
Abstract
Background
Apoptosis plays pivotal roles in organ development and tissue homeostasis, with its major function to remove unhealthy cells that may compromise the fitness of the organism. Toll signaling, with the ancient evolutionary origin, regulates embryonic dorsal–ventral patterning, axon targeting and degeneration, and innate immunity. Using Drosophila as a genetic model, we characterized the role of Toll signaling in apoptotic cell death.
Results
We found that gain of Toll signaling is able to trigger caspase-dependent cell death in development. In addition, JNK activity is required for Toll-induced cell death. Furthermore, ectopic Toll expression induces the activation of JNK pathway. Moreover, physiological activation of Toll signaling is sufficient to produce JNK-dependent cell death. Finally, Toll signaling activates JNK-mediated cell death through promoting ROS production.
Conclusions
As Toll pathway has been evolutionarily conserved from Drosophila to human, this study may shed light on the mechanism of mammalian Toll-like receptors (TLRs) signaling in apoptotic cell death.
Similar content being viewed by others
Background
The type I trans-membrane receptor Toll was first identified in Drosophila for its role in establishing the dorsal–ventral axis at the early embryonic stage [1], and was subsequently determined as a key component of the innate immune response [2]. To date, nine Toll family members have been identified in fly and thirteen Toll-like receptors (TLRs) in mammals [3,4,5,6]. In Drosophila, Toll is activated by the cleaved cytokine Spätzle, and proceeds to the phosphorylation and degradation of the IκB factor Cactus through the MyD88-Tube-Pelle complex, eventually results in the release and translocation of two NF-kB factors Dorsal and Dorsal-related immunity factor (Dif) from cytoplasm to nucleus to activate the transcription of target genes [7]. Dorsal is required for the dorsal–ventral patterning during embryonic development, Dif is essential for the innate immunity in the adulthood, whereas both Dorsal and Dif are involved in the larval immune response [8,9,10].
The c-Jun N-terminal kinase (JNK) is a member of the highly conserved MAPKs family that plays pivotal roles in various cellular processes including apoptosis [11,12,13,14,15]. basket (bsk) encodes the sole Drosophila JNK that is phosphorylated and activated by the conserved upstream MAPK cascade, including the JNKK kinase dTAK1 and the JNK kinase Hemipterous (Hep) [16, 17]. puckered (puc), a target gene of JNK signaling, encodes a phosphatase that dephosphorylates and negatively regulates JNK activity [18]. In fly, JNK signaling plays an important role in programmed cell death [19], which is initiated by one or more of the pro-apoptotic genes reaper (rpr), head involution defective (hid) and grim [20], whose protein products bind to dIAP1 (Drosophila IAP-1) to release the initiator caspase Dronc (Drosophila NEDD2-like caspase) [21], which in turn activates the effector caspases Dcp-1 (Decapping protein 1) and Drice (Death related ICE-like caspase) [20]. JNK signaling can be activated by various extrinsic and intrinsic stress stimuli including oxidative stress generated by reactive oxygen species (ROS) [22,23,24], which is generated from partial reduction of oxygen, including hydroxyl radical, superoxide and hydrogen peroxide [25].
Besides the well-documented functions of Toll/NF-kB signaling in development and immunity, several reports suggest that Toll pathway is also required for cell death triggered by tumor necrosis factor (TNF) [26] or chromosomal instability (CIN) [27], yet the mechanism underlies Toll-induced cell death remain elusive. In this work, we employed Drosophila as an in vivo system and characterized that Toll signaling induces JNK-dependent apoptotic cell death via ROS production. Firstly, activation of Toll signaling induces apoptotic cell death in the developing wings and eyes. Secondly, depletion of JNK signaling suppresses Toll-induced apoptosis. Moreover, Toll signaling is able to trigger JNK pathway activation. Finally, Toll elicits JNK-dependent apoptosis via promoting ROS production.
Results
Toll signaling triggers cell death in Drosophila wing development
Ectopic expression of Toll10B, an activated form of Toll, driven by patched (ptc)-Gal4 along the A/P compartment border (Additional file 1: Figure S1a) [28] (ptc>Toll10B) produces a loss of anterior cross vein (ACV) phenotype in the adult wings (Fig. 1a, b and quantified in Fig. 1i), which resembles the phenotype generated by expressing the cell death gene grim [26], implying a potential role of Toll signaling in promoting cell death in development. To validate this assumption, we performed Acridine Orange (AO) staining assay that detects dying cells [29], and observed massive cell death along the anterior/posterior (A/P) compartment boundary in 3rd instar larval wing discs (Fig. 1a′, b′ and quantified in Fig. 1j). Toll10B-induced loss-of-ACV phenotype and cell death were notably inhibited by expressing two independent RNAi lines of dorsal (Fig. 1d, e, d′, e′), which encodes the Drosophila NF-kB factor operating in the Toll pathway [30], but not GFP (Fig. 1c, c′). Furthermore, expression of Toll10B in the wing pouch driven by Scalloped (Sd)-Gal4 (Additional file 1: Figure S1b) [31] results in enhanced cell death (Fig. 2a, b, p), which was suppressed by RNAi-mediated knockdown of dorsal (Fig. 2d, e), while GFP RNAi served as a negative control (Fig. 2c). A quantitative reverse transcription polymerase chain reaction (qRT-PCR) assay was performed to verify the knockdown efficiencies of the two dorsal RNAi lines (Additional file 1: Figure S2a). Consistently, over-expression of Dorsal produces a similar loss-of-ACV phenotype in the adult wing and cell death in the wing disc (Fig. 1g, g′), indicating that ectopic Toll-induced cell death depends on the canonical NF-kB pathway. Importantly, depletion of the IκB gene cactus also results in the loss of ACV and cell death (Fig. 1h, h′), suggesting a physiological function of the Toll/NF-kB pathway in developmental cell death.

