
Abstract
Background
Altered cerebrovascular function and accumulation of amyloid-β (Aβ) after traumatic brain injury (TBI) can contribute to chronic neuropathology and increase the risk for Alzheimer’s disease (AD). TBI due to a blast-induced shock wave (bTBI) adversely affects the neurovascular unit (NVU) during the acute period after injury. However, the chronic effects of bTBI and Aβ on cellular components of the NVU and capillary network are not well understood.
Methods
We exposed young adult (age range: 76–106 days) female transgenic (Tg) APP/PS1 mice, a model of AD-like Aβ amyloidosis, and wild type (Wt) mice to a single bTBI (~ 138 kPa or ~ 20 psi) or to a Sham procedure. At 3-months or 12-months survival after exposure, we quantified neocortical Aβ load in Tg mice, and percent contact area between aquaporin-4 (AQP4)-immunoreactive astrocytic end-feet and brain capillaries, numbers of PDGFRβ-immunoreactive pericytes, and capillary densities in both genotypes.
Results
The astroglia AQP4-capillary contact area in the Tg-bTBI group was significantly lower than in the Tg-Sham group at 3-months survival. No significant changes in the AQP4-capillary contact area were observed in the Tg-bTBI group at 12-months survival or in the Wt groups. Capillary density in the Tg-bTBI group at 12-months survival was significantly higher compared to the Tg-Sham control and to the Tg-bTBI 3-months survival group. The Wt-bTBI group had significantly lower capillary density and pericyte numbers at 12-months survival compared to 3-months survival. When pericytes were quantified relative to capillary density, no significant differences were detected among the experimental groups, for both genotypes.
Conclusion
In conditions of high brain concentrations of human Aβ, bTBI exposure results in reduced AQP4 expression at the astroglia-microvascular interface, and in chronic capillary proliferation like what has been reported in AD. Long term microvascular changes after bTBI may contribute to the risk for developing chronic neurodegenerative disease later in life.
Similar content being viewed by others

Background
Traumatic brain injury (TBI) is considered a risk factor for chronic neurodegenerative disorders including Alzheimer’s disease (AD), Parkinson disease (PD), and chronic traumatic encephalopathy (CTE) [37, 75, 77, 86]. The pathobiological changes underlying this phenomenon are not well-understood. One hypothesis is that TBI induces or accelerates accumulation of aggregation-prone molecules such as amyloid-β (Aβ) peptides, hyper-phosphorylated tau protein, alpha-synuclein, and transactive response DNA-binding protein 43 (TDP-43) [55, 60, 130]. Pertaining to the risk for AD following TBI, an injury-induced imbalance between Aβ production and clearance can promote accumulation of Aβ in the brain [20, 58, 109] which can contribute to, and be enhanced by, vascular changes involving impaired blood–brain barrier (BBB) function and dysregulation of cerebral blood flow [3, 53, 54, 57, 67, 118, 136]. Because Aβ- and vascular changes could contribute to poor recovery after a TBI and, according to the two-hit vascular hypothesis, promote chronic neurodegeneration in AD [92, 136], they are attractive targets for development of therapy interventions and biomarkers in both conditions [23, 34, 54, 66, 104, 120, 122].
Cerebrovascular homeostasis is regulated by the combined actions of endothelial cells, mural cells (capillary pericytes and smooth muscle cells), glial cells, and neuronal activity [54, 74, 123]. Pericytes have multiple roles in microvascular function [132] including modulation of capillary diameter and cerebral microcirculation [41, 45, 68, 69, 91, 99] and maintenance of endothelial junctional proteins that form the BBB [16, 123]. The barrier function of endothelial cells and pericytes is influenced by astrocytes which secrete (and uptake) cytokines, growth factors, and neurotransmitters [1, 54, 123], and interact with pericytes in regulating neurovascular coupling [54, 88]. In the aftermath of a brain injury, both pericytes and astrocytes are involved in reparatory processes [9, 21, 23] and are capable of clearing Aβ by an apolipoprotein E/low-density lipoprotein receptor-related protein 1 (LRP-1) mediated mechanism [6, 76, 82, 124, 126] and secretion of Aβ-degrading enzymes [108]. Thus, brain injury-induced dysfunction or loss of either cell type could impair these mechanisms and contribute to brain Aβ accumulation in conjunction with microvascular dysregulation.
Clinical studies, and experimental studies in animals and in vitro models, reported that exposure to explosive blasts can result in acute dysfunction or loss of pericytes and astrocytes as well as altered BBB permeability [2, 5, 7, 13, 30, 35, 36, 38, 39, 47,48,49,50,51,52, 61, 63,64,65, 71,72,73, 78, 79, 81, 83, 84, 105, 112, 116, 129, 134]. However, the effects of blast TBI on brain Aβ concentration and deposition have been assessed in only a few studies and at acute time points after injury exposure. In wild type rodents, endogenous murine Aβ was lower acutely in animals exposed to a single low-level blast TBI (bTBI) compared to sham animals [27], while transgenic AD mice exposed to repetitive low-level bTBI with 4 weeks survival had lower levels of soluble and insoluble Aβ but no change in Aβ plaque load compared to sham mice [100]. These findings were unexpected due to previous reports of increased Aβ concentration and deposition in brains of young adults within hours to days after severe TBI [29, 58], and higher Aβ concentrations at 3 weeks after controlled cortical impact injury in human Aβ knock-in mice compared to sham mice [3], suggesting that blast TBI and blunt force TBI may have differential effects on Aβ production, accumulation and/or clearance. To gain insight into the unexplored chronic effects of blast TBI on Aβ accumulation and microvascular changes, the current study exposed transgenic (Tg) APP/PS1 mice to bTBI at the age prior to their onset of Aβ deposits, and at 3 months and 12 months after injury evaluated Aβ deposition, pericyte numbers, astrocytic end-foot/capillary interactions, and capillary densities. We hypothesized that bTBI would produce chronic alterations in the capillary network, pericytes, and perivascular astrocytes in both genotypes, with Tg mice showing exacerbated changes and enhanced burden of Aβ pathology.
Methods
Experimental animal groups, genetics, and interventions
The Iowa City Department of Veterans Affairs and the University of Pittsburgh Institutional Animal Care and Use Committees approved all investigative procedures. The study used adult female transgenic APPswe, PS1ΔE9 (B6C3Tg(APPswe,PSEN1dE9)85Dbo/Mmjax, The Jackson Laboratory, MMRRC Stock No: 34829-JAX | APP/PS1) double mutant mice (referred to as APP/PS1 or Tg mice in this report) and their littermate B6C3F1/J (Stock No: 100010, Jackson) wild-type controls (Wt). The APP/PS1 mice harbor a ‘humanized’ APP gene driven by the prion promoter, with two mutations which are linked to familial AD [25, 90]. The Swedish mutation enhances β-secretase activity while the human presenilin gene with the 9th exon removed enhances gamma secretase processing of APP. These two mutations and constitutive APP overexpression result in enhanced amyloidogenic APP processing, overproduction of Aβ, and progressive Aβ deposition in brain parenchyma and vasculature. The APP/PS1 mice exhibit Aβ pathology at 4 to 6 months of age, and progressively develop more severe parenchymal and vascular amyloid deposits at older ages [59]. This pathology appears to be driven by overall increases in Aβ peptides as well as a shift in the Aβ40:Aβ42 ratio due to larger increases in Aβ42 compared to Aβ40 forms [59]. The Wt mice used in our study are the adult female founder mice of the APP/PS1 transgenics and harbor murine APP and PS1 genes that are driven by their endogenous promoters; these Wt mice do not have human APP and do not exhibit Aβ accumulation and therefore serve as the amyloid-negative control.
Each genotype was randomly stratified into two exposure groups: anesthesia plus blast exposure (referred to as bTBI) or anesthesia without bTBI exposure (referred to as Sham). Sham or bTBI exposure occurred at approximately 3 months of age (range: 76–106 days) for all mice included in the experiment. The mice were then sacrificed after either 3-months survival (around 6 months of age; range: 165–189 days) or 12-months survival (around 15 months of age; range: 442–455 days). We subsequently define age for each experimental group as the post-exposure “survival age” for the remainder of the present study. Accordingly, there were 24 mice in the study, with 3 mice analyzed in each of the 8 experimental groups (Tg bTBI and Tg Sham at 3-months and 12-months survival, and Wt bTBI and Wt Sham at 3-months and 12-months survival).
Blast injury induction
The blast pressure wave was generated in an enclosed blast chamber which was divided in two parts, with a 13-cm diameter opening between the chamber halves, as described previously [43, 44, 89]. A Mylar membrane (Mylar A, 0.00142 gauge; Country Plastics, Ames, IA) was placed over the opening between the two parts of the blast chamber. One side of the tank remained unpressurized and contained a padded polyvinyl chloride (PVC) protective restraint for positioning of an anesthetized mouse 30 cm from the Mylar membrane [43, 44, 89]. To create the blast wave, compressed room air was pumped into the pressurized side of the tank to 20 psi, the pressure at which the membrane ruptures. Using this model, a complex blast wave without a negative pressure component was produced as described [43] with the following characteristics: 137.8 ± 1.3 kPa (~ 20 psi) peak overpressure at the point of exposure (i.e., the head, the body of the animal was shielded from the blast wind) and a 10- to 15-ms blast duration [43, 89]. The pressure was calculated by using a sensor 1 cm in diameter placed directly below the head of the mouse [89]. Prior to blast wave induction, mice were anesthetized by an intraperitoneal (IP) injection of a combination of ketamine (0.03 mg/g body weight) and xylazine (0.005 mg/g body weight). Mice were positioned within the unpressurized half of the blast chamber with the left side of the head oriented toward the source of the blast wave. Only the head of the mouse was exposed to the blast wave, with the rest of the body shielded from the blast wind. The head of the mouse was unrestrained during the blast wave exposure, allowing for free rotation without contact with the cradle. Sham mice underwent the same anesthesia procedure and were placed in the blast chamber but did not receive a blast exposure. After bTBI exposure or Sham procedure, mice were placed on a heating pad to facilitate recovery from general anesthesia and to maintain a body temperature of 37 ± 0.5 °C. Xylazine anesthesia was reversed with yohimbine chloride (0.001 mg/g, IP) to facilitate the recovery from anesthesia. Mice in the Sham and bTBI groups received analgesic via subcutaneous injection (0.1 mL/20 g body weight) of buprenorphine (0.003 mg/mL) immediately after recovery from either procedure.
