
Abstract
Background
Quantitative PCR (qPCR) is one of the most common and accurate methods of gene expression analysis. However, the biggest challenge for this kind of examinations is normalization of the results, which requires the application of dependable internal controls. The selection of appropriate reference genes (RGs) is one of the most crucial points in qPCR data analysis and for correct assessment of gene expression. Because of the fact that many reports indicate that the expression profiles of typically used RGs can be unstable in certain experimental conditions, species or tissues, reference genes with stable expression levels should be selected individually for each experiment. In this study, we analysed a set of ten candidate RGs for wheat seedlings under short-term drought stress. Our tests included five ‘traditional’ RGs (GAPDH, ACT, UBI, TUB, and TEF1) and five novel genes developed by the RefGenes tool from the Genevestigator database.
Results
Expression stability was assessed using five different algorithms: geNorm, NormFinder, BestKeeper, RefFinder and the delta Ct method. In the final ranking, we identified three genes: CJ705892, ACT, and UBI, as the best candidates for housekeeping genes. However, our data indicated a slight variation between the different algorithms that were used. We revealed that the novel gene CJ705892, obtained by means of in silico analysis, showed the most stable expression in the experimental tissue and condition.
Conclusions
Our results support the statement, that novel genes selected for certain experimental conditions have a more stable level of expression in comparison to routinely applied RGs, like genes encoding actin, tubulin or GAPDH. Selected CJ705892 gene can be used as a housekeeping gene in the expression analysis in wheat seedlings under short-term drought. The results of our study will be useful for subsequent analyses of gene expression in wheat tissues subjected to drought.
Similar content being viewed by others

Background
Quantitative PCR (qPCR, real-time PCR) is a widely applied method in the analysis of gene expression due to its high sensitivity, high specificity and good reproducibility [1,2,3]. However, for proper analysis of gene expression involving qPCR, a normalization step is necessary. The most common strategy is based on reference genes (RGs), also called ‘housekeeping genes’, which are internal controls with stable expression levels in the tested material under the experimental conditions. Therefore, the selection of appropriate RGs is one of the most crucial points in qPCR data analysis and for correct assessment of gene expression [4, 5]. Numerous housekeeping genes, such as actin (ACT), tubulin (TUB), and 18S ribosomal RNA (18S rRNA), that are necessary for proper cellular metabolism are widely used as RGs in many studies. Nevertheless, many reports indicate that the expression profiles of these genes can be unstable in certain experimental conditions, species or tissues [1, 4, 6]. Many authors suggest that there is no universal RG for all experimental subjects [e.g. 2, 6,7,8,9]. Each experiment requires the selection of an ideal RG [10]. Many studies have been conducted on the selection of appropriate RGs for Arabidopsis thaliana [11], soya [12], peach [13], rice [14], cotton [15, 16], and poplar [1]. However, there are still few studies concerning wheat [2, 3, 17], especially in response to abiotic stresses such as drought. There are data for wheat infected by Puccinia spp. [18], by BYDV-PAV and BYDV-PAS viruses [19] or under different farming conditions (nitrogen fertilization and type of system) [2]. For rice in water shortage conditions, the gene encoding ubiquitin was identified as a RG [20]. However, the selection of RG for gene expression analysis under drought conditions in wheat remains a major challenge. Most of the reports concerning RG tests have been focused on validating a set of commonly used reference genes. Currently, many studies show that the identification of ideal RGs can be based on in silico analysis, such as the Genevestigator database and the RefGenes tool [21]. The Genevestigator database contains a large set of systematically annotated and quality-controlled microarray data from several organisms [22], and RefGenes is an online tool that utilizes this database to enable users to search for genes that exhibit minimal expression variance across a chosen set of arrays. This method ensures the identification of genes with more stable expression than the standard genes [1, 2, 16, 23, 24]. For the analysis of the results and selection of the best RG, numerous platforms using different algorithms have been developed, including geNorm [25], NormFinder [26], BestKeeper [27] and RefFinder [28].
