
Abstract
Background
The purpose of this study was to explore the potential molecular targets of hyperlipidaemia and the related molecular mechanisms.
Methods
The microarray dataset of GSE66676 obtained from patients with hyperlipidaemia was downloaded. Weighted gene co-expression network (WGCNA) analysis was used to analyse the gene expression profile, and the royal blue module was considered to have the highest correlation. Gene Ontology (GO) functional and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were implemented for the identification of genes in the royal blue module using the Database for Annotation, Visualization and Integrated Discovery (DAVID) online tool (version 6.8; http://david.abcc.ncifcrf.gov). A protein–protein interaction (PPI) network was established by using the online STRING tool. Then, several hub genes were identified by the MCODE and cytoHubba plug-ins in Cytoscape software.
Results
The significant module (royal blue) identified was associated with TC, TG and non-HDL-C. GO and KEGG enrichment analyses revealed that the genes in the royal blue module were associated with carbon metabolism, steroid biosynthesis, fatty acid metabolism and biosynthesis pathways of unsaturated fatty acids. SQLE (degree = 17) was revealed as a key molecule associated with hypercholesterolaemia (HCH), and SCD was revealed as a key molecule associated with hypertriglyceridaemia (HTG). RT-qPCR analysis also confirmed the above results based on our HCH/HTG samples.
Conclusions
SQLE and SCD are related to hyperlipidaemia, and SQLE/SCD may be new targets for cholesterol-lowering or triglyceride-lowering therapy, respectively.
Similar content being viewed by others


Background
With the development of markedly improved living standards throughout society, coronary artery disease (CAD) has emerged as a leading factor of mortality, functional deterioration, skyrocketing healthcare expenditure, disability and morbidity. CAD contributes to roughly 30% of all the deaths globally. It is estimated that its incidence will continue to rise in the coming decades [1,2,3]. Prior research has proved that the occurrence of CAD was the result of numerous factors comprising of genetic background, blood lipid levels, lifestyle, environmental exposures as well as the interactions between these factors [4, 5]. Coronary atherosclerosis is usually considered to be the initial step of CAD [6], which is due to the dysregulation of lipid metabolism and abnormal accumulation of cholesterol in the subintima layer of the coronary arteries [7, 8]. Hyperlipidaemia (HLP) acts as a critical risk factor that gives rise to CAD and its complications. Several investigations have demonstrated that for every 2% decrease in high-density lipoprotein cholesterol (HDL-C) levels, there is a resultant increase in CAD risk by 1%. Similarly, every 1% decrease in low-density lipoprotein cholesterol (LDL-C) levels results in reducing CAD risk by 1% [9, 10]. Several compelling studies have also demonstrated that combined effect in reducing the triglyceride (TG) [11], LDL-C [12] and total cholesterol (TC) [11] levels yielded higher decreases in cardiovascular risk compared to reduction of LDL-C levels alone [13]. The “6 percent effect” of statins refers to the fact that doubling the dose of statins only decreases LDL-C levels by 6.4%, and PCSK9 inhibitors combined with statins are recommended for patients with acute coronary syndrome (ACS) with a high risk of cardiovascular events [14]. Thus, the identification of novel therapeutic targets for HLP is expected to further reduce the risk of cardiovascular disease.
Microarray analysis might serve as a novel and practical approach to identify susceptibility genes associated with HLP [15]. However, the reproducibility and sensitivity of microarray analysis based on differentially expressed genes may be limited [16, 17]. Gene co-expression network-based methods have been widely used in processing microarray data and have especially been used to identify meaningful functional modules [18, 19]. Weighted gene co-expression network analysis (WGCNA) is one of the most effective methods of gene co-expression network analysis. Instead of simply identifying the differentially expressed genes, a scale-free network of gene–gene interactions is generated by WGCNA, and several significant modules composed of genes with similar functions could be identified by WGCNA; in addition, it can be used to further analyse the correlation between modules and phenotypes or clinical characteristics [20]. Therefore, WGCNA could be utilized to construct a co-expression network and identify significant modules in the network, which may help us to illuminate the intrinsic characteristics of HLP and provide new insights into potential genetic biomarkers, signalling pathways and molecular mechanisms involved in HLP.