Tissue processing
At the end of the experiment, mice were anesthetized and killed by transcardial perfusion with 0.9% saline which also washed blood from the brain vasculature. After perfusion, the skull was opened to extract the brain, and the left hemisphere was placed into 4% paraformaldehyde fixative for 48 h at 4 °C after which it was infiltrated with graded sucrose solutions to allow for frozen sectioning. The brain hemisphere was then cut in the coronal plane using a freezing sledge microtome to create 40 µm-thick tissue sections. Sections were stored in a cryopreservation solution [131] at − 20 °C until use. Two coronal sections at the level of the dorsal hippocampus were selected randomly and included in the analysis of each mouse.
Immunohistochemistry and histology
Tissue sections were placed free-floating in 0.1 M potassium phosphate-buffered solution (KPBS), pH 7.4, for 24 h to wash off the cryoprotection solution. Sections were then washed in Tris-buffered saline solution (TBS) before incubation in 0.5% sodium borohydride made in TBS for 20 min. After again washing in TBS, the sections were incubated in 3% normal goat serum (NGS, Sigma, catalogue #G9023) in TBS at room temperature for 30 min, followed by two rinses in 1% NGS in TBS at room temperature for 10 min each. The sections were then incubated with primary antibodies specific to the experimental arm. Sections being analyzed for pericytes were incubated at 4 °C for 24 h in mouse monoclonal IgG raised against PDGFRβ (clone D-6, Santa Cruz, catalogue #sc-374573, lot #B2118) diluted 1:250 in 1% NGS made in TBS with Triton, and Lycopersicon Esculentum (Tomato) Lectin (LEL, Vector Laboratories, catalogue #DL-1177, lot #ZE0131) conjugated to a 594 nm-emitting fluorophore and diluted 1:200 in 1% NGS TBS with Triton [10, 103, 110]. Sections included in the astrocytic end-feet analysis were processed under the same conditions as the pericyte marker, but used a mouse monoclonal IgG generated against AQP4 diluted 1:1000 in 1% NGS made in TBS with Triton (clone 4/18, Santa Cruz, catalogue #sc-32739, lot #D2318), and Solanum Tuberosum (Potato) Lectin (STL, Vector, catalogue # B-1165-2, lot #ZC0519) conjugated to biotin and diluted 1:200 in 1% NGS made in TBS with Triton [46, 93]. After primary antibody/lectin cocktail incubation, sections were rinsed in TBS and incubated in goat anti-mouse IgG secondary antibodies (to mark primary antibodies) and streptavidin conjugated to a 488 nm-emitting Alexa fluorophore (streptavidin-Alexa488, to mark STL; Life Technologies, Eugene, OR, catalogue #S323356, lot #1902487) diluted 1:250 in 1% NGS made in TBS with Triton for 90 min at room temperature. The secondary antibodies included goat anti-mouse IgG conjugated to Alexa488 (pericyte studies, Jackson ImmunoResearch, catalogue #115-545-146, lot #138610) and goat anti-mouse IgG conjugated to a 594 nm-emitting Alexa fluorophore (astrocytic end-feet study, Jackson, catalogue #115-585-146, lot #136295) in 1% NGS made in TBS with Triton. Finally, the sections were rinsed in TBS, mounted on glass slides, and coverslipped using 4′,6-diamidino-2-phenylindole (DAPI) based medium (Vector, catalogue #H-1500) and stored in darkness at 4 °C until imaging [62]. The choice of fluorophore combinations for the multi-fluorescence studies was determined empirically. For immunohistochemical staining of Aβ, sections were rinsed in TBS and then incubated in 80% formic acid for two minutes. Endogenous peroxidase activity was inhibited by incubating the tissue in 1.5% H2O2 made in TBS for 30 min. Sections were then rinsed in TBS and incubated in 5% NGS made in TBS, rinsed, and incubated overnight in mouse monoclonal IgG raised against Aβ diluted 1:3000 in 1% goat serum made in TBS (clone 6E10, BioLegend catalogue #803002, lot #B198896) at 4 °C. Sections were then washed in TBS and incubated in biotinylated goat anti-mouse IgG (Jackson, catalogue #115-065-146, lot #128250) in 1% goat serum made in TBS for 1 h, washed in TBS, and reacted using the avidin–biotin method (ABC Elite kit, Vector Laboratories). The color reaction was developed using a 0.05% solution of nickel-enhanced 3,3-diaminobenzidine tetra-hydrochloride and 0.03% H2O2. Sections were then dehydrated in alcohols, cleared in xylenes, and coverslipped with Permount (Fisher). In all experiments using mouse monoclonal antibodies, control tissue sections were processed as described above but without the primary antibody (primary delete). No non-specific staining was observed in these control sections (not shown).
Cyano-PiB (2-(4′-methylaminophenyl)-6-cyanobenzothiazole) histofluorescence was performed as described previously [56]. Tissue sections were incubated in 10 μM cyano-PiB for 45 min in dark conditions at room temperature. Sections were then dipped three times in potassium phosphate buffer, incubated for 1 min in fresh potassium phosphate buffer, and coverslipped with Fluoromount-G (SouthernBiotech, Birmingham, AL, catalogue #0100-01).
Random sampling scheme of the cerebral cortex
Image stacks were acquired using an Olympus BX-51WI upright microscope equipped with an Olympus DSU spinning disk confocal, a super-corrected 65× Olympus Plan Apo N 1.42 numerical aperture oil immersion objective, a MBF CX9000 front mounted digital camera (MBF Bioscience), a BioPrecision2 XYZ motorized stage with linear XYZ encoders (Ludl), and filters for laser wavelength output of 405 nm (to visualize DAPI), 488 nm (to visualize the Alexa 488 fluorophore), and 594 nm (to visualize the Alexa 594 fluorophore). All z-stacks were obtained using an optical section separation interval (z-interval) of 0.25 µm. Two 25 µm-thick (post-processing shrinkage of original 40 µm-thick sections) sections were chosen per mouse using the previously described criteria. Using unbiased stereological principles on StereoInvestigator (MBF), five optical disectors per hemi-section were examined within the cortex using the central sulcus and rhinal fissure as dorsal and ventral borders, respectively.
Amyloid burden (% area)
Brain sections were processed for total Aβ load using Aβ immunohistochemistry as described above and imaged using light microscopy (see Fig. 1). A set of adjacent sections was processed for mature fibrillar amyloid deposits (avoiding the formic acid pre-treatment) using the fluorescent amyloid binding dye cyano-Pittsburgh compound-B (cyano-PiB) [56] and imaged using epifluorescence microscopy. Digital images of stained sections were acquired at 4× magnification to create a composite of the cerebral cortex and hippocampus for each section. Percent area coverage of labeled deposits was then calculated by thresholding positive signal and determining the percent of cerebral cortical tissue occupied by plaques using modified algorithms in the FIJI Open-Source image analysis software adapted from ImageJ [114].