Activated Toll signaling triggers cell death in wing development. Light micrographs of Drosophila adult wings (a–h) and fluorescence micrographs of third instar larval wing discs (a′–h′, a′′–h′′) are shown. Compared with the ptc-Gal4 controls (a–a′′), ptc > Toll10B induces a loss-of-ACV phenotype in adult wings (b), massive cell death (b′) and apoptosis (b′′) in third instar wing discs, all of which are suppressed by expressing two independent dorsal RNAi (d–d′′, e–e′′) or DroncDN (f–f′′), but not GFP RNAi (c–c′′). Expression of Dorsal or depletion of cactus also results in the loss-of-ACV phenotype in adult wings (g, h), and increased apoptotic cell death in third instar wing discs (g′, g′′, h′, h′′). The lower panels show high magnification view of the boxed areas in upper panels (a–h). For all wings, anterior is to the left and distal up. i–k Statistical analysis of the ACV phenotype in adult wings (i, n = 50 for each genotype), AO positive cell number (j, n = 9) and cleaved caspase-3 (CC-3) activity (k, n = 10) in wing discs are shown. Error bar indicates standard deviation. One-way ANOVA test was used to compute P-values, ****P < 0.0001, ***P < 0.001, **P < 0.01. ptc>Dorsal flies were reared at 20 °C to avoid lethality at 25 °C, while ptc>cactus-IR were reared at 29 °C to enhance the expression of RNAi. See Additional file 1 for detailed genotypes. Scale bar: 100 μm

Toll signaling promotes apoptotic cell death. Fluorescence micrographs (a–e, k–o) and light micrographs (f–j) of third instar larval wing discs are shown. Compared with the controls (a, f, k), ectopic expression of Toll10B in the wing pouch induces massive cell death (b), activates rpr transcription detected by X-Gal staining of a rpr-LacZ reporter (g), and promotes caspase activation indicated by cleaved caspase-3 (CC-3) antibody staining (l). These phenotypes are suppressed by depletion of dorsal (d, e, i, j, n, o), but not GFP (c, h, m). p–r Statistical analysis of cell death number (p, n = 8), X-Gal staining (q, n = 8) and CC-3 activity in wing discs (r, n = 9) are shown. Error bar indicates standard deviation. One-way ANOVA test was used to compute P-values, ****P < 0.0001, ***P < 0.001, **P < 0.01, ns is no significant difference. See Additional file 1 for detailed genotypes. Scale bar: 100 μm
Toll signaling promotes caspase-mediated apoptotic cell death
Apoptosis in Drosophila is triggered by transcriptional up-regulation of one or more of three pro-apoptotic genes (hid, rpr and grim), and is mediated by the cleavage and activation of a group of cysteine proteases, termed caspases [32, 33]. ptc>Toll10B triggers apoptosis visualized by anti-cleaved caspase-3 (CC-3) antibody staining (Fig. 1a′′, b′′ and quantified in Fig. 1k), which was suppressed by the expression of two independent dorsal RNAi (Fig. 1d′′, e′′), but remained unaffected by that of GFP RNAi (Fig. 1c′′). Moreover, expression of Dorsal or depletion of cactus also promotes apoptotic cell death (Fig. 1g′′, h′′). Intriguingly, Toll10B-induced apoptotic cell death and loss-of-ACV phenotype were efficiently blocked by expressing a dominant-negative form of the initiator caspase Dronc (DroncDN) (Fig. 1f–f′′), suggesting Toll signaling induces caspase-dependent cell death. Consistently, expression of Toll10B by Scalloped (Sd)-Gal4 up-regulates the transcription of the pro-apoptotic gene rpr, revealed by X-gal staining of a rpr-LacZ reporter, accompanied by enhances caspase activity (Fig. 2f, g, k, l, quantified in Fig. 2q, r) [34]. Both phenotypes were significantly impeded by the expression of dorsal RNAi (Fig. 2i, j, n, o), but not that of GFP (Fig. 2h, m). Thus, we conclude that Toll triggers NF-kB-mediated apoptotic cell death in Drosophila.
