zhu, X., Sun, X. et al. Slik maintains tissue homeostasis by preventing JNK-mediated apoptosis. Cell Div 18, 16 (2023). https://doi.org/10.1186/s13008-023-00097-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13008-023-00097-4