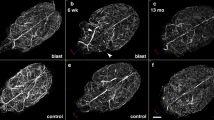
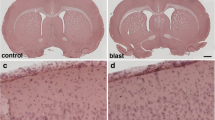
Cortical Aβ load after bTBI in APP/PS1 mice at 3-months and 12-months survival. A–E A whole hemisphere coronal section stained for Nissl substance (obtained from the Allen Brain Atlas) illustrates the cytoarchitecture of the cortical area targeted (A; purple cresyl violet signal) and Aβ-immunostained sections illustrating Aβ pathology load (B–E) in transgenic mice undergoing Sham or bTBI exposure and evaluated at 3-months survival (B and C, respectively) or at 12-months survival (D and E, respectively). F, G Graphs showing percent area coverage of Aβ-immunoreactive deposits (F) and cyano-PiB labeled amyloid pathology (G). Wild type mice are not shown as they were free of Aβ immunolabeling and cyano-PiB fluorescence. CTX, neocortex; HPC, hippocampus; THAL, thalamus. N = 3 mice/group. One-way ANOVA: F (3, 8) = 229.9, P < 0.0001 [Aβ (6E10) immunohistochemistry]; F (3, 8) = 94.10, P < 0.0001 (cyano-PiB). Brackets indicate differences of P < 0.05
Capillary density estimation
We defined cortical capillaries as microvessels measuring 10 µm in diameter or less. Our technical approach to measuring total number of capillaries [Ntot(cap)] is adapted from published reports [80, 94, 96]. The number of intersecting points (nodes) in each capillary network created a “node-segment network” where a single capillary was defined as the vessel segment between two nodes. Each node has a valence (n) defined by the number of vessel segments that intersect at a node. In each individual optical dissector, we first calculated the number of capillaries (Ncap) as a function of the number of nodes (Nnode) containing each valence (n with range 3–4), as described in Eq. 1:
To determine the Ntot(cap) in each optical dissector, we calculated the sum of all Ncap of each valence using correction factors as previously described [80, 94, 96], and detailed below. In the present study, we counted capillaries in two cerebral hemi-sections selected from a series through the rostral-caudal extent of the mouse brain. These selected hemi-sections represented a known fraction of total sections which we determined to be 1/53.5 (ssf) based on tissue thickness and the rostral-caudal extent of the mouse brain. Our randomly generated optical dissector consisted of a 143.95 µm × 111.52 µm counting frame with an area (Aframe) equal to 16,052.65 µm2. The average distance between positions of the optical dissector (Astep) varied per mouse, due to differences in brain volume, and ranged from 732,656 to 1,911,707 µm in both x and y dimensions. Thus, the counting frame also represented a known fraction of the total brain hemi-section area (asf), defined as (Aframe/Astep). Finally, we examined a known fraction of the total tissue thickness using the confocal microscope (tsf) defined as the average height of imaging depth per mouse (h) divided by the total tissue thickness (t) accounting for shrinkage with processing, where h varied per mouse (range: 9.61–23.70 µm) and t equals 25 µm. Adjusting for these variations in fraction of sections, area, and thickness, Ntot(cap) is presented in Eq. 2:
Pericyte estimation
A similar quantification method was used to estimate the total number of pericytes using the optical dissector probe, adapted from a previous study [94], in two cerebral hemi-sections. To identify pericytes situated amongst the capillary basement membrane, we performed dual-labeling using fluorophore-conjugated LEL and the pericyte-specific PDGFRβ antibody visualized using a fluorophore-conjugated secondary antibody. As seen in Fig. 3, the clear bumps along the LEL-labeled basement membrane, coined “ghost-like cell bodies” [94], co-localize with PDGFRβ and DAPI. Using our optical dissector, we counted these “ghost-bodies” to estimate the total number of pericytes (see Fig. 3). Using the same parameters as the capillary analysis, total numbers of pericytes (Ntot peri) were calculated using the sum of pericytes in each optical dissector (Q) as listed in Eq. 3:
Astrocytic end-feet contacts estimation
To estimate the contact surface of astrocytic end-feet interfacing with the endothelial basement membrane, we identified percentage overlap of AQP4-immunoreactive end-feet and STL immunosignal as measured with 2D voxels corresponding to fluorescence intensities in FIJI. We first masked the surrounding tissue around the capillary vessel to create several “iso-surfaces” of capillary per optical dissector [40]. This allowed for detection of AQP4-labeled interaction at the level of the capillary iso-surface which could be quantified as both red- and green-fluorescent signal. Then, we took the percentage of AQP4-immunoreactive astrocytic end-feet as a function of STL (vessel) co-localizing within the given unmasked iso-surface, and created an average surface area overlap using a 2D reconstruction, as described in Eq. 4:
Statistical analysis
We report all data as mean ± SEM. Excel (Microsoft) and Prism software (GraphPad, San Diego, CA) were used in generating statistics and graphs for all data. The mean value per randomly sampled optical dissector was used in all statistical analyses. One-way ANOVA with Tukey’s post hoc testing was used to assess the plaque load data. Two-way ANOVA with Sidak’s multiple test comparison were used to analyze the astrocyte, capillary, and pericyte data. Using an alpha of 0.05, results were considered significant when p < 0.05.
Results
Cortical Aβ plaque load in Tg mice after bTBI or Sham surgery
Tg-bTBI and Tg-Sham groups had statistically significantly higher Aβ pathology loads at 12-months survival (i.e., 15-month-old mice) compared to 3-months survival (i.e., 6-month-old mice) when assessed by Aβ immunohistochemistry (Fig. 1A–F) or cyano-PiB histofluorescence stain of fibrillar Aβ deposits (Fig. 1G). No statistically significant differences in Aβ-immunoreactive plaque load in the cerebral cortex were detected between the two Tg groups at 3-months survival (Tg-bTBI/3-months: 0.2706 ± 0.0757; Tg-Sham/3-months: 0.1688 ± 0.0850) or at 12-months survival (Tg-bTBI/12-months: 5.8820 ± 0.3933; Tg-Sham/12-months: 5.4390 ± 0.5910) (Fig. 1F). Similarly, no statistically significant differences in cyano-PiB positive plaque load in the cerebral cortex were detected between the Tg-bTBI and Tg-Sham groups at 3-months survival (Tg-bTBI/3-months: 0.2087 ± 0.0326; Tg-Sham/3-months: 0.1352 ± 0.0501) or at 12-months survival (Tg-bTBI/12-months: 5.0539 ± 0.6457; Tg-Sham/12-months: 4.4579 ± 0.6931) (Fig. 1G).
Aβ-immunoreactive and cyano-PiB plaque loads in the hippocampus were higher at 12-months survival compared to 3-months survival (Aβ: Tg-Sham/3-months: 0.4797 ± 0.1040; Tg-Sham/12-months: 5.0546 ± 1.2075; Tg-bTBI/3-months: 0.4285 ± 0.1665; Tg-bTBI/12-months: 4.8861 ± 0.7188; cyano-PiB: Tg-Sham/3-months: 0.4279 ± 0.2631; Tg-Sham/12 months: 4.7037 ± 0.9840; Tg-bTBI/3-months: 0.5441 ± 0.3897; Tg-bTBI/12 months: 4.5529 ± 0.8166). No statistically significant differences in hippocampal Aβ-immunoreactive or cyano-PiB plaque loads were detected between the Tg-bTBI and Tg-Sham groups at either survival interval.
No Aβ immunoreactive or cyano-PiB fluorescent plaques were detected in brain tissue sections from Wt mice exposed to bTBI or to the Sham procedure.
Astrocyte AQP4-immunoreactive end-foot contacts with microvasculature in Wt and Tg mice after bTBI or Sham exposure
AQP4 immunofluorescence was observed primarily in contact with capillary endothelium, indicating its preferential localization to astrocytic end-feet (Fig. 2A–F). Quantification of the interface between AQP4-immunofluorescence at the level of capillary endothelium showed statistically significantly lower values in the Tg-bTBI group compared to the Tg-Sham group at 3-months survival (Fig. 2G) but not at 12-months survival. We did not observe differences in AQP4 among experimental groups of Wt mice at either survival interval (Fig. 2G).

Reduced perivascular AQP4-expressing astrocytic end-feet after bTBI in APP/PS1 mice. A, B Maximum intensity projection of a confocal image stack demonstrating aquaporin-4 immunofluorescence signal (AQP4, red) contacting capillaries labeled with a fluorescent lectin (STL, green) in representative Tg-Sham (A–C) and Tg-bTBI (D–F) mice at 3-months survival (n = 3 mice/group). Merged images (C, F) have an overlay of DAPI-labeled nuclei (blue). G Quantification of percent area coverage of sampled AQP4 immunofluorescence interfacing with the STL-labeled endothelium. N = 3 mice/group. Two-way ANOVA: F (1, 16) = 5.816, P = 0.0343. Brackets indicate differences of P < 0.05. Scale bar = 20 µm (A–F)
Numbers of pericytes in Wt and Tg mice after bTBI or Sham exposure
Figure 3 illustrates representative images of immunofluorescently labeled pericytes (anti-PDGFRβ, green fluorescence, panels A, D1, E1, D3, E3) located within bump-like protrusions of the basement membrane (LEL, red fluorescence, B, D2, E2, D3 E3). There were no injury- or genotype-related effects on pericyte numbers at 3-months survival (Fig. 3F).

Blast TBI affects pericytes and capillaries differently in APP/PS1 mice and wild type mice. A–E Maximum intensity projection of a confocal image stack demonstrating PDGFR-β immunofluorescent signal (A, D1, E1, green) associated with capillaries labeled with a fluorescent lectin (B, D2, E2, LEL, red). Merged pairs of images with DAPI-stained nuclei are shown in panels C, D3, and E3. Asterisks in panels A–C mark the position of two pericytes shown at higher magnification in panels D1–D3 and E1–E3 where arrows demarcate the area occupied by each pericyte. F–H Graphs showing total numbers of pericytes (F), pericytes per capillary length (G), and capillary densities (H) in experimental groups. N = 3 mice/group. Two-way ANOVA (pericytes): F (3, 16) = 22.15, p < 0.0001 (row). Two-way ANOVA (capillaries): F (3, 16) = 38.98, P < 0.0001 (row); F (1, 16) = 3.299; P = 0.0297 (column); F (3, 16) = 8.448, P = 0.0014 (interaction). Brackets indicate differences of P < 0.05. Scale bar = 25 µm (A-C) 5 µm (D1-D3, E1-E3)
The Wt-bTBI group at 12-months survival had statistically significantly lower numbers of pericytes compared to the Wt-bTBI group at 3-months survival (Fig. 3F). In Tg mice, pericyte numbers were not different between the Tg-bTBI group and the Tg-Sham group at 3-months or 12-months survival. There were no differences when the Tg-TBI and Tg-Sham groups at 3-months survival were compared to the same groups at 12-months survival (Fig. 3F). However, significantly greater numbers of pericytes were observed in Tg mice compared to Wt mice for both the bTBI and Sham groups at 12-months survival (Fig. 3F).
Pericytes relative to capillary density in Wt and Tg mice after bTBI or Sham exposure
Figure 3G shows the total number of pericytes relative to capillary density within each experimental group. No differences in pericyte numbers were observed between experimental groups when adjusted for capillary densities (Fig. 3G).
Capillary densities in Wt and Tg mice after bTBI or Sham exposure
In Wt mice, the bTBI and Sham groups did not differ by capillary density at 3-months survival or at 12-months survival (Fig. 3H). The Wt-bTBI group at the 12-months survival had statistically significantly lower capillary density when compared to the Wt-bTBI group at 3-months survival (Fig. 3H).
In Tg mice, capillary density at 12-months survival was statistically significantly higher in the bTBI group than the Sham group (Fig. 3H). The Tg-bTBI group had significantly higher capillary density at 12-months survival than at 3-months survival (Fig. 3H).
Numbers of capillaries did not differ between genotypes at 3-months survival (Fig. 3H). At 12-months survival, both the Tg-bTBI group and the Tg-Sham group had statistically significantly higher numbers of capillaries compared to the Wt-bTBI group and the Wt-Sham group, respectively (Fig. 3H).