In this study, we conducted an analysis using geNorm, NormFinder, BestKeeper and RefFinder to select the most suitable RG for wheat plants in the seedling stage when tested under drought conditions. Moreover, a method of directly delta Ct analysis based on comparisons between each RG and the other RGs within each sample and calculation the average standard deviation against the other RGs [29] was performed.
Results
Selection of candidate reference genes using the RefGenes tool
RefGenes is an in silico method enabling the identification of genes with high expression stability within microarray libraries of wheat subjected to drought. Using this tool to examine normalized and well-annotated microarray experiments, we found 20 candidate RGs. Among these genes, we selected five candidates with stable expression levels under drought conditions. The candidate RGs obtained in this analysis were used for validation in qPCR (Table 1).
Expression levels of the reference genes
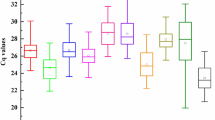
To identify the most stable housekeeping genes, cDNA of all tested lines (stress imposed and control) was used in qPCR. The specificity of the primers was estimated by qPCR melting curve analysis. A single peak of the melting curve was observed for most of the tested primers (8 of 10 primer pairs), confirming the specificity of the amplicons. Only for two primer pairs (for the TEF1 and CA596223.1 genes) unspecific products of expression were observed, and because of that fact, they were excluded from further analysis. Moreover, no signal was detected in the NTC samples. We used the standard curve method with a pool of all the cDNAs to determine the PCR efficiency (E) and the correlation coefficient (R2) for each primer pair. The obtained results were analysed according to Bustin et al. [10], and the results indicated that the acceptable range of efficiency was from 80 to 120%. According to Tyburski et al. [30], a slope equal to − 3.32 is evidence of high reaction efficiency, and R2 = 1 indicates that the same expression level was observed in the calibrator and tested sample. We obtained E values varying between 83.01% and 112.75% and R2 from 0.83 to 1 (Table 2). The raw quantification cycle (Cq) values were estimated for determination of the gene expression levels. The Cq values for analysed samples ranged between 20.16 and 37.60 (Fig. 1).

Cq values for eight candidate reference genes across experimental samples. A line across the box is depicted as the median. The box indicates the 25th and 75th percentiles, the whiskers represent the maximum and minimum values
Expression stability of the reference genes
The expression stability of selected RGs was estimated using five different algorithms: geNorm, NormFinder, BestKeeper, RefFinder and the delta Ct method. For each platform, eight RGs were ranked from the most stable to the least stable. In the results generated by the software packages, differences were observed. The ranking of RGs using geNorm was mostly in agreement with that of NormFinder. We found that the first three genes with the most stable expression and the gene with the least stable expression were the same for these two platforms. RefFinder and delta Ct analysis gave the same rankings among all 8 RGs. However, the results of BestKeeper and RefFinder showed different rankings for the most and least stable candidate genes (Table 3).
geNorm analysis
geNorm analysis indicated that the stability of gene expression (M-value) varied between 0.550 for the most stable gene and 0.975 for the least stable gene (Table 3). According to this algorithm, genes with the lowest M-value were considered to be the most stable, whereas genes with the highest M-value were considered to be the least stable [31]. Based on geNorm software results, we identified a CJ705892 gene as the most stable in the tested wheat lines. Among a set of commonly used housekeeping genes, actin (0.575) and ubiquitin (0.600) were assessed as the most stable. The rest of the genes obtained from the Genevestigator database indicated a low level of expression stability. The least stable was CA728440 (0.975).