Toll-induced cell death depends on JNK activity
Previous studies have suggested that the JNK signaling plays a critical role in the caspase-dependent cell death [19, 35,36,37,38]. To investigate whether JNK is required for Toll-induced cell death, we blocked JNK activity by expressing a dominant negative form of Drosophila JNK Bsk (BskDN) or Puc, an inhibitor of JNK kinase activity [18]. We found that Sd>Toll10B-induced adult wing blade reduction and apoptotic cell death in 3rd instar larval wing discs were soundly suppressed by expressing BskDN or Puc (Fig. 3a–d, a′–d′, a′′–d′′ and quantified in Fig. 3i–k). Notably, depletion of cactus by two independent RNAi (Additional file 1: Figure S2b) was also sufficient to generate Bsk-dependent small wing phenotype and apoptotic cell death (Fig. 3e–h, e′–h′, e′′–h′′). Furthermore, ptc>Toll10B- or ptc>cactus-IR-triggered loss-of-ACV phenotype was fully suppressed by blocking JNK activity (Additional file 1: Figure S3). Taken together, these data proved that JNK activity is indispensable for ectopic or physiological Toll/NF-kB pathway-induced cell death.

JNK signaling is required for Toll pathway-triggered cell death. Micrographs showing Drosophila adult wings (a–h) and third instar larval wing discs (a′–h′, a′′–h′′). Compared with the controls (a–a′′), ectopic expression of Toll10B driven by Sd-Gal4 results in a small wing phenotype in adults (b), extensive cell death (b′) and apoptosis (b′′) in larval wing discs, which are suppressed by expressing a dominant-negative form of Bsk (c–c′′) or Puc (d–d′′). Depletion of cactus also produces a small wing phenotype in adults (e, g), massive cell death (e′, g′) and apoptosis (e′′, g′′) in larval wing discs, which are suppressed by expressing BskDN (f–f′′, h–h′′). i–k Statistical analysis of the adult wing size/wild type (WT) (i, n = 8), cell death number (j, n = 10) and apoptosis (k, n = 10) in wing discs are shown. One-way ANOVA was used to compute P-values, ****P < 0.0001, ***P < 0.001. See Additional file 1 for detailed genotypes. Scale bar: 100 μm
Elevated Toll signaling activates JNK pathway
Given that JNK activity is required for Toll-induced cell death, we hypothesized that Toll may induce the activation of JNK pathway. To monitor JNK pathway activation, we examined the expression of puc-LacZ, a widely accepted reporter for JNK signaling [18]. In support of the assumption, we found that puc-LacZ expression was significantly activated by ectopic Toll10B expression driven by ptc-Gal4 or Sd-Gal4 in a Bsk-dependent manner (Fig. 4a–f and quantified in Fig. 4g). Moreover, depletion of cactus also up-regulated puc-LacZ expression (Additional file 1: Figure S4), suggesting both ectopically and physiologically elevated Toll signaling are sufficient to trigger JNK pathway activation, which in turn activates apoptotic cell death.

Toll activates JNK signaling. Light micrographs of third instar larval wing discs with X-Gal staining (a–f) are shown. Compared with the ptc-Gal4 and Sd-Gal4 controls (a, d), ectopic expression of Toll10B up-regulates puc-LacZ expression (b, e), which is blocked by expressing BskDN (c, f). g Statistical analysis of X-Gal staining (n = 8) is shown. One-way ANOVA was used to compute P-values, ****P < 0.0001. See Additional file 1 for detailed genotypes. Scale bar: 100 μm
JNK is required for Toll-induced cell death in Drosophila eye development
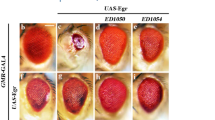
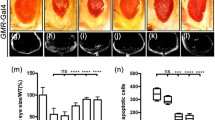
To investigate whether Toll signaling triggers JNK-dependent cell death in other cellular contexts, we expressed Toll10B by Glass Multimer Reporter (GMR)-Gal4, which expresses in all cell types posterior to the morphogenetic furrow (MF) in the developing eye (Additional file 1: Figure S1c) [39]. We observed extensive cell death posterior to the MF in third instar larval eye discs (Fig. 5e, f and quantified in Fig. 5h), and size reduction of adult eyes (Fig. 5a, b and quantified in Fig. 5d). Both phenotypes were suppressed by expressing BskDN (Fig. 5c, g), suggesting that Toll induces JNK-mediated cell death in a non-tissue-specific manner.

Toll triggers JNK-dependent cell death in eye development. Light micrographs of Drosophila adult eyes (a–c) and fluorescent micrographs of third instar larval eye discs (e–g) are shown. Compared with the GMR-Gal4 controls (a, e), Toll10B-induced small eye phenotype (b) and cell death (f) are suppressed by expressing BskDN (c, g). d, h Statistical analysis of the adult eye size/wild type (WT) (n = 10) and cell death number in eye discs (e, n = 10; f, n = 8; g, n = 10) are shown. One-way ANOVA was used to compute P-values, ****P < 0.0001. See Additional file 1 for detailed genotypes. Scale bar: 100 μm
Toll activates JNK-mediated cell death through ROS
JNK signaling could be activated by reactive oxygen species (ROS)-mediated oxidative stress in Drosophila [23]. To address whether ROS is involved in Toll-induced JNK-dependent cell death, we examined ROS level in 3rd instar wing discs. Compared with the Sd-Gal4 control (Fig. 6a), Toll10B expression strongly raised ROS production (Fig. 6b and quantified in Fig. 6i), which was suppressed by expression of Sod (Fig. 6c), a superoxide dismutase enzymes that eliminates oxygen radicals [23], but not by BskDN (Fig. 6d), suggesting Toll-induced ROS production is independent of JNK. To verify the results, we assessed the expression of gstD1 (Glutathione S transferase D1), which encodes a detoxification enzyme that responds to oxidative stress [40, 41]. We found that ectopic Toll10B was sufficient to increase the gstD1 mRNA level as measured by qRT-PCR, which was suppressed by expressing Sod, but not BskDN (Additional file 1: Figure S5a). Intriguingly, Toll10B-induced puc-LacZ expression were significantly impeded by expressing Sod (Fig. 6e–g and quantified in Fig. 6j), while BskDN served as a positive control (Fig. 6h). Collectively, our data suggest that Toll signaling promotes ROS production, which activates JNK-mediated apoptotic cell death.