Discussion
This study demonstrates chronic changes in the cerebral cortical microvasculature after a single bTBI (~ 138 kPa/~ 20 psi) in adult female transgenic (Tg) APP/PS1 human Aβ over-producing mice compared to non-transgenic wild type (Wt) mice which have physiological levels of murine Aβ. We observed that compared to the Sham procedure, bTBI resulted in lower AQP4-expression at contact areas between astrocyte end-feet and the capillary network in the cerebral cortex of Tg, but not Wt, mice at 3-months survival. At 12-months survival, Tg mice exposed to bTBI had greater capillary densities in the cerebral cortex than Tg mice exposed to the Sham procedure (12-months survival) as well as Tg mice exposed to bTBI and assessed at 3-months survival. In contrast, Wt mice exposed to bTBI had statistically significant lower capillary densities at 12-months survival than at 3-months survival. Changes in microvascular densities paralleled changes in numbers of pericytes in both Tg and Wt mice, resulting in stable numbers of pericytes per capillary density at both chronic survival intervals after bTBI or Sham procedure.
Our previous study found that APP/PS1 Tg mice subjected to a bTBI at 2–3 months of age and assessed after a 2-months survival had no significant changes in cortical Aβ plaque load relative to their Sham-exposed Tg counterparts [44]. Similarly, in the current study using the same bTBI injury procedure, we did not detect statistically significant changes in Aβ load after a 3-months survival in APP/PS1 Tg-bTBI mice compared to Tg-Sham mice. These results are in agreement with a recent report that Aβ plaque load is not significantly different in 5-months-old APP/PS1 Tg mice exposed to repetitive low-level bTBI compared to sham APP/PS1 Tg mice when assessed after 3–4 months survival [100]. We extend these findings by showing that at a longer chronic (12-months) survival interval, bTBI has no significant effect on Aβ load in APP/PS1 Tg mice. Interestingly, a study of bTBI in wild type rodents with acute (24 h and 1 week) survival reported no changes in endogenous Aβ concentration when the level of blast exposure was similar to our study (116.7 kPa), while reduced Aβ42 concentrations were detected when using lower level (36.6 kPa and 74.5 kPa) blast exposures [27]. The same group reported that in APP/PS1 Tg mice, low-level (34.5 kPa) blast exposure had no effect on soluble and insoluble Aβ42 at 1 week survival while Aβ42 levels were reduced at 1-month survival [100]. Collectively, these studies indicate that the level/severity of bTBI, in addition to other variables such as the number and frequency of blast exposures and the length of survival, may differentially influence Aβ concentrations in brain. However, the progressive transgene-driven Aβ deposition in Tg mice could mask the potentially subtle injury-related changes in cortical Aβ deposits. It will be important that future studies examine time points between 3 and 12 months.
While bTBI significantly altered AQP4 expression at the astrocytic end-foot/endothelium interface in Tg mice, we did not observe such changes in the age-matched Wt mice undergoing the same injury protocol. This observation suggests that high concentrations of human Aβ present in the Tg mouse brain confers susceptibility for chronic dysfunction of perivascular astrocytes in the context of bTBI exposure. Astrocyte dysfunction has been reported in multiple models of CNS insult. For example, after TBI, ischemia, and in neurodegenerative disease, astrocytes enter a hypertrophic state characteristic of astrogliosis [31, 117], and this process can contribute to impairments in gliovascular connections, BBB permeability, and cerebral blood circulation [21, 97, 98]. Several studies reported adverse effects of bTBI on perivascular astrocytes in adult male rats. Acutely after single or repetitive bTBI using an open-ended shock tube model (peak pressure of 150 kPa), Uzunalli and colleagues reported reactive astrogliosis accompanied by early signs of astrocytic end-foot displacement from blood vessels, and a trend for reduced AQP4 expression [127]. Another bTBI study applied a series of three low-energy blast exposures of 74.5 kPa (10.8 psi) intensity and reported reduced levels of vascular-associated astrocytic GFAP, coupled with swelling of perivascular astrocytes and degeneration of their end-feet, 6-weeks after the last impact [36]. Our current data suggest that in wild type animals which do not accumulate human Aβ, astrocyte pathology, reported previously at acute and subacute time points after TBI, may resolve over chronic time intervals after injury. Alternatively, sex or species differences may account for discrepancies between studies. When bTBI occurs in individuals with ongoing Aβ accumulation as a predisposition for developing AD-like amyloid pathology (modeled in adult Tg APP/PS1 mice exposed to bTBI), perivascular astrocyte dysfunction is either more severe or damaged astrocytes undergo slower recovery relative to astrocytes in Wt mice. The consequences of chronic astrocyte changes (pathological changes and recovery) on microvascular circulation and BBB permeability after bTBI require further exploration.
The current study finding that the longer bTBI survival in Wt mice (12-months compared to 3-months) is associated with lower numbers of PDGFR-β immunolabeled pericytes could reflect a progressive loss of pericytes lengthily after bTBI. Loss of PDGFR-β expression was reported over 24 h after a bTBI in rats [2]. However, single or repetitive exposure of rats to bTBI did not affect numbers of PDGFR-β-immunoreactive pericytes at 1–3 days survival [127]. Using CD-13 and desmin immunofluorescence as alternative markers of pericytes, the same study confirmed stability of pericyte numbers after a single bTBI exposure but detected a transient increase in pericyte numbers after repetitive bTBI [127]. These data indicate that different models of bTBI and use of different markers of pericytes can result in different and even opposite observations when pericyte numbers are assessed acutely after bTBI in rodents. In other models of TBI, loss of pericytes, a disrupted (permeable) BBB, and gliosis within days after TBI (i.e., acutely) were reported in wild type C57Bl/6 mice exposed to closed head mild controlled cortical impact (CCI) [133] and in rats with fluid percussion injury to the exposed brain dura [12]. Another study, using the model of CCI injury over the exposed brain dura of wild type C57Bl/6 mice, reported loss of pericytes within 6–12 h after injury followed by a recovery and proliferation of pericytes by 5 days after injury [135]. Loss or dysfunction of pericytes can affect microvascular flow [15] and BBB permeability [11]. It has been hypothesized that pericyte loss “would disrupt optimized flow patterns and cause imbalanced perfusion and oxygen delivery” [45]. This imbalance could be exacerbated by injury- or disease-induced elevations in reactive oxygen species, which cause pericyte contraction [45], or by exposure to high levels of human Aβ forms [45] which has been associated with pericyte contraction in AD and mouse models of the disease [8, 70, 95]. In regard to BBB permeability, pericytes influence the transcription of tight junction proteins by endothelial cells [11, 26]. Changes in BBB permeability coinciding with loss of expression of the tight junction protein claudin-5 in the hippocampus of adult male C57Bl/6J mice were reported up to 72 h after a 20-psi bTBI [78], and loss of occludin acutely was reported in rat models of single bTBI [63] and repeated bTBI [48]. Another bTBI study in rat [2] reported diminished expression of tight junction proteins in parallel with loss of PDGFR-β-immunoreactivity. Though analysis of tight junction proteins was outside the scope of the current study, whether such changes persist lengthily and are influenced by changes in pericytes are important avenues of investigation.
In addition to impairing the NVU, loss or dysfunction of pericytes after bTBI exposure could also influence Aβ pathology. For example, when the Tg APPsw mouse model of human Aβ peptide overproduction was crossed with pericyte-deficient PDGFR-β+/− mice, accelerated Aβ accumulation in brain parenchyma and in blood vessel walls was observed [111]. We did not see statistically significant effects of bTBI on pericyte numbers in our Tg APP/PS1 mice compared to Tg-Sham mice. This was surprising when considering previous reports of pericyte depletion, dysfunction, and/or disrupted pericyte-endothelial cell signaling with exposure to soluble toxic Aβ peptides due to their impaired clearance and accumulation [15, 19, 24, 28, 111]. In relation to Aβ pathology in postmortem AD brains, several studies reported reduced numbers of pericytes and pericyte coverage of capillaries which corresponded to greater Aβ deposition and BBB impairment in these cases [42, 87, 115], while others reported no significant changes in pericyte numbers [32, 33, 119]. The lack of a difference in the number of pericytes per capillary density when comparing bTBI and Sham groups of our Tg mice at 12-months survival was likely due to higher pericyte frequencies and capillary densities, as both could be stimulated by Aβ. Proliferation of pericytes contributes to increased angiogenesis [106] and, alternatively, angiogenesis could influence recruitment of pericytes to newly formed capillaries [18]. It will be important to determine if these cells function normally, particularly in a brain environment with high concentrations of Aβ which causes capillary constriction at pericyte locations [95].
Our observations of higher capillary densities at 12-months survival than 3-months survival in Tg mice exposed to bTBI, and no net change in pericyte numbers per capillary density are reminiscent of the results from stereological analyses in the frontal cortex of AD patients, reporting a 24% increase in capillary density and no change in numbers of pericytes per capillary segment [33]. Other studies also reported that Aβ pathology is associated with higher microvascular density in AD brain [14, 22, 33, 85, 101, 107] and in aged transgenic mice with human tau-overexpression [17]. This could be due, in part, to angiogenesis which plays a role in compensatory vascular remodeling after cerebral hypoperfusion and hypoxia and/or an inflammatory process activated by AD pathology [102, 128]. Angiogenic signaling factors are increased in brains of people with AD and vascular dementia [17, 102, 125], and multiple serum biomarkers of vascular functions including angiogenesis (vascular endothelial growth factor; von Willebrand factor) are elevated after experimental TBI [7, 73, 112]. Thus, angiogenesis may underlie the increased microvascular densities observed in Tg mice in our bTBI study and reported previously in AD, but this change may not be successful in countering long-term brain hypoxia because, similar to the aftermath of stroke and in AD, newly formed microvascular networks can have impaired vessel reactivity and reduced lumen diameter which manifests as greater vasoconstriction, thus limiting oxygen delivery [22, 125]. From a therapeutic standpoint, stimulation of angiogenesis by brain implantation of human umbilical cord perivascular cells after fluid percussion TBI in rats normalized (the reduced) capillary densities and pericyte coverage of capillaries, resulting in lower BBB permeability and less axonal injury [12]. In contrast to the findings in Tg mice, we observed that Wt mice with bTBI had lower capillary density at 12-months survival than after 3-months survival, possibly reflecting an effect of age on capillary density in Wt mice as has been reported previously [113], or lack of high Aβ concentrations which could stimulate angiogenesis and pericyte proliferation in Tg mice.