NormFinder analysis
The stability of the eight selected RGs was further analysed using the NormFinder platform. The NormFinder software analyses datasets and estimates stability based on intra-group and inter-group variation. The genes with lower stability values were considered to be the most stable RGs, whereas the genes with higher stability values were ranked as the least stable [32]. Based on the NormFinder algorithm, we found that the CJ705892 gene (stability value: 0.072) was the most stable gene, followed by ACT (0.084) and UBI (0.131). The least stable was CA728440 (0.300) (Table 3). We observed that the results of NormFinder and geNorm were slightly different. However, both algorithms indicated that the CJ705892 gene and CA728440 were the most and least stable genes, respectively. Therefore, based on the geNorm and NormFinder analysis and previous data concerning the BestKeeper and RefFinder platforms, we concluded that a novel gene, CJ705892, developed by the RefGenes tool, was found to be the most stable RG in tested wheat lines under short-term drought.
BestKeeper analysis
The obtained data were also analysed using the BestKeeper algorithm. BestKeeper software is usually employed by assessing the correlation coefficients of each individual gene with the geometric mean of all genes (the BestKeeper Index) [33]. These results were different from those of NormFinder and geNorm. According to BestKeeper, the most stable gene was ACT (0.468), followed by CJ705892 (0.526). The least stable was CA728440 (1.032) (Table 3).
RefFinder analysis
The RefFinder platform requires only raw Cq values without any option to include PCR efficiency. The ranking obtained by this algorithm is based on the standard deviations of the RG Cq values [33]. The results obtained by geNorm and NormFinder were not provided by the RefFinder output. RefFinder assessed ACT (1.00) and UBI (1.861) as the most stable genes. The gene CJ705892 (2.711) was in third place (Table 3).
Delta Ct method
The results obtained by the delta Ct method showed the same results as we observed using the RefFinder software. The most stable genes were ACT (0.868) and UBI (0.887), followed by CJ705892 (0.895) (Table 3). Analysis of all datasets suggests that the results obtained by the NormFinder, geNorm and BestKeeper methods indicated that the gene CJ705892 is on top of the RG rankings, with some slight differences in the rankings. All statistical algorithms showed CA728440 as the least stable gene.
Discussion
Analysis of gene expression patterns is the basis for the development of knowledge about the mechanisms involved in the initial reaction of plants to stress. The most accurate technique for expression analysis is qPCR, and a selection of the best RG is a crucial step to avoid experimental errors and incorrect interpretation of results. The ideal internal control has stable expression in the tested material under experimental conditions. In the present study, we analysed 10 potential RGs for wheat substitution lines under short-term drought conditions. We combined four algorithms (RefFinder, BestKeeper, geNorm, and NormFinder) and the delta Ct method to estimate the best RG. In the final ranking, we identified three genes, CJ705892, ACT, and UBI, as the best candidates. However, our data indicated a slight variation between the different algorithms that were used. According to the geNorm and NormFinder platforms, the CJ705892 gene had the most stable expression, while the BestKeeper and RefFinder programs showed this gene in second and third place, respectively. The obtained results are confirmed by numerous studies suggesting that variation is caused by the use of different algorithms [9, 33].
As suggested by previous studies, the most reliable tools for RG stability estimation are geNorm and NormFinder [33]. Many reports based only on these two algorithms have been used for the identification of RGs, e.g., in berry [34], rice [14, 35], tomato [36], soy [37], cotton [16] or wheat [17, 23, 38]. Based on these data, the results generated by geNorm and NormFinder were crucial for RG selection in our study.
The data obtained by BestKeeper demonstrated slight differences compared to those from geNorm and NormFinder. As the best RG, BestKeeper showed actin, followed by the CJ705892 gene and UBI. Thus, the ranking order was not identical; however, the three first genes, which were considered the most stable under the given experimental conditions, were the same. A similar variance was described for apple under postharvest conditions [39], Caragana intermedia under osmotic stress [8] and Actinidia deliciosa infected by Pseudomonas syringae pv. actinidiae [9]. Based on previous studies, we suggest that these variances are caused by the BestKeeper algorithm, which employs correlation analyses between the candidate gene Cq and an index derived from the candidate geometric mean. In contrast, the algorithms of the geNorm and NormFinder platforms use variation measures to calculate the stability of gene transcription [39].