Materials and methods
Hyperlipidaemia microarray datasets
The microarray dataset obtained from patients with HLP (GSE66676) was downloaded from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) database, which is based on the platform of the GPL6244 Affymetrix Human Gene 1.0 ST Array. Gene expression value matrices were obtained from the original files in CEL format after normalizing the expression values by using RMA methods in R software (version 4.0.0). [21]. Then, the Bioconductor package was used to transform the probe identification numbers (IDs) into gene symbols [22]. When multiple probe IDs corresponded to the same gene, the average expression value was used as the expression value.
Construction of the weighted gene co-expression network
WGCNA is a widely used systems biology method that is usually used to establish a scale-free network based on gene expression data profiles [18]. The co-expression network was constructed by selecting the genes whose variance was greater than all the quartiles of variance. After the sample cluster tree was constructed, cut height = 35 was used to screen the samples for subsequent studies. To ensure the reliability of the results of the network construction, the outlier samples were eliminated, and the samples in cluster 1 were selected to build the sample dendrogram and trait heatmap. The appropriate soft threshold power (soft power = 9) was selected according to the standard scale-free networks, and the adjacency values between all differentially expressed genes were calculated using a power function. Then, the adjacency values were transformed into a topological overlap matrix (TOM), and the corresponding dissimilarity (1-TOM) values were calculated. Module identification was accomplished with the dynamic tree cut method by hierarchically clustering genes using 1-TOM as the distance measure with a minimum size cut-off of 30 and a deep split value of 2 for the resulting dendrogram. To verify the stability of the identified modules, a module preservation function was used to calculate module preservation and quality statistics in the WGCNA package [23].
Identification of the module of interest and functional annotation
Pearson correlation analysis was used to assess the correlations between modules and clinical characteristics to identify biologically meaningful modules. All genes associated with the significant module were subjected to Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses by using the Database for Annotation, Visualization and Integrated Discovery (DAVID) online tool (version 6.8; http://david.abcc.ncifcrf.gov). P < 0.05 was set as the cut-off criterion.
Hub gene analysis
The degree of module membership (MM) was defined as the correlation between the gene expression profile and the module eigengenes (Mes). The degree of gene significance (GS) was defined as the absolute value of the correlation between the gene and external traits. In general, modules with increased MS and GS values among all the identified modules were selected for further analysis of their biological function [24]. The protein–protein interaction (PPI) network of genes in the selected module was constructed by the Search Tool for the Retrieval of Interacting Genes database (version 11.0; http://www.string-db.org) [25] and then visualized using Cytoscape software [26]. Molecular complex detection (MCODE) [27] was used to identify the most valuable clustering module. An MCODE score > 4 was the threshold for inclusion in further analysis. CytoHubba, a Cytoscape plugin, was used to identify hub genes in the PPI network; it provides 11 methods to explore important nodes in biological networks, of which degree has a better performance [28].
Sample verification and diagnostic criteria
A total of 462 (229 males, 49.57%; 233 females, 50.43%) unrelated participants with normal lipid levels and 485 (236 males, 48.66%; 249 females, 51.34%) unrelated subjects with hypercholesterolaemia (HCH, TC > 5.17 mmol/l) and 474 (232 males, 49.16%; 241 females, 50.84%) unrelated participants with hypertriglyceridaemia (HTG, TG > 1.70 mmol/l) were randomly recruited from the Physical Examination Center of the Affiliated Hospital of Guizhou Medical University. The age ranged from 24 to 82 years. There was no difference in age distribution or sex ratio between the control and HCH or HTG groups. Patients suffering from HCH did not have a history of HTG, and patients suffering from HTG did not have a history of HCH. All participants were basically healthy and had no history of myocardial infarction, CAD, type 2 diabetes mellitus (T2DM) or ischaemic stroke. They were not taking any medicines that could alter serum lipid levels. All subjects had signed written informed consent. The research protocol was approved by the Ethics Committee of the Affiliated Hospital of Guizhou Medical University.