Some limitations of the current study should be considered. The number of mice per experimental group was small, however we used an unbiased stereological approach with five optical dissectors per brain hemi-section and, overall, 10 sites per mouse and did not observe large variations within groups. Nevertheless, we recognize that the study was not sufficiently powered to draw definitive conclusions. The high complexity of the NVU requires that future studies of bTBI in mouse models of human neurogenerative diseases analyze additional components of the microvasculature and BBB including the endothelial tight junction proteins. TBI can impair the ability of astrocytes and pericytes to crosstalk with endothelial cells and this can lead to loss of tight junction proteins and further impairment of the BBB permeability and capillary flow [10, 11, 54, 121]. Studies of human Aβ-overexpressing transgenic mice with longer survival intervals after bTBI will further define chronic changes in the microvasculature, permeability of the BBB, and cerebral blood circulation in relation to progressive accumulation of Aβ as a risk of developing chronic neurodegeneration.
Conclusions
Human and experimental studies of brain injury report changes in BBB permeability, induction of perivascular inflammation, altered endothelial-immune cell interactions, and dysfunction of endothelial cells and pericytes [4]. While most of these changes are reported within the acute and subacute period, the current study contributes to a better understanding of the chronic phase of bTBI in relation to a risk of developing sustained cerebral vascular dysfunction and AD-related Aβ pathology. We demonstrate that at the level of the microvasculature, bTBI results in significant long-term changes (i.e. loss of pericytes and lower capillary density) in Wt mice while more complex detrimental and potentially compensatory chronic changes occur in a transgenic mouse model with AD-relevant brain accumulation of human Aβ. In the latter model, we observed that bTBI resulted in chronic loss of astrocytic AQP4 expression at the level of microvessels which could be due to AQP4 redistribution from end-feet to the soma, retraction of astrocytic end-feet, or lack of coverage of newly formed vessels. Our observation that chronic bTBI in transgenic AD mice is associated with increased capillary density, with no significant change in numbers of pericytes relative to capillary density, is similar to what has been reported in brains of AD patients [33]. Further efforts to clarify the mechanisms responsible for chronic changes in components of the microvasculature after bTBI in relation to increased Aβ accumulation may help identify areas for early or delayed therapeutic intervention.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- Aβ:
-
Amyloid beta
- AD:
-
Alzheimer’s disease
- APP:
-
Amyloid precursor protein
- AQP4:
-
Aquaporin-4
- BBB:
-
Blood–brain barrier
- bTBI:
-
Blast-induced traumatic brain injury
- CTE:
-
Chronic traumatic encephalopathy
- IP:
-
Intraperitoneal
- LEL:
-
Lycopersicon Esculentum
- NVU:
-
Neurovascular unit
- PDGF:
-
Platelet derived growth factor
- PDGFRβ:
-
Platelet derived growth factor receptor beta
- PiB:
-
Pittsburgh compound B
- PS1:
-
Presenilin 1
- STL:
-
Solanum Tuberosum
- Tg:
-
Transgenic
- TBI:
-
Traumatic brain injury
- Wt:
-
Wild type
References
Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood–brain barrier. Nat Rev Neurosci. 2006;7:41–53.
Abdul-Muneer PM, Schuetz H, Wang F, Skotak M, Jones J, Gorantla S, Zimmerman MC, Chandra N, Haorah J. Induction of oxidative and nitrosative damage leads to cerebrovascular inflammation in an animal model of mild traumatic brain injury induced by primary blast. Free Radic Biol Med. 2013;60:282–91.
Abrahamson EE, Foley LM, Dekosky ST, Hitchens TK, Ho C, Kochanek PM, Ikonomovic MD. Cerebral blood flow changes after brain injury in human amyloid-beta knock-in mice. J Cereb Blood Flow Metab. 2013;33:826–33.
Abrahamson EE, Ikonomovic MD. Brain injury-induced dysfunction of the blood brain barrier as a risk for dementia. Exp Neurol. 2020;328: 113257.
Abutarboush R, Gu M, Kawoos U, Mullah SH, Chen Y, Goodrich SY, Lashof-Sullivan M, McCarron RM, Statz JK, Bell RS, Stone JR, Ahlers ST. Exposure to blast overpressure impairs cerebral microvascular responses and alters vascular and astrocytic structure. J Neurotrauma. 2019;36:3138–57.
Acioglu C, Li L, Elkabes S. Contribution of astrocytes to neuropathology of neurodegenerative diseases. Brain Res. 2021;1758: 147291.
Ahmed F, Plantman S, Cernak I, Agoston DV. The temporal pattern of changes in serum biomarker levels reveals complex and dynamically changing pathologies after exposure to a single low-intensity blast in mice. Front Neurol. 2015;6:114.
Alcendor DJ. Interactions between amyloid-beta proteins and human brain pericytes: implications for the pathobiology of Alzheimer’s disease. J Clin Med. 2020;9:1490.
Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–23.
Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215.
Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, Johansson BR, Betsholtz C. Pericytes regulate the blood–brain barrier. Nature. 2010;468:557–61.
Barretto TA, Park E, Telliyan T, Liu E, Gallagher D, Librach C, Baker A. Vascular dysfunction after modeled traumatic brain injury is preserved with administration of umbilical cord derived mesenchymal stromal cells and is associated with modulation of the angiogenic response. J Neurotrauma. 2021;38(19):2747–62.
Bauman RA, Ling G, Tong L, Januszkiewicz A, Agoston D, Delanerolle N, Kim Y, Ritzel D, Bell R, Ecklund J, Armonda R, Bandak F, Parks S. An introductory characterization of a combat-casualty-care relevant swine model of closed head injury resulting from exposure to explosive blast. J Neurotrauma. 2009;26:841–60.
Bell MA, Ball MJ. Morphometric comparison of hippocampal microvasculature in ageing and demented people: diameters and densities. Acta Neuropathol. 1981;53:299–318.
Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic BV. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron. 2010;68:409–27.
Bennett HC, Kim Y. Pericytes across the lifetime in the central nervous system. Front Cell Neurosci. 2021;15: 627291.
Bennett RE, Robbins AB, Hu M, Cao X, Betensky RA, Clark T, Das S, Hyman BT. Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer’s disease. Proc Natl Acad Sci USA. 2018;115:E1289–98.
Berthiaume AA, Grant RI, McDowell KP, Underly RG, Hartmann DA, Levy M, Bhat NR, Shih AY. Dynamic remodeling of pericytes in vivo maintains capillary coverage in the adult mouse brain. Cell Rep. 2018;22:8–16.
Bhowmick S, D’Mello V, Caruso D, Wallerstein A, Abdul-Muneer PM. Impairment of pericyte-endothelium crosstalk leads to blood–brain barrier dysfunction following traumatic brain injury. Exp Neurol. 2019;317:260–70.
Bird SM, Sohrabi HR, Sutton TA, Weinborn M, Rainey-Smith SR, Brown B, Patterson L, Taddei K, Gupta V, Carruthers M, Lenzo N, Knuckey N, Bucks RS, Verdile G, Martins RN. Cerebral amyloid-beta accumulation and deposition following traumatic brain injury—a narrative review and meta-analysis of animal studies. Neurosci Biobehav Rev. 2016;64:215–28.
Burda JE, Bernstein AM, Sofroniew MV. Astrocyte roles in traumatic brain injury. Exp Neurol. 2016;275(Pt 3):305–15.
Burke MJ, Nelson L, Slade JY, Oakley AE, Khundakar AA, Kalaria RN. Morphometry of the hippocampal microvasculature in post-stroke and age-related dementias. Neuropathol Appl Neurobiol. 2014;40:284–95.
Cheng J, Korte N, Nortley R, Sethi H, Tang Y, Attwell D. Targeting pericytes for therapeutic approaches to neurological disorders. Acta Neuropathol. 2018;136:507–23.
Choi YK, Maki T, Mandeville ET, Koh SH, Hayakawa K, Arai K, Kim YM, Whalen MJ, Xing C, Wang X, Kim KW, Lo EH. Dual effects of carbon monoxide on pericytes and neurogenesis in traumatic brain injury. Nat Med. 2016;22:1335–41.
Crook R, Verkkoniemi A, Perez-Tur J, Mehta N, Baker M, Houlden H, Farrer M, Hutton M, Lincoln S, Hardy J, Gwinn K, Somer M, Paetau A, Kalimo H, Ylikoski R, Poyhonen M, Kucera S, Haltia M. A variant of Alzheimer’s disease with spastic paraparesis and unusual plaques due to deletion of exon 9 of presenilin 1. Nat Med. 1998;4:452–5.
Daneman R, Prat A. The blood–brain barrier. Cold Spring Harb Perspect Biol. 2015;7: a020412.
De Gasperi R, Gama Sosa MA, Kim SH, Steele JW, Shaughness MC, Maudlin-Jeronimo E, Hall AA, Dekosky ST, McCarron RM, Nambiar MP, Gandy S, Ahlers ST, Elder GA. Acute blast injury reduces brain abeta in two rodent species. Front Neurol. 2012;3:177.