The most significant differences were observed using RefFinder software. This program indicated ACT and UBI as the most stable genes, followed by CJ705892. De Spiegelaere et al. [33] investigated differences between all four algorithms that were used in our study. The authors explained that RefFinder ranking is based on the standard deviations of the RG Cq values and that the analysis requires only non-corrected raw Cq values. Moreover, De Spiegelaere et al. [33] suggested that the RefFinder system is applied in many studies of RG validation because it is free and performs a quick analysis using the three most popular algorithms. However, this platform has not been well validated yet and has no scientific basis. Thus, we suggest that RefFinder software should be used as a complementary tool in the analysis of RG stability.
Among the ‘traditional’ RGs, we found that ACT had the most stable expression level. This result was obtained by three software packages: geNorm, NormFinder, and BestKeeper. Our results confirmed a previous study conducted on Chinese Spring wheat treated with different abiotic (nutrient deprivation, hormone application) and biotic (rust infection) stress factors. This analysis performed with the geNorm, NormFinder and BestKeeper packages also showed actin as the best RG.
Our results indicated that a novel gene obtained using the RefGenes tool from the Genevestigator database was the most stable among all the tested genes. Many studies have demonstrated that novel genes selected for experimental conditions have a more stable level of expression. Marcolino-Gomes et al. [24] analysed a number of genes, including genes widely used as references (GAPDH, TUB, β-actin, etc.) and additional genes developed with Genevestigator and RNA-seq libraries. The authors found that some novel genes obtained by in silico analysis indicated stable expression profiles in soy under drought. Similar observations were described for Triticum aestivum in different tissues under temperature stress.
Despite slight differences between the rankings obtained by the four different programs, the results of geNorm and NormFinder overlapped and showed CJ705892 as the best RG. Based on previous studies suggesting that these two algorithms are the most reliable, we suggest that CJ705892 can be used as a housekeeping gene in the expression analysis of wheat seedlings under short-term drought. The results of our study provide new information that will be useful in molecular studies of wheat response to water deficiency.
Conclusions
In our study, we combined analysis based on standard RG and novel genes obtained via RefGenes tool from Genevestigator database in order to identify the optimal RG for common wheat (Triticum aestivum L.) seedlings subjected to short-time osmotic stress. qPCR results were analyzed using four different algorithms. Our study allowed for the identification of the novel gene showed the most stable expression level in tested lines of Triticum aestivum L. under water deficit, which can be used as RG for subsequent experiments based on similar plant material and conditions–wheat seedlings subjected to water deficit stress.
Methods
Plant materials and stress induction
In our study, the set of 18 inter-varietal single chromosome substitution lines (ISCSLs) of Triticum aestivum L. were used. ISCSLs based on the drought-tolerant cultivar ‘Saratovskaya 29’ (S29) as a recipient and the drought-sensitive cultivar ‘Janetzkis Probat’ (JP) as a donor were used in the study.
For the induction of germination, sterilized kernels were incubated at 4 °C for 48 h. Then, the kernels were germinated in Petri dishes containing filter papers soaked in distilled water in the dark at 24 °C. After 2 days, seedlings were transferred into plastic pots containing full-strength Murashige Skoog (MS) medium. Plants were grown under controlled conditions in a hydroponic culture in a phytotron greenhouse for 5 days under control conditions (light/dark regime of 16/8 h at 25 ± 3 °C, relative humidity of 50 ± 10%, and the light intensity during the day time was 350 μmol m−2 s−1). Seven-day-old seedlings were treated with 10% polyethylene glycol (PEG-6000) dissolved in MS solution to induce drought stress. Seedlings without roots were collected after 1, 3 and 6 h of stress treatment. Plants growing in MS medium without PEG were used as a control.
Total RNA isolation
RNA extraction was performed after 0, 1, 3 and 6 h of exposure to stress. After harvesting, plant material was immediately frozen in liquid nitrogen and grind to a fine powder with mortar and pestle. Total RNA was isolated using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. The quality and quantity of RNA samples were assessed on 2% agarose gel electrophoresis and spectrophotometrically using the DeNovix DS-11 (DeNovix).