Toll induces ROS-dependent JNK activation and apoptosis. Third instar larval wing discs showing ROS level (a–d) and puc-LacZ expression (e–h). Compared with the control (a), ectopic expression of Toll10B promotes ROS production (b), which is suppressed by expressing Sod (c), but not BskDN (d). Compared with the control (e), Toll10B-induced JNK pathway activation (f) is suppressed by expressing Sod (g) or BskDN (h). i, j Statistical analysis of the ROS spots (a, n = 10; b, n = 7; c, n = 10; d, n = 9) and X-Gal expression (n = 8) are shown. Kruskal–Wallis test and one-way ANOVA were used to compute P-values, ****P < 0.0001, *P < 0.05 and ns indicates not significant. See Additional file 1 for detailed genotypes. Scale bar: 100 μm
Discussion
Drosophila has been widely accepted as an excellent model organism to dissect the roles of various signaling pathways in regulating the apoptotic program for the last two decades [42, 43]. While the functions of Toll pathway in embryonic dorsal–ventral patterning and innate immunity have been extensively studied, much less is known about its cell death functions in development, and the underlying mechanisms remain largely elusive. Necroptosis is a type of programmed cell death, characterized by membrane swell and rupture, which is mediated by receptor-interacting protein kinase 1 (RIPK1) and RIPK3, but independent of caspases activity [44]. In vitro studies show activation of mammalian Toll-like receptors (TLRs) can trigger necroptotic cell death through RIPK1, RIPK3, and pseudokinase mixed lineage kinase-domain-like (MLKL) complex [45, 46]. Intriguingly, inhibition of JNK with SP600125 restricts TLRs-induced necroptosis in macrophages, whereas loss of JNK by short-interfering RNA (siRNA) augments TLRs-induced necroptotic cell death, suggesting a dual role for JNK in regulating necroptosis [47]. Recent studies in Drosophila suggest that Toll signaling, consisting of the Toll ligand Spätzle, several Toll-related receptors and NF-kB factors, is required for apoptotic cell death of loser cell elimination during epithelial cell competition, which up-regulates the expression of pro-apoptotic genes, yet this function of Toll signaling is independent of the JNK activity [48,49,50,51]. In addition, Toll signaling also plays pivotal roles in cell number plasticity during the nervous system development [52]. Drosophila neurotrophin (NT) ligands (including Spätzle, Spätzle2 and Spätzle5), combined with distinct Toll receptors, can switch from promoting cell survival to death in the central nervous system (CNS) via a three-tier mechanism [53]. For example, Toll-6 promotes neuronal survival via MyD88/NF-κB in the embryonic CNS but neuronal death via Wek/Sarm/JNK in the pupal CNS. However, whether NTs function in cooperation with TLRs to mediate neuronal survival/death in mammals is still unclear. Previous study also suggested that Toll-JNK is required for chromosomal instability (CIN)-triggered cell death in proliferating Drosophila larval tissue [27], yet whether activated Toll signaling is sufficient to promote JNK-mediated cell death, and the underlying mechanism from Toll to JNK remains unknown.
Toll signaling can be activated in response to Gram positive bacteria, fungi and viruses in both Drosophila and mammals [2, 54,55,56,57,58]. In the present study, we first found that ectopic expression of Toll triggers JNK-mediated apoptotic cell death, yet in this scenario, Toll is expressed at a much higher level than that induced by injection of Staphylococcus aureus (S. aureus), a Gram-positive bacterium (Additional file 1: Figure S6a). To overcome this artificial activation which may not implicate a physiological relevance, we depleted cactus encoding the Drosophila IκB factor, and confirmed that physiological activation of Toll signaling is sufficient to induce JNK-dependent apoptosis in development. Furthermore, we show that Toll signaling triggers JNK activation via promoting the production of reactive oxygen species (ROS), yet the mechanism by which Toll signaling activates ROS production remains unclear, which merits further investigation. Consistent with our findings, mammalian TLRs was reported to induce the production of pro-inflammatory mediators upon pathogen invasion, which act as secondary messengers to regulate oxidative stress [59]. Up-regulated expression of inflammatory regulators, such as inducible nitric oxide synthase (iNOS), resulted in high levels of ROS [60]. Given that ROS production is closely associated with mitochondria dysfunction, a recent study found that TLRs recruited mitochondria to macrophage phagosomes and augmented ROS production [61].