DeGracia DJ, Kreipke CW, Kayali FM, Rafols JA. Brain endothelial HSP-70 stress response coincides with endothelial and pericyte death after brain trauma. Neurol Res. 2007;29:356–61.
DeKosky ST, Abrahamson EE, Ciallella JR, Paljug WR, Wisniewski SR, Clark RS, Ikonomovic MD. Association of increased cortical soluble abeta42 levels with diffuse plaques after severe brain injury in humans. Arch Neurol. 2007;64:541–4.
Elder GA, Gama Sosa MA, De Gasperi R, Stone JR, Dickstein DL, Haghighi F, Hof PR, Ahlers ST. Vascular and inflammatory factors in the pathophysiology of blast-induced brain injury. Front Neurol. 2015;6:48.
Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, Steinhauser C, Volterra A, Carmignoto G, Agarwal A, Allen NJ, Araque A, Barbeito L, Barzilai A, Bergles DE, Bonvento G, Butt AM, Chen WT, Cohen-Salmon M, Cunningham C, Deneen B, De Strooper B, Diaz-Castro B, Farina C, Freeman M, Gallo V, Goldman JE, Goldman SA, Gotz M, Gutierrez A, Haydon PG, Heiland DH, Hol EM, Holt MG, Iino M, Kastanenka KV, Kettenmann H, Khakh BS, Koizumi S, Lee CJ, Liddelow SA, MacVicar BA, Magistretti P, Messing A, Mishra A, Molofsky AV, Murai KK, Norris CM, Okada S, Oliet SHR, Oliveira JF, Panatier A, Parpura V, Pekna M, Pekny M, Pellerin L, Perea G, Perez-Nievas BG, Pfrieger FW, Poskanzer KE, Quintana FJ, Ransohoff RM, Riquelme-Perez M, Robel S, Rose CR, Rothstein JD, Rouach N, Rowitch DH, Semyanov A, Sirko S, Sontheimer H, Swanson RA, Vitorica J, Wanner IB, Wood LB, Wu J, Zheng B, Zimmer ER, Zorec R, Sofroniew MV, Verkhratsky A. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. 2021;24:312–25.
Farkas E, De Jong GI, de Vos RA, Jansen Steur EN, Luiten PG. Pathological features of cerebral cortical capillaries are doubled in Alzheimer’s disease and Parkinson’s disease. Acta Neuropathol. 2000;100:395–402.
Fernandez-Klett F, Brandt L, Fernandez-Zapata C, Abuelnor B, Middeldorp J, Sluijs JA, Curtis M, Faull R, Harris LW, Bahn S, Hol EM, Priller J. Denser brain capillary network with preserved pericytes in Alzheimer’s disease. Brain Pathol. 2020;30:1071–86.
Franzblau M, Gonzales-Portillo C, Gonzales-Portillo GS, Diamandis T, Borlongan MC, Tajiri N, Borlongan CV. Vascular damage: a persisting pathology common to Alzheimer’s disease and traumatic brain injury. Med Hypotheses. 2013;81:842–5.
Gama Sosa MA, De Gasperi R, Janssen PL, Yuk FJ, Anazodo PC, Pricop PE, Paulino AJ, Wicinski B, Shaughness MC, Maudlin-Jeronimo E, Hall AA, Dickstein DL, McCarron RM, Chavko M, Hof PR, Ahlers ST, Elder GA. Selective vulnerability of the cerebral vasculature to blast injury in a rat model of mild traumatic brain injury. Acta Neuropathol Commun. 2014;2:67.
Gama Sosa MA, De Gasperi R, Perez Garcia GS, Perez GM, Searcy C, Vargas D, Spencer A, Janssen PL, Tschiffely AE, McCarron RM, Ache B, Manoharan R, Janssen WG, Tappan SJ, Hanson RW, Gandy S, Hof PR, Ahlers ST, Elder GA. Low-level blast exposure disrupts gliovascular and neurovascular connections and induces a chronic vascular pathology in rat brain. Acta Neuropathol Commun. 2019;7:6.
Gardner RC, Byers AL, Barnes DE, Li Y, Boscardin J, Yaffe K. Mild TBI and risk of Parkinson disease: a chronic effects of neurotrauma consortium study. Neurology. 2018;90:e1771–9.
Garman RH, Jenkins LW, Switzer RC 3rd, Bauman RA, Tong LC, Swauger PV, Parks SA, Ritzel DV, Dixon CE, Clark RS, Bayir H, Kagan V, Jackson EK, Kochanek PM. Blast exposure in rats with body shielding is characterized primarily by diffuse axonal injury. J Neurotrauma. 2011;28:947–59.
Ghai V, Fallen S, Baxter D, Scherler K, Kim TK, Zhou Y, Meabon JS, Logsdon AF, Banks WA, Schindler AG, Cook DG, Peskind ER, Lee I, Wang K. Alterations in plasma microRNA and protein levels in war veterans with chronic mild traumatic brain injury. J Neurotrauma. 2020;37:1418–30.
Giuliani A, Sivilia S, Baldassarro VA, Gusciglio M, Lorenzini L, Sannia M, Calza L, Giardino L. Age-related changes of the neurovascular unit in the cerebral cortex of Alzheimer disease mouse models: a neuroanatomical and molecular study. J Neuropathol Exp Neurol. 2019;78:101–12.
Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, O’Farrell FM, Buchan AM, Lauritzen M, Attwell D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature. 2014;508:55–60.
Halliday MR, Rege SV, Ma Q, Zhao Z, Miller CA, Winkler EA, Zlokovic BV. Accelerated pericyte degeneration and blood–brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J Cereb Blood Flow Metab. 2016;36:216–27.
Harper MM. Author response: pressure wave dosimetry for “retinal ganglion cell damage in an experimental rodent model of blast-mediated traumatic brain injury.” Invest Ophthalmol Vis Sci. 2014;55:1350–1.
Harper MM, Hedberg-Buenz A, Herlein J, Abrahamson EE, Anderson MG, Kuehn MH, Kardon RH, Poolman P, Ikonomovic MD. Blast-mediated traumatic brain injury exacerbates retinal damage and amyloidosis in the APPswePSENd19e mouse model of Alzheimer’s disease. Invest Ophthalmol Vis Sci. 2019;60:2716–25.
Hartmann DA, Berthiaume AA, Grant RI, Harrill SA, Koski T, Tieu T, McDowell KP, Faino AV, Kelly AL, Shih AY. Brain capillary pericytes exert a substantial but slow influence on blood flow. Nat Neurosci. 2021;24:633–45.
Hawkes CA, Michalski D, Anders R, Nissel S, Grosche J, Bechmann I, Carare RO, Hartig W. Stroke-induced opposite and age-dependent changes of vessel-associated markers in co-morbid transgenic mice with Alzheimer-like alterations. Exp Neurol. 2013;250:270–81.
Heyburn L, Abutarboush R, Goodrich S, Urioste R, Batuure A, Statz J, Wilder D, Ahlers ST, Long JB, Sajja V. Repeated low-level blast overpressure leads to endovascular disruption and alterations in TDP-43 and Piezo2 in a rat model of blast TBI. Front Neurol. 2019;10:766.
Heyburn L, Abutarboush R, Goodrich S, Urioste R, Batuure A, Wheel J, Wilder DM, Arun P, Ahlers ST, Long JB, Sajja VS. Repeated low-level blast acutely alters brain cytokines, neurovascular proteins, mechanotransduction, and neurodegenerative markers in a rat model. Front Cell Neurosci. 2021;15: 636707.
Huber BR, Meabon JS, Hoffer ZS, Zhang J, Hoekstra JG, Pagulayan KF, McMillan PJ, Mayer CL, Banks WA, Kraemer BC, Raskind MA, McGavern DB, Peskind ER, Cook DG. Blast exposure causes dynamic microglial/macrophage responses and microdomains of brain microvessel dysfunction. Neuroscience. 2016;319:206–20.
Hue CD, Cao S, Dale Bass CR, Meaney DF, Morrison B 3rd. Repeated primary blast injury causes delayed recovery, but not additive disruption, in an in vitro blood-brain barrier model. J Neurotrauma. 2014;31:951–60.
Hue CD, Cao S, Haider SF, Vo KV, Effgen GB, Vogel E 3rd, Panzer MB, Bass CR, Meaney DF, Morrison B 3rd. Blood–brain barrier dysfunction after primary blast injury in vitro. J Neurotrauma. 2013;30:1652–63.
Hue CD, Cho FS, Cao S, Nicholls RE, Vogel Iii EW, Sibindi C, Arancio O, Dale Bass CR, Meaney DF, Morrison IB. Time course and size of blood–brain barrier opening in a mouse model of blast-induced traumatic brain injury. J Neurotrauma. 2016;33:1202–11.
Iadecola C. Cerebrovascular effects of amyloid-beta peptides: mechanisms and implications for Alzheimer’s dementia. Cell Mol Neurobiol. 2003;23:681–9.
Iadecola C. The neurovascular unit coming of age: a journey through neurovascular coupling in health and disease. Neuron. 2017;96:17–42.
Ikonomovic MD, Abrahamson EE, Carlson SW, Graham SH, Dixon CE. Novel therapies for combating chronic neuropathological sequelae of TBI. Neuropharmacology. 2019;145:160–76.
Ikonomovic MD, Buckley CJ, Abrahamson EE, Kofler JK, Mathis CA, Klunk WE, Farrar G. Post-mortem analyses of PiB and flutemetamol in diffuse and cored amyloid-beta plaques in Alzheimer’s disease. Acta Neuropathol. 2020;140:463–76.
Ikonomovic MD, Mi Z, Abrahamson EE. Disordered APP metabolism and neurovasculature in trauma and aging: combined risks for chronic neurodegenerative disorders. Ageing Res Rev. 2017;34:51–63.