Reverse transcription
Reverse transcription PCR was performed with an iScript™ cDNA Synthesis Kit (Bio-Rad) following the manufacturer’s instructions. RT-PCRs were carried out in a total volume of 30 µl containing 1.5 µg of the total RNA, 6 μl 5× iScript Reaction Mix containing blend of oligo(dT) and random hexamer primers and 1.5 μl of iScript Reverse Transcriptase, which is modified Moloney murine leukemia virus (MMLV) reverse transcriptase. The thermal conditions applied were as follows: priming for 5 min at 25 °C; reverse transcription for 20 min at 46 °C and inactivation for 1 min at 95 °C. Obtained cDNA was stored in -25 °C.
Selection of candidate reference genes
To identify the gene with the most stable expression in common wheat under drought treatment, a set of ten genes were selected and tested. Five genes commonly used as internal controls in wheat were obtained from previous expression studies and included TUB, ACT, GAPDH, UBI and TEF1 [40,41,42,43]. Five novel genes were identified as potential references via the RefGenes in silico tool from the Genevestigator platform [https://www.genevestigator.com/gv/plant.jsp] [21] (Table 1). The Genevestigator database provides normalized and well-annotated microarray tests. The RefGenes tool enables searching for genes with minimal expression variance across a chosen set of arrays on the Genevestigator platform [22, 24].
Design of qPCR primers and amplification efficiency testing
The sequences of all tested gene transcripts were obtained from the NCBI database. Primers for qPCR were designed using the Primer-BLAST tool [44] (Table 1). The same tool was used for determination of the primer’s specificity in silico. The PCR amplification efficiency was determined for each primer pair by the analysis of the slope obtained from a standard curve generated from a serial dilution of pooled cDNA as reported previously [32]. The amplification efficiency (E) and correlation coefficient (R2) of the primers were calculated according to the equation [10(1/−S)−1] × 100%, where S represents the slope of the linear regression.
Quantitative PCR (qPCR) conditions
Quantitative PCR (qPCR) was performed based on SYBR Select Master Mix (Applied Biosystems) according to the manufacturer’s instructions. PCRs were carried out in a total volume of 20 µl containing 800 ng of cDNA, 1 × SYBR Select Master Mix (Applied Biosystems) based on AmpliTaq® Fast DNA Polymerase and 400 nM of each primer. qPCR was performed on a LightCycler® 96 System (Roche) under the following thermal conditions: 2 min at 50 °C; 10 min at 95 °C; 40 cycles of 15 s at 95 °C and 1 min at 60 °C. Each reaction was carried out in three technical replicates along with a no template control (NTC). To confirm the amplification specificity and lack of primer dimer formation, each run was performed with a melting curve analysis. Each sample was analyzed in two full biological and three technical replications at the qPCR level.
Analysis of gene expression stability
The raw data of qPCR was processed by means of LightCycler® 96 software v. 1.1 (Roche). The expression stability of the ten selected housekeeping genes (TUB, ACT, GAPDH, UBI, TEF1, BJ254354, CA596223, CA728440, CJ705892, and CA644093) in wheat seedlings under drought conditions were analysed using the RefFinder [28], geNorm [25], BestKeeper [27] and NormFinder [26] software packages and the delta Ct (dCt) method [29]. Raw Cq values were used in the BestKeeper and delta Ct algorithms. For the geNorm and NormFinder analysis, raw Cq values were transformed into relative quantities.
Availability of data and materials
All data generated or analysed during this study are included in this published article and its supplementary information file.
References
Brunner AM, Yakovlev IA, Strauss SH. Validating internal controls for quantitative plant gene expression studies. BMC Plant Biol. 2004;4:14. https://doi.org/10.1186/1471-2229-4-14.