Conclusions
In this study, we report that Toll signaling induces ROS-mediated JNK-dependent apoptotic cell death in vivo. First, we characterized a physiological function of the Toll/NF-kB signaling in developmental cell death. In addition, Toll-induced cell death depends on JNK activity. Furthermore, Toll signaling is sufficient to trigger JNK pathway activation. Finally, we provide evidence that Toll activates JNK-mediated apoptosis through ROS production. Thus, our study provided the first in vivo evidence that Toll signaling promotes JNK-dependent apoptosis via ROS production.
Materials and methods
Drosophila strains
Flies were raised on a standard cornmeal and ager medium at 25 °C unless otherwise indicated. Fly strains used in this work include: ptc-Gal4, Sd-Gal4, GMR-Gal4, UAS-GFP [62], act-Gal4, UAS-GFP-IR [63], UAS-Toll10B [26], UAS-DroncDN, UAS-BskDN, UAS-Puc, pucE69 and rpr-LacZ [64] were previously described. UAS-dorsal-IR (27650) and UAS-Sod1 (24750) were obtained from the Bloomington stock center, UAS-dorsal-IR (45998) was obtained from the VDRC center, UAS-cactus-IR-1 (5848R-3) and UAS-cactus-IR-2 (5848R-1) were obtained from the NIG-FLY center, and dld05894 was obtained from the Exelixis collection at Harvard.
Microbial injection
Staphylococcus aureus (S. aureus) strain was obtained from Prof. Wei Zuo at Tongji University. For experiment, the bacterial culture diluted with PBS to 1 × 1013 cells/ml. Third instar larvae were washed with PBS. l μl bacterial suspension of S. aureus was injected into the larva body with a sharp needle. Treated larvae were placed on the medium at 25 °C. mRNA level was monitored by qRT-PCR 4 h post infection.
qRT-PCR
Eastep Super (Shanghai Promega) was used to isolate total RNA from ten third instar larvae of indicated genotypes, and qRT-PCR was performed using SYBR Green PCR Premix Kit (TaKaRa). Primers used were as follows:
For rp49 FP: TCTCCTTGCGCTTCTTGGA
RP: TACAGGCCCAAGATCGTGAA
For dl FP: ATCCGTGTGGATCCGTTTAA
RP: AATCGCACCGAATTCAGATC
For cactus FP: CTCACTAGCCACTAGCGGTAA
RP: CCCGAATCACTGGTTTCGTTT
For Toll FP: AATCCCACGTTTAGGCTAACCA
RP: CCTCACCGATCCGCAACTT
For gstD1 FP: CGCGCCATCCAGGTGTATTT
RP: CTGGTACAGCGTTCCCATGT
AO staining
Eye and wing discs were dissected from third-instar larvae in 0.1% PBST (phosphate-buffered saline (PBS) + 0.1% Tween-20) and incubated in 1 × 10−5 M AO for 5 min at room temperature prior to imaging [65].
Immunostaining
Antibody staining was performed by standard procedures for imaginal discs [66]. Rabbit anti-Cleaved Caspase-3 (1:400, Cell Signaling Technology, CST, Cat # 9661, Danvers, MA, USA) was used as a primary antibody, and goat anti-rabbit CY3 (1:1000, Life technologies, Cat # A10520) was used as a secondary antibody.
X-gal staining
Wing discs were dissected from third instar larvae in PBST (PBS + 0.1% Tween-20) and stained for β-galactosidase as described [67].
ROS detection
The level of ROS was measured by CellROX (Life Technologies, C10443). Wing discs were dissected from third instar larvae, incubated in 5 μM CellROX for 30 min at 37 °C, rinsed in PBS, fixed in 3.7% formaldehyde for 5 min, and mounted in PBS for imaging [68].
Image and quantification of fly eyes and wings
Images of fly eyes and wings were prepared as described [69]. Briefly, 3-day-old flies were collected and frozen at − 80 °C for more than 12 h. When taking pictures, flies were unfrozen at room temperature and placed on 1% agarose plate. Light images of eye were taken by OLYMPUS stereo microscope SZX16 (Olympus Corporation, Shinjuku, Tokyo, Japan). Wings were dissected and placed on slide with alcohol/glycerol (1:1) medium. Light images of wing were taken by OLYMPUS BX51 microscope. Adobe Photoshop 2014 was used to measure the size of fly wings and eyes on the images.
Availability of data and materials
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
- JNK:
-
c-Jun N-terminal kinase
- MAPK:
-
Mitogen-activated protein kinase
- ROS:
-
Reactive oxygen species
- NF-kB:
-
Nuclear factor kB
- TLR:
-
Toll-like receptor
- ACV:
-
Anterior cross vein
- CIN:
-
Chromosomal instability
- qRT-PCR:
-
Quantitative reverse transcription polymerase chain reaction
References
Hashimoto C, Hudson KL, Anderson KV. The Toll gene of Drosophila, required for dorsal-ventral embryonic polarity, appears to encode a transmembrane protein. Cell. 1988;52(2):269–79.
Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86(6):973–83.