Ikonomovic MD, Uryu K, Abrahamson EE, Ciallella JR, Trojanowski JQ, Lee VM, Clark RS, Marion DW, Wisniewski SR, DeKosky ST. Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp Neurol. 2004;190:192–203.
Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–70.
Johnson VE, Stewart W, Arena JD, Smith DH. Traumatic brain injury as a trigger of neurodegeneration. Adv Neurobiol. 2017;15:383–400.
Kabu S, Jaffer H, Petro M, Dudzinski D, Stewart D, Courtney A, Courtney M, Labhasetwar V. Blast-associated shock waves result in increased brain vascular leakage and elevated ROS levels in a rat model of traumatic brain injury. PLoS ONE. 2015;10: e0127971.
Kapuscinski J. DAPI: a DNA-specific fluorescent probe. Biotech Histochem. 1995;70:220–33.
Kawoos U, Abutarboush R, Gu M, Chen Y, Statz JK, Goodrich SY, Ahlers ST. Blast-induced temporal alterations in blood–brain barrier properties in a rodent model. Sci Rep. 2021;11:5906.
Kawoos U, Abutarboush R, Zarriello S, Qadri A, Ahlers ST, McCarron RM, Chavko M. N-Acetylcysteine amide ameliorates blast-induced changes in blood–brain barrier integrity in rats. Front Neurol. 2019;10:650.
Kawoos U, Gu M, Lankasky J, McCarron RM, Chavko M. Effects of exposure to blast overpressure on intracranial pressure and blood–brain barrier permeability in a rat model. PLoS ONE. 2016;11: e0167510.
Kenney K, Amyot F, Haber M, Pronger A, Bogoslovsky T, Moore C, Diaz-Arrastia R. Cerebral vascular injury in traumatic brain injury. Exp Neurol. 2016;275(Pt 3):353–66.
Kisler K, Nelson AR, Montagne A, Zlokovic BV. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci. 2017;18:419–34.
Kisler K, Nelson AR, Rege SV, Ramanathan A, Wang Y, Ahuja A, Lazic D, Tsai PS, Zhao Z, Zhou Y, Boas DA, Sakadzic S, Zlokovic BV. Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat Neurosci. 2017;20:406–16.
Kisler K, Nikolakopoulou AM, Sweeney MD, Lazic D, Zhao Z, Zlokovic BV. Acute ablation of cortical pericytes leads to rapid neurovascular uncoupling. Front Cell Neurosci. 2020;14:27.
Korte N, Nortley R, Attwell D. Cerebral blood flow decrease as an early pathological mechanism in Alzheimer’s disease. Acta Neuropathol. 2020;140:793–810.
Kovesdi E, Gyorgy AB, Kwon SK, Wingo DL, Kamnaksh A, Long JB, Kasper CE, Agoston DV. The effect of enriched environment on the outcome of traumatic brain injury; a behavioral, proteomics, and histological study. Front Neurosci. 2011;5:42.
Kuriakose M, Younger D, Ravula AR, Alay E, Rama Rao KV, Chandra N. Synergistic role of oxidative stress and blood–brain barrier permeability as injury mechanisms in the acute pathophysiology of blast-induced neurotrauma. Sci Rep. 2019;9:7717.
Kwon SK, Kovesdi E, Gyorgy AB, Wingo D, Kamnaksh A, Walker J, Long JB, Agoston DV. Stress and traumatic brain injury: a behavioral, proteomics, and histological study. Front Neurol. 2011;2:12.
Lecrux C, Hamel E. The neurovascular unit in brain function and disease. Acta Physiol. 2011;203:47–59.
Li Y, Li Y, Li X, Zhang S, Zhao J, Zhu X, Tian G. Head injury as a risk factor for dementia and Alzheimer’s disease: a systematic review and meta-analysis of 32 observational studies. PLoS ONE. 2017;12: e0169650.
Liu CC, Hu J, Zhao N, Wang J, Wang N, Cirrito JR, Kanekiyo T, Holtzman DM, Bu G. Astrocytic LRP1 mediates brain abeta clearance and impacts amyloid deposition. J Neurosci. 2017;37:4023–31.
LoBue C, Wadsworth H, Wilmoth K, Clem M, Hart J Jr, Womack KB, Didehbani N, Lacritz LH, Rossetti HC, Cullum CM. Traumatic brain injury history is associated with earlier age of onset of Alzheimer disease. Clin Neuropsychol. 2017;31:85–98.
Logsdon AF, Meabon JS, Cline MM, Bullock KM, Raskind MA, Peskind ER, Banks WA, Cook DG. Blast exposure elicits blood–brain barrier disruption and repair mediated by tight junction integrity and nitric oxide dependent processes. Sci Rep. 2018;8:11344.
Logsdon AF, Schindler AG, Meabon JS, Yagi M, Herbert MJ, Banks WA, Raskind MA, Marshall DA, Keene CD, Perl DP, Peskind ER, Cook DG. Nitric oxide synthase mediates cerebellar dysfunction in mice exposed to repetitive blast-induced mild traumatic brain injury. Sci Rep. 2020;10:9420.
Lokkegaard A, Nyengaard JR, West MJ. Stereological estimates of number and length of capillaries in subdivisions of the human hippocampal region. Hippocampus. 2001;11:726–40.
Lucke-Wold BP, Logsdon AF, Smith KE, Turner RC, Alkon DL, Tan Z, Naser ZJ, Knotts CM, Huber JD, Rosen CL. Bryostatin-1 restores blood brain barrier integrity following blast-induced traumatic brain injury. Mol Neurobiol. 2015;52:1119–34.
Ma Q, Zhao Z, Sagare AP, Wu Y, Wang M, Owens NC, Verghese PB, Herz J, Holtzman DM, Zlokovic BV. Blood–brain barrier-associated pericytes internalize and clear aggregated amyloid-beta42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol Neurodegener. 2018;13:57.
Meabon JS, Cook DG, Yagi M, Terry GE, Cross DJ, Muzi M, Pagulayan KF, Logsdon AF, Schindler AG, Ghai V, Wang K, Fallen S, Zhou Y, Kim TK, Lee I, Banks WA, Carlson ES, Mayer C, Hendrickson RC, Raskind MA, Marshall DA, Perl DP, Keene CD, Peskind ER. Chronic elevation of plasma vascular endothelial growth factor-A (VEGF-A) is associated with a history of blast exposure. J Neurol Sci. 2020;417: 117049.
Meabon JS, Huber BR, Cross DJ, Richards TL, Minoshima S, Pagulayan KF, Li G, Meeker KD, Kraemer BC, Petrie EC, Raskind MA, Peskind ER, Cook DG. Repetitive blast exposure in mice and combat veterans causes persistent cerebellar dysfunction. Sci Transl Med. 2016;8: 321ra326.
Meier-Ruge W, Ulrich J, Stahelin HB. Morphometric investigation of nerve cells, neuropil and senile plaques in senile dementia of the Alzheimer type. Arch Gerontol Geriatr. 1985;4:219–29.
Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR, Alosco ML, Solomon TM, Nowinski CJ, McHale L, Cormier KA, Kubilus CA, Martin BM, Murphy L, Baugh CM, Montenigro PH, Chaisson CE, Tripodis Y, Kowall NW, Weuve J, McClean MD, Cantu RC, Goldstein LE, Katz DI, Stern RA, Stein TD, McKee AC. Clinicopathological evaluation of chronic traumatic encephalopathy in players of American football. JAMA. 2017;318:360–70.
Miners JS, Schulz I, Love S. Differing associations between Abeta accumulation, hypoperfusion, blood–brain barrier dysfunction and loss of PDGFRB pericyte marker in the precuneus and parietal white matter in Alzheimer’s disease. J Cereb Blood Flow Metab. 2018;38:103–15.
Mishra A, Reynolds JP, Chen Y, Gourine AV, Rusakov DA, Attwell D. Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat Neurosci. 2016;19:1619–27.
Mohan K, Kecova H, Hernandez-Merino E, Kardon RH, Harper MM. Retinal ganglion cell damage in an experimental rodent model of blast-mediated traumatic brain injury. Invest Ophthalmol Vis Sci. 2013;54:3440–50.
Mullan M, Crawford F, Axelman K, Houlden H, Lilius L, Winblad B, Lannfelt L. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992;1:345–7.
Nelson AR, Sagare MA, Wang Y, Kisler K, Zhao Z, Zlokovic BV. Channelrhodopsin excitation contracts brain pericytes and reduces blood flow in the aging mouse brain in vivo. Front Aging Neurosci. 2020;12:108.
Nelson AR, Sweeney MD, Sagare AP, Zlokovic BV. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim Biophys Acta. 2016;1862:887–900.
Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP. Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci. 1997;17:171–80.
Nikolajsen GN, Kotynski KA, Jensen MS, West MJ. Quantitative analysis of the capillary network of aged APPswe/PS1dE9 transgenic mice. Neurobiol Aging. 2015;36:2954–62.
Nortley R, Korte N, Izquierdo P, Hirunpattarasilp C, Mishra A, Jaunmuktane Z, Kyrargyri V, Pfeiffer T, Khennouf L, Madry C, Gong H, Richard-Loendt A, Huang W, Saito T, Saido TC, Brandner S, Sethi H, Attwell D. Amyloid beta oligomers constrict human capillaries in Alzheimer’s disease via signaling to pericytes. Science. 2019;365(6450):eaav9518.
Nyengaard JR, Bendtsen TF, Gundersen HJ. Stereological estimation of the number of capillaries, exemplified by renal glomeruli. APMIS Suppl. 1988;4:92–9.
Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–34.
Pekny M, Pekna M, Messing A, Steinhauser C, Lee JM, Parpura V, Hol EM, Sofroniew MV, Verkhratsky A. Astrocytes: a central element in neurological diseases. Acta Neuropathol. 2016;131:323–45.