Tenea GN, Peres Bota A, Cordeiro Raposo F, Maquet A. Reference genes for gene expression studies in wheat flag leaves grown under different farming conditions. BMC Res Notes. 2011;4:373. https://doi.org/10.1186/1756-0500-4-373.
Li Y, Chen W, Wang Q, Wang N, Wu YF. Assessment of reference genes for quantitative real-time PCR gene expression normalization in periwinkle during Wheat Blue Dwarf phytoplasma infection. Austr Plant Pathol. 2014;43(4):477–85. https://doi.org/10.1007/s13313-014-0288-5.
Bustin SA. Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J Mol Endocrinol. 2002;29(1):23–39. https://doi.org/10.1677/jme.0.0290023.
Huggett J, Dheda K, Bustin SA, Zumla A. Real-time RT-PCR normalisation; strategies and considerations. Gene Immun. 2005;6(4):279–84. https://doi.org/10.1038/sj.gene.6364190.
Nakayama T, Okada N, Yoshikawa M, Asaka D, Kuboki A, Kojima H, et al. Assessment of suitable reference genes for RT-qPCR studies in chronic rhinosinusitis. Sci Rep. 2018;8:1568. https://doi.org/10.1038/s41598-018-19834-9.
Mafra V, Kubo KS, Alves-Ferreira M, Ribeiro-Alves M, Stuart RM, Boava LP, et al. Reference genes for accurate transcript normalization in citrus genotypes under different experimental conditions. PLoS ONE. 2012;7(2):e31263. https://doi.org/10.1371/journal.pone.0031263.
Zhu J, Zhang L, Li W, Han S, Yang W, Qi L. Reference gene selection for quantitative real-time PCR normalization in Caragana intermedia under different abiotic stress conditions. PLoS ONE. 2013;8:e53196. https://doi.org/10.1371/journal.pone.0053196.
Petriccione M, Mastrobuoni F, Zampella L, Scortichini M. Reference gene selection for normalization of RT-qPCR gene expression data from Actinidia deliciosa leaves infected with Pseudomonas syringae pv. actinidiae. Sci Rep. 2015;5:16961. https://doi.org/10.1038/srep16961.
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55(4):611–22. https://doi.org/10.1373/clinchem.2008.112797.
Remans T, Smeets K, Opdenakker K, Mathijsen D, Vangronsveld J, Cuypers A. Normalisation of real-time RT-PCR gene expression measurements in Arabidopsis thaliana exposed to increased metal concentrations. Planta. 2008;227(6):1343–9. https://doi.org/10.1007/s00425-008-0706-4.
Hu R, Fan C, Li H, Zhang Q, Fu YF. Evaluation of putative reference genes for gene expression normalization in soybean by quantitative real-time RT-PCR. BMC Mol Biol. 2009;10:93. https://doi.org/10.1186/1471-2199-10-93.
Tong Z, Gao Z, Wang F, Zhou J, Zhang Z. Selection of reliable reference genes for gene expression studies in peach using real-time PCR. BMC Mol Biol. 2009;10(71):1–13. https://doi.org/10.1186/1471-2199-10-71.
Jain M, Nijhawan A, Tyagi AK, Khurana JP. Validation of housekeeping genes as internal control for studying gene expression in rice by quantitative real-time PCR. Biochem Biophys Res Commun. 2006;345(2):646–51. https://doi.org/10.1016/j.bbrc.2006.04.140.
Tu L, Zhang XL, Liu DQ, Jin SX, Cao JL, Zhu LF, et al. Suitable internal control genes for qRT-PCR normalization in cotton fiber development and somatic embryogenesis. Chin Sci Bull. 2007;52:3110–7. https://doi.org/10.1007/s11434-007-0461-0.
Artico S, Nardeli SM, Brilhante O, Grossi-de-Sa MF, Alves-Ferreira M. Identification and evaluation of new reference genes in Gossypium hirsutum for accurate normalization of real-time quantitative RT-PCR data. BMC Plant Biol. 2010;10:49. https://doi.org/10.1186/1471-2229-10-49.