Rock FL, Hardiman G, Timans JC, Kastelein RA, Bazan JF. A family of human receptors structurally related to Drosophila Toll. Proc Natl Acad Sci USA. 1998;95(2):588–93.
Singh BP, Chauhan RS, Singhal LK. Toll-like receptors and their role in innate immunity. Curr Sci. 2003;85(8):1156–64.
Shi ZC, Cai ZY, Sanchez A, Zhang TT, Wen S, Wang J, et al. A novel Toll-like receptor that recognizes vesicular stomatitis virus. J Biol Chem. 2011;286(6):4517–24.
Valanne S, Wang JH, Ramet M. The Drosophila Toll signaling pathway. J Immunol. 2011;186(2):649–56.
Minakhina S, Steward R. Nuclear factor-kappa B pathways in Drosophila. Oncogene. 2006;25(51):6749–57.
Manfruelli P, Reichhart JM, Steward R, Hoffmann JA, Lemaitre B. A mosaic analysis in Drosophila fat body cells of the control of antimicrobial peptide genes by the Rel proteins Dorsal and DIF. EMBO J. 1999;18(12):3380–91.
Meng XJ, Khanuja BS, Ip YT. Toll receptor-mediated Drosophila immune response requires Dif, an NF-kappa B factor. Genes Dev. 1999;13(7):792–7.
Rutschmann S, Jung AC, Hetru C, Reichhart JM, Hoffmann JA, Ferrandon D. The Rel protein DIF mediates the antifungal but not the antibacterial host defense in Drosophila. Immunity. 2000;12(5):569–80.
Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103(2):239–52.
Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Genet Dev. 2002;12(1):14–21.
Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene. 2008;27(48):6245–51.
Li P, Huang P, Li X, Yin D, Ma Z, Wang H, et al. Tankyrase mediates K63-linked ubiquitination of JNK to confer stress tolerance and influence lifespan in Drosophila. Cell Rep. 2018;25(2):437–48.
Wu C, Li Z, Ding X, Guo X, Sun Y, Wang X, et al. Snail modulates JNK-mediated cell death in Drosophila. Cell Death Dis. 2019;10(12):893.
Glise B, Bourbon H, Noselli S. Hemipterous encodes a novel Drosophila map kinase kinase, required for epithelial-cell sheet movement. Cell. 1995;83(3):451–61.
Sluss HK, Han ZQ, Barrett T, Davis RJ, Ip YT. A JNK signal transduction pathway that mediates morphogenesis and an immune response in Drosophila. Genes Dev. 1996;10(21):2745–58.
Martin-Blanco E, Gampel A, Ring J, Virdee K, Kirov N, Tolkovsky AM, et al. puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev. 1998;12(4):557–70.
Moreno E, Yan MH, Basler K. Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr Biol. 2002;12(14):1263–8.
Hay BA, Guo M. Caspase-dependent cell death in Drosophila. Annu Rev Cell Dev Biol. 2006;22:623–50.
Wilson R, Goyal L, Ditzel M, Zachariou A, Baker DA, Agapite J, et al. The DIAP1 RING finger mediates ubiquitination of Dronc and is indispensable for regulating apoptosis. Nat Cell Biol. 2002;4(6):445–50.
Ip YT, Davis RJ. Signal transduction by the c-Jun N-terminal kinase (JNK)—from inflammation to development. Curr Opin Cell Biol. 1998;10(2):205–19.
Santabarbara-Ruiz P, Lopez-Santillan M, Martinez-Rodriguez I, Binagui-Casas A, Perez L, Milan M, et al. ROS-induced JNK and p38 signaling is required for unpaired cytokine activation during Drosophila regeneration. PLoS Genet. 2015;11(10):e1005595.
Pinal N, Martin M, Medina I, Morata G. Short-term activation of the Jun N-terminal kinase pathway in apoptosis-deficient cells of Drosophila induces tumorigenesis. Nat Commun. 2018;9(1):1541.
Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279(6):L1005–28.
Wu C, Chen C, Dai J, Zhang F, Chen Y, Li W, et al. Toll pathway modulates TNF-induced JNK-dependent cell death in Drosophila. Open Biol. 2015;5(7):140171.
Liu D, Shaukat Z, Saint RB, Gregory SL. Chromosomal instability triggers cell death via local signalling through the innate immune receptor Toll. Oncotarget. 2015;6(36):38552–65.
Phillips RG, Roberts IJ, Ingham PW, Whittle JR. The Drosophila segment polarity gene patched is involved in a position-signalling mechanism in imaginal discs. Development. 1990;110(1):105–14.
Abrams JM, White K, Fessler LI, Steller H. Programmed cell death during Drosophila embryogenesis. Development. 1993;117(1):29–43.
Wu LP, Anderson KV. Regulated nuclear import of Rel proteins in the Drosophila immune response. Nature. 1998;392(6671):93–7.
Milan M, Campuzano S, Garcia-Bellido A. Developmental parameters of cell death in the wing disc of Drosophila. Proc Natl Acad Sci USA. 1997;94(11):5691–6.
Thornberry NA, Lazebnik Y. Caspases: enemies within. Science. 1998;281(5381):1312–6.
Steller H. Regulation of apoptosis in Drosophila. Cell Death Differ. 2008;15(7):1132–8.