Epidemiological analysis
Universally standardized methods and protocols were used to conduct the epidemiological survey [29]. Detailed lifestyle and demographic characteristics were collected with a standard set of questionnaires. Alcohol consumption (0 (non-drinker), < 25 g/day and ≥ 25 g/day) and smoking status (0 (non-smoker), < 20 cigarettes/day and ≥ 20 cigarettes/day) were divided into three different subgroups. Waist circumference, BMI, height, blood pressure and weight were measured as previously described [30].
Biochemical assays
Fasting venous blood samples of 5 ml were collected from each subject. A portion of the sample (2 ml) was placed in a tube and used to measure serum lipid levels. The remaining sample (3 ml) was collected in a glass tube containing anticoagulants (14.70 g/L glucose, 13.20 g/L trisodium citrate, 4.80 g/L citric acid) and utilized to extract deoxyribonucleic acid (DNA). The methods for performing serum ApoA1, HDL-C, ApoB, TG, LDL-C and TC measurements were described in a previous study [31]. All determinations were conducted using an autoanalyser (Type 7170A; Hitachi Ltd., Tokyo, Japan) in the Clinical Science Experiment Center of the Affiliated Hospital of Guizhou Medical University.
Quantitative real-time PCR
Peripheral blood monocytes (PBMCs) were isolated from blood samples with TRIzol reagent, which was used to extract the total RNA that was then reverse-transcribed into cDNA by using the PrimeScript RT reagent kit (Takara Bio, Japan). The obtained cDNA was used as a template for RT-qPCR. Table 1 shows that specific primer sequences, which were designed by Sangon Biotech (Shanghai, China), were used to detect the 2 hub genes. Quantitative RT-PCR was performed using a Taq PCR Master Mix Kit (Takara) on an ABI Prism 7500 sequence-detection system (Applied Biosystems, USA) using RT Reaction Mix in a total volume of 20 μL with the following reaction conditions: pre-denaturation at 95 °C for 30 s, then 40 cycles of 95 °C for 30 s and 60 °C for 30 s.
Diagnostic criteria
The values of serum ApoB (0.80–1.05 g/L), HDL-C (1.16–1.42 mmol/L), ApoA1 (1.20–1.60 g/L), TC (3.10–5.17 mmol/L), TG (0.56–1.70 mmol/L), the ApoA1/ApoB ratio (1.00–2.50) and LDL-C (2.70–3.10 mmol/L) were defined as normal at our Clinical Science Experiment Center. Subjects with TG > 1.70 mmol/L were defined as having hypertriglyceridaemia, and TC > 5.17 mmol/L was defined as having hypercholesterolaemia [32]. Participants with a fasting plasma (blood) glucose value ≥ 7.0 mmol/L were defined as having diabetes [33]. The diagnostic criteria of hypertension [34], obesity, normal weight and overweight were described in our previous study [35].
Statistical analyses
SPSS (Version 22.0) was used to process the research data. The results are presented as the mean ± SD except for TG levels, which are presented as medians and interquartile ranges. The differences in the general characteristics except for TG between HCH/HTG patients and controls were analysed by independent-samples t tests. The Kruskal–Wallis and Mann–Whitney nonparametric tests were used to detect the difference in TG levels between patients with HCH/HTG and controls. The chi-square test was utilized to assess the differences in the proportion of smokers, age distribution and alcohol consumption between patients with HCH/HTG and controls. Heat mapping of the correlation models and bioinformatic analysis were performed in R software (version 4.0.0). A P value < 0.05 was considered to be statistically significant.
Results
Data pre-processing
Gene expression profiles were obtained after normalization of the data and removing the outliers, and a total of 20,284 gene symbols were identified from 67 samples. Additional details about the gene expression profile and the sample phenotypes are presented in Additional file 1: Tables S1 and S2.
Weighted gene co‑expression networks