Peppiatt CM, Howarth C, Mobbs P, Attwell D. Bidirectional control of CNS capillary diameter by pericytes. Nature. 2006;443:700–4.
Perez Garcia G, De Gasperi R, Tschiffely AE, Gama Sosa MA, Abutarboush R, Kawoos U, Statz JK, Ciarlone S, Reed E, Jeyarajah T, Perez GM, Otero-Pagan A, Pryor D, Hof PR, Cook DG, Gandy S, Elder GA, Ahlers ST. Repetitive low-level blast exposure improves behavioral deficits and chronically lowers Abeta42 in an Alzheimer disease transgenic mouse model. J Neurotrauma. 2021;38:3146–73.
Perlmutter LS, Chui HC, Saperia D, Athanikar J. Microangiopathy and the colocalization of heparan sulfate proteoglycan with amyloid in senile plaques of Alzheimer’s disease. Brain Res. 1990;508:13–9.
Pogue AI, Lukiw WJ. Angiogenic signaling in Alzheimer’s disease. NeuroReport. 2004;15:1507–10.
Porter GA, Palade GE, Milici AJ. Differential binding of the lectins Griffonia simplicifolia I and Lycopersicon esculentum to microvascular endothelium: organ-specific localization and partial glycoprotein characterization. Eur J Cell Biol. 1990;51:85–95.
Ramos-Cejudo J, Wisniewski T, Marmar C, Zetterberg H, Blennow K, de Leon MJ, Fossati S. Traumatic brain injury and Alzheimer’s disease: the cerebrovascular link. EBioMedicine. 2018;28:21–30.
Readnower RD, Chavko M, Adeeb S, Conroy MD, Pauly JR, McCarron RM, Sullivan PG. Increase in blood–brain barrier permeability, oxidative stress, and activated microglia in a rat model of blast-induced traumatic brain injury. J Neurosci Res. 2010;88:3530–9.
Ribatti D, Nico B, Crivellato E. The role of pericytes in angiogenesis. Int J Dev Biol. 2011;55:261–8.
Richard E, van Gool WA, Hoozemans JJ, van Haastert ES, Eikelenboom P, Rozemuller AJ, van de Berg WD. Morphometric changes in the cortical microvascular network in Alzheimer’s disease. J Alzheimers Dis. 2010;22:811–8.
Ries M, Sastre M. Mechanisms of abeta clearance and degradation by glial cells. Front Aging Neurosci. 2016;8:160.
Roberts GW, Allsop D, Bruton C. The occult aftermath of boxing. J Neurol Neurosurg Psychiatry. 1990;53:373–8.
Robertson RT, Levine ST, Haynes SM, Gutierrez P, Baratta JL, Tan Z, Longmuir KJ. Use of labeled tomato lectin for imaging vasculature structures. Histochem Cell Biol. 2015;143:225–34.
Sagare AP, Bell RD, Zhao Z, Ma Q, Winkler EA, Ramanathan A, Zlokovic BV. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat Commun. 2013;4:2932.
Sajja VS, Tenn C, McLaws LJ, Vandevord PJ. A temporal evaluation of cytokines in rats after blast exposure. Biomed Sci Instrum. 2012;48:374–9.
Schager B, Brown CE. Susceptibility to capillary plugging can predict brain region specific vessel loss with aging. J Cereb Blood Flow Metab. 2020;40:2475–90.
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–82.
Sengillo JD, Winkler EA, Walker CT, Sullivan JS, Johnson M, Zlokovic BV. Deficiency in mural vascular cells coincides with blood–brain barrier disruption in Alzheimer’s disease. Brain Pathol. 2013;23:303–10.
Skotak M, Wang F, Alai A, Holmberg A, Harris S, Switzer RC, Chandra N. Rat injury model under controlled field-relevant primary blast conditions: acute response to a wide range of peak overpressures. J Neurotrauma. 2013;30:1147–60.
Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35.
Soto-Rojas LO, Pacheco-Herrero M, Martinez-Gomez PA, Campa-Cordoba BB, Apatiga-Perez R, Villegas-Rojas MM, Harrington CR, de la Cruz F, Garces-Ramirez L, Luna-Munoz J. The neurovascular unit dysfunction in Alzheimer’s disease. Int J Mol Sci. 2021;22:2022.
Stewart PA, Hayakawa K, Akers MA, Vinters HV. A morphometric study of the blood–brain barrier in Alzheimer’s disease. Lab Invest. 1992;67:734–42.
Sullivan DR. A cerebrovascular hypothesis of neurodegeneration in mTBI. J Head Trauma Rehabil. 2019;34:E18–27.
Sweeney MD, Ayyadurai S, Zlokovic BV. Pericytes of the neurovascular unit: key functions and signaling pathways. Nat Neurosci. 2016;19:771–83.
Sweeney MD, Montagne A, Sagare AP, Nation DA, Schneider LS, Chui HC, Harrington MG, Pa J, Law M, Wang DJJ, Jacobs RE, Doubal FN, Ramirez J, Black SE, Nedergaard M, Benveniste H, Dichgans M, Iadecola C, Love S, Bath PM, Markus HS, Salman RA, Allan SM, Quinn TJ, Kalaria RN, Werring DJ, Carare RO, Touyz RM, Williams SCR, Moskowitz MA, Katusic ZS, Lutz SE, Lazarov O, Minshall RD, Rehman J, Davis TP, Wellington CL, Gonzalez HM, Yuan C, Lockhart SN, Hughes TM, Chen CLH, Sachdev P, O’Brien JT, Skoog I, Pantoni L, Gustafson DR, Biessels GJ, Wallin A, Smith EE, Mok V, Wong A, Passmore P, Barkof F, Muller M, Breteler MMB, Roman GC, Hamel E, Seshadri S, Gottesman RF, van Buchem MA, Arvanitakis Z, Schneider JA, Drewes LR, Hachinski V, Finch CE, Toga AW, Wardlaw JM, Zlokovic BV. Vascular dysfunction—the disregarded partner of Alzheimer’s disease. Alzheimers Dement. 2019;15:158–67.
Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood–brain barrier: from physiology to disease and back. Physiol Rev. 2019;99:21–78.
Tachibana M, Yamazaki Y, Liu CC, Bu G, Kanekiyo T. Pericyte implantation in the brain enhances cerebral blood flow and reduces amyloid-beta pathology in amyloid model mice. Exp Neurol. 2018;300:13–21.
Tarkowski E, Issa R, Sjogren M, Wallin A, Blennow K, Tarkowski A, Kumar P. Increased intrathecal levels of the angiogenic factors VEGF and TGF-beta in Alzheimer’s disease and vascular dementia. Neurobiol Aging. 2002;23:237–43.
Thal DR. The role of astrocytes in amyloid beta-protein toxicity and clearance. Exp Neurol. 2012;236:1–5.
Uzunalli G, Herr S, Dieterly AM, Shi R, Lyle LT. Structural disruption of the blood–brain barrier in repetitive primary blast injury. Fluids Barriers CNS. 2021;18:2.
Vagnucci AH Jr, Li WW. Alzheimer’s disease and angiogenesis. Lancet. 2003;361:605–8.
Walls MK, Race N, Zheng L, Vega-Alvarez SM, Acosta G, Park J, Shi R. Structural and biochemical abnormalities in the absence of acute deficits in mild primary blast-induced head trauma. J Neurosurg. 2016;124:675–86.
Washington PM, Villapol S, Burns MP. Polypathology and dementia after brain trauma: does brain injury trigger distinct neurodegenerative diseases, or should they be classified together as traumatic encephalopathy? Exp Neurol. 2016;275(Pt 3):381–8.
Watson RE Jr, Wiegand SJ, Clough RW, Hoffman GE. Use of cryoprotectant to maintain long-term peptide immunoreactivity and tissue morphology. Peptides. 1986;7:155–9.
Winkler EA, Sagare AP, Zlokovic BV. The pericyte: a forgotten cell type with important implications for Alzheimer’s disease? Brain Pathol. 2014;24:371–86.
Wu Y, Wu H, Zeng J, Pluimer B, Dong S, Xie X, Guo X, Ge T, Liang X, Feng S, Yan Y, Chen JF, Sta Maria N, Ma Q, Gomez-Pinilla F, Zhao Z. Mild traumatic brain injury induces microvascular injury and accelerates Alzheimer-like pathogenesis in mice. Acta Neuropathol Commun. 2021;9:74.
Yeoh S, Bell ED, Monson KL. Distribution of blood–brain barrier disruption in primary blast injury. Ann Biomed Eng. 2013;41:2206–14.
Zehendner CM, Sebastiani A, Hugonnet A, Bischoff F, Luhmann HJ, Thal SC. Traumatic brain injury results in rapid pericyte loss followed by reactive pericytosis in the cerebral cortex. Sci Rep. 2015;5:13497.
Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12:723–38.
Acknowledgements
The authors thank Ms. Lan Shao for expert technical assistance.
Funding
Supported by Department of Veterans Affairs Grant RX000952 (MDI, MMH), NIH R01 AG052528 (MDI), T32 MH019986 (ATC), and the University of Pittsburgh Scholarly Research Project for Medical Students (ATC). The content is solely the responsibility of the authors and does not necessarily represent the official views of the Department of Veterans Affairs or the United States Government.
Author information
Authors and Affiliations
Contributions
MDI and MMH created the concept of the study. MDI, MMH, ATC, and EEA carried out the experiments, performed statistical analyses, contributed to the interpretation of the results, and drafted the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Clark, A.T., Abrahamson, E.E., Harper, M.M. et al. Chronic effects of blast injury on the microvasculature in a transgenic mouse model of Alzheimer’s disease related Aβ amyloidosis. Fluids Barriers CNS 19, 5 (2022). https://doi.org/10.1186/s12987-021-00301-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12987-021-00301-z