Fan Y, Bergmann A. The cleaved-Caspase-3 antibody is a marker of Caspase-9-like DRONC activity in Drosophila. Cell Death Differ. 2010;17(3):534–9.
Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, et al. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science. 2000;288(5467):870–4.
Igaki T, Kanda H, Yamamoto-Goto Y, Kanuka H, Kuranaga E, Aigaki T, et al. Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J. 2002;21(12):3009–18.
McEwen DG, Peifer M. Puckered, a Drosophila MAPK phosphatase, ensures cell viability by antagonizing JNK-induced apoptosis. Development. 2005;132(17):3935–46.
Luo X, Puig O, Hyun J, Bohmann D, Jasper H. Foxo and Fos regulate the decision between cell death and survival in response to UV irradiation. EMBO J. 2007;26(2):380–90.
Ellis MC, O’Neill EM, Rubin GM. Expression of Drosophila glass protein and evidence for negative regulation of its activity in non-neuronal cells by another DNA-binding protein. Development. 1993;119(3):855–65.
Sawicki R, Singh SP, Mondal AK, Benes H, Zimniak P. Cloning, expression and biochemical characterization of one Epsilon-class (GST-3) and ten Delta-class (GST-1) glutathione S-transferases from Drosophila melanogaster, and identification of additional nine members of the Epsilon class. Biochem J. 2003;370(Pt 2):661–9.
Sykiotis GP, Bohmann D. Keap1/Nrf2 signaling regulates oxidative stress tolerance and lifespan in Drosophila. Dev Cell. 2008;14(1):76–85.
Hay BA, Huh JR, Guo M. The genetics of cell death: approaches, insights and opportunities in Drosophila. Nat Rev Genet. 2004;5(12):911–22.
Nakajima YI, Kuranaga E. Caspase-dependent non-apoptotic processes in development. Cell Death Differ. 2017;24(8):1422–30.
Weinlich R, Oberst A, Beere HM, Green DR. Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol. 2017;18(2):127–36.
Newton K, Manning G. Necroptosis and inflammation. Annu Rev Biochem. 2016;85:743–63.
He S, Liang Y, Shao F, Wang X. Toll-like receptors activate programmed necrosis in macrophages through a receptor-interacting kinase-3-mediated pathway. Proc Natl Acad Sci USA. 2011;108(50):20054–9.
Cao M, Chen F, Xie N, Cao MY, Chen P, Lou Q, et al. c-Jun N-terminal kinases differentially regulate TNF- and TLRs-mediated necroptosis through their kinase-dependent and -independent activities. Cell Death Dis. 2018;9(12):1140.
Meyer SN, Amoyel M, Bergantinos C, de la Cova C, Schertel C, Basler K, et al. An ancient defense system eliminates unfit cells from developing tissues during cell competition. Science. 2014;346(6214):1258236.
Alpar L, Bergantinos C, Johnston LA. Spatially restricted regulation of Spatzle/Toll signaling during cell competition. Dev Cell. 2018;46(6):706–719.e5.
Germani F, Hain D, Sternlicht D, Moreno E, Basler K. The Toll pathway inhibits tissue growth and regulates cell fitness in an infection-dependent manner. Elife. 2018;7:e39939.
Byun PK, Zhang C, Yao B, Wardwell-Ozgo J, Terry D, Jin P, et al. The Taiman transcriptional coactivator engages Toll signals to promote apoptosis and intertissue invasion in Drosophila. Curr Biol. 2019;29(17):2790–2800.e4.
Anthoney N, Foldi I, Hidalgo A. Toll and Toll-like receptor signalling in development. Development. 2018;145(9):dev156018.
Foldi I, Anthoney N, Harrison N, Gangloff M, Verstak B, Nallasivan MP, et al. Three-tier regulation of cell number plasticity by neurotrophins and Tolls in Drosophila. J Cell Biol. 2017;216(5):1421–38.
Medzhitov R, Preston-Hurlburt P, Janeway CA Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388(6640):394–7.
Krutzik SR, Sieling PA, Modlin RL. The role of Toll-like receptors in host defense against microbial infection. Curr Opin Immunol. 2001;13(1):104–8.
Silverman N, Maniatis T. NF-kappaB signaling pathways in mammalian and insect innate immunity. Genes Dev. 2001;15(18):2321–42.
Thoma-Uszynski S, Stenger S, Takeuchi O, Ochoa MT, Engele M, Sieling PA, et al. Induction of direct antimicrobial activity through mammalian toll-like receptors. Science. 2001;291(5508):1544–7.
Hoffmann JA, Reichhart JM. Drosophila innate immunity: an evolutionary perspective. Nat Immunol. 2002;3(2):121–6.
Li Y, Deng SL, Lian ZX, Yu K. Roles of Toll-like receptors in nitroxidative stress in mammals. Cells. 2019;8(6):576.
Lugrin J, Rosenblatt-Velin N, Parapanov R, Liaudet L. The role of oxidative stress during inflammatory processes. Biol Chem. 2014;395(2):203–30.
West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, et al. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature. 2011;472(7344):476–80.