Paolacci A, Tanzarella O, Porceddu E, Ciaffi M. Identification and validation of reference genes for quantitative RT-PCR normalization in wheat. BMC Mol Biol. 2009;10(1):11. https://doi.org/10.1186/1471-2199-10-11.
Scholtz JJ, Visser B. Reference gene selection for qPCR gene expression analysis of rust-infected wheat. Physiol Mol Plant Pathol. 2013;81:22–5. https://doi.org/10.1016/j.pmpp.2012.10.006.
Jarosová J, Kundu JK. Validation of reference genes as internal control for studying viral infections in cereals by quantitative real-time RT-PCR. BMC Plant Biol. 2010;10:146. https://doi.org/10.1186/1471-2229-10-146.
Pabuayon IM, Yamamoto N, Trinidad JL, Longkumer T, Raorane ML, Kohli A. Reference genes for accurate gene expression analyses across different tissues, developmental stages and genotypes in rice for drought tolerance. Rice. 2011;9(1):32. https://doi.org/10.1186/s12284-016-0104-7.
Hruz T, Wyss M, Docquier M, Pfaffl MW, Masanetz S, Borghi L, et al. RefGenes: identification of reliable and condition specific reference genes for RT-qPCR data normalization. BMC Genomics. 2011;21(12):156. https://doi.org/10.1186/1471-2164-12-156.
Hruz T, Laule O, Szabo G, Wessendorp F, Bleuler S, Oertle L, et al. Genevestigator V3: a reference expression database for the meta-analysis of transcriptomes. Adv Bioinform. 2008. https://doi.org/10.1155/2008/420747.
Long XY, Wang JR, Ouellet T, Rocheleau H, Wei YM, Pu ZE, et al. Genome-wide identification and evaluation of novel internal control genes for q-PCR based transcript normalization in wheat. Plant Mol Biol. 2010;74(3):307–11. https://doi.org/10.1007/s11103-010-9666-8.
Marcolino-Gomes J, et al. Transcriptome-wide identification of reference genes for expression analysis of soybean responses to drought stress along the day. PLoS ONE. 2015;10(9):1–16. https://doi.org/10.1371/journal.pone.0139051.
Vandesompele J. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002. https://doi.org/10.1186/gb-2002-3-7-research0034.
Andersen CL, Jensen JL, Orntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004;64(15):5245–50. https://doi.org/10.1158/0008-5472.CAN-04-0496.
Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: bestKeeper-Excel-based tool using pair-wise correlations. Biotechnol Lett. 2004;26:509–15.
Xie G, Kato H, Imai R. Biochemical identification of the OsMKK6–OsMPK3 signalling pathway for chilling stress tolerance in rice. Biochem J. 2012;443:95–102. https://doi.org/10.1042/BJ20111792.
Silver N, Best S, Jiang J, Thein SL. Selection of housekeeping genes for gene expression studies in human reticulocytes using real-time PCR. BMC Mol Biol. 2006;7:33. https://doi.org/10.1186/1471-2199-7-33.
Tyburski J, Studzińska A, Daca P, Tretyn A. PCR w czasie rzeczywistym. Metody analizy danych. Biotechnologia. 2008;1(80):86–96.
Sinha P, Saxena RK, Singh VK, Krishnamurthy L, Varshney RK. Selection and validation of housekeeping genes as reference for gene expression studies in pigeonpea (Cajanus cajan) under heat and salt stress conditions. Front Plant Sci. 2015;6:1071. https://doi.org/10.3389/fpls.2015.01071.
Sinha P, Singh VK, Suryanarayana V, Krishnamurthy L, Saxena RK, Varshney RK. Evaluation and validation of housekeeping genes as reference for gene expression studies in pigeonpea (Cajanus cajan) under drought stress conditions. PLoS ONE. 2015;10(4):e0122847. https://doi.org/10.1371/journal.pone.0122847.