Zhang S, Guo X, Chen C, Chen Y, Li J, Sun Y, et al. dFoxO promotes Wingless signaling in Drosophila. Sci Rep. 2016;6:22348.
Wu C, Chen Y, Wang F, Chen C, Zhang S, Li C, et al. Pelle modulates dFoxO-mediated cell death in Drosophila. PLoS Genet. 2015;11(10):e1005589.
Ma XJ, Huang JH, Yang LX, Yang Y, Li WZ, Xue L. NOPO modulates Egr-induced JNK-independent cell death in Drosophila. Cell Res. 2012;22(2):425–31.
Huang JH, Feng Y, Chen XH, Li WZ, Xue L. Myc inhibits JNK-mediated cell death in vivo. Apoptosis. 2017;22(4):479–90.
Ma X, Xu W, Zhang D, Yang Y, Li W, Xue L. Wallenda regulates JNK-mediated cell death in Drosophila. Cell Death Dis. 2015;6:e1737.
Xue L, Noll M. Dual role of the Pax gene paired in accessory gland development of Drosophila. Development. 2002;129(2):339–46.
Ren P, Li W, Xue L. GLYAT regulates JNK-mediated cell death in Drosophila. Sci Rep. 2017;7(1):5183.
Sun Y, Zhang D, Li C, Huang J, Li W, Qiu Y, et al. Lic regulates JNK-mediated cell death in Drosophila. Cell Prolif. 2019;52(3):e12593.
Acknowledgements
We thank the Bloomington Drosophila Stock Center, Vienna Drosophila Research Center, NIG-FLY center and the Exelixis collection at Harvard for fly stocks, Prof. Wei Zuo at Tongji University for S.aureus strain and members of Xue lab for discussion and critical comments.
Funding
This work was supported by the National Natural Science Foundation of China (31771595, 31701244), the Fundamental Research Funds for the Central Universities (20002150001, 22120180549), Natural Science Fund of Hebei Province of China (C2018209119), Scientific and Technological Research Project of Higher Education of Hebei Province (BJ2019040), and Doctoral Scientific Research Foundation of North China University of Science and Technology (BS2017063).
Author information
Authors and Affiliations
Contributions
ZL and LX conceived and designed the experiments. ZL, CW, XD and WL conducted experiments. LX and WL supervised/advised on the study, ZL, CW and LX analyzed the data and wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors have read and agreed to the final version of the manuscript.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
The expression patterns of ptc-Gal4, Sd-Gal4 and GMR-Gal4. Fluorescence micrographs of third instar larval wing (a, b) and eye discs (c) are shown. Expression region of ptc-Gal4 (a), Sd-Gal4 (b) and GMR-Gal4 (c) are labeled by the UAS-GFP reporter. Scale bar: 100 μm. Figure S2. The knock-down efficacies of dorsal and cactus RNAi lines. (a and b) Expression of two independent dorsal RNAi and cactus RNAi significantly decrease the level of dorsal mRNA and cactus mRNA, as measured by qRT-PCR. Total RNA of Drosophila third instar larval wing discs (n = 10, in each group) was extracted and normalized for cDNA synthesis. Error bar indicates standard deviation. One-way ANOVA test was used to compute P-values, ****P < 0.0001, ***P < 0.001, **P < 0.01, *P < 0.05. Figure S3. JNK is required for Toll/NF-kB signaling impaired ACV development. Light micrographs of Drosophila adult wings (a-f) are shown. Compared with the ptc-Gal4 control (a), Toll10B-induced loss-of-ACV phenotype in adult wings (b), is blocked by expressing BskDN (c) or Puc (d). Depletion of cactus also produces a loss-of-ACV phenotype (e), which is suppressed by expressing BskDN (f). The lower panels show high magnification view of the boxed areas in upper panels (a-f). (g) Statistical analysis of ACV phenotype in adult wings (n = 45 for each genotype) is shown. Error bar indicates standard deviation. One-way ANOVA test was used to compute P-values, ****P < 0.0001. Scale bar: 100 μm. Figure S4. JNK pathway is up-regulated by physiological activation of Toll signaling. Light micrographs of third instar wing discs with X-Gal staining (a-c) are shown. Compared with the Sd-Gal4 control (a), elevating endogenous Toll signaling by knockdown of cactus (b and c) up-regulates puc-LacZ expression. (d) Statistical analysis of X-Gal staining (n = 8) is shown. One-way ANOVA was used to compute P-values, ****P < 0.0001. Scale bar: 100 μm. Figure S5. Toll regulates the stress response gene gstD1. (a) Histogram showing the level of gstD1 mRNA as measured by qRT-PCR. Error bar represents standard deviation from three independent experiments. One-way ANOVA was used to compute P-values, ***P < 0.001, ns indicates not significant. Figure S6. Evaluate the level of Toll expression. (a) Histogram showing the level of Toll mRNA as measured by qRT-PCR. Error bar represents standard deviation from three independent experiments. One-way ANOVA was used to compute P-values, ****P < 0.0001, **P < 0.01.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, Z., Wu, C., Ding, X. et al. Toll signaling promotes JNK-dependent apoptosis in Drosophila. Cell Div 15, 7 (2020). https://doi.org/10.1186/s13008-020-00062-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13008-020-00062-5