De Spiegelaere W, Dern-Wieloch J, Weigel R, Schumacher V, Schorle H, Nettersheim D, et al. Reference gene validation for RT-QPCR, a note on different available software packages. PLoS ONE. 2015;10(3):e0122515. https://doi.org/10.1371/journal.pone.0122515.
Reid KE, Olsson N, Schlosser J, Peng F, Lund ST. An optimized grapevine RNA isolation procedure and statistical determination of reference genes for real-time RT-PCR during berry development. BMC Plant Biol. 2006;6:27. https://doi.org/10.1186/1471-2229-6-27.
Li QF, Sun SSM, Yuan DY, Yu HX, Gu MH, Liu QQ. Validation of candidate reference genes for the accurate normalization of real-time quantitative RT-PCR data in rice during seed development. Plant Mol Biol Rep. 2010;28:49–57. https://doi.org/10.1007/s11105-009-0124-1.
Expósito-Rodríguez M, Borges AA, Borges-Pérez A, Pérez JÁ. Selection of internal control genes for quantitative real-time RT-PCR studies during tomato development process. BMC Plant Biol. 2008;8:131. https://doi.org/10.1186/1471-2229-8-131.
Libault M, Thibivilliers S, Bilgin DD, Radwan O, Benitez M, Clough SJ, et al. Identification of four soybean reference genes for gene expression normalization. Plant Genome. 2008;1(1):44–54. https://doi.org/10.3835/plantgenome2008.02.0091.
Lekshmy S, Jha SK. Selection of reference genes suitable for qRT-PCR expression profiling of biotic stress, nutrient deficiency and plant hormone responsive genes in bread wheat. Ind J Plant Physiol. 2017;22(1):101–6. https://doi.org/10.1007/s40502-017-0282-3.
Storch TT, Pegoraro C, Finatto T, Quecini V, Rombaldi CV, Girardi CL. Identification of a novel reference gene for apple transcriptional profiling under postharvest conditions. PLoS ONE. 2015;10(3):e0120599. https://doi.org/10.1371/journal.pone.0120599.
Rampino P, Pataleo S, Gerardi C, Mita G, Perrotta C. Drought stress response in wheat: physiological and molecular analysis of resistant and sensitive genotypes. Plant, Cell Environ. 2006;29(12):2143–52.
Li YC, Meng FR, Zhang CY, Ming NZ, Jiang SS, Hong PR, et al. Comparative analysis of water stress-responsive transcriptomes in drought-susceptible and -tolerant wheat (Triticum aestivum L.). J Plant Biol. 2012;55(5):349–60. https://doi.org/10.1007/s12374-011-0032-4.
Liu F, Guo J, Bai P, Duan Y, Wang X, Cheng Y, et al. Wheat TaRab7 GTPase is part of the signaling pathway in responses to stripe rust and abiotic stimuli. PLoS ONE. 2012;7(5):1–9. https://doi.org/10.1371/journal.pone.0037146.
Wei L, Wang L, Yang Y, Wang P, Guo T, Kang G. Abscisic acid enhances tolerance of wheat seedlings to drought and regulates transcript levels of genes encoding ascorbate-glutathione biosynthesis. Front Plant Sci. 2015;6:1–11. https://doi.org/10.3389/fpls.2015.00458.
Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformat. 2012;13:134. https://doi.org/10.1186/1471-2105-13-134.
Acknowledgements
Not applicable.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
KD, MN, KK, AB and AK designed the experiment and methodology of analysis. AB provided the plant material for analyses. KD and MS performed the gene expression analyses. KD and MN performed the in silico analyses of obtained results. KD and HS wrote the manuscript. MN and KK reviewed and edited the manuscript text. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Dudziak, K., Sozoniuk, M., Szczerba, H. et al. Identification of stable reference genes for qPCR studies in common wheat (Triticum aestivum L.) seedlings under short-term drought stress. Plant Methods 16, 58 (2020). https://doi.org/10.1186/s13007-020-00601-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13007-020-00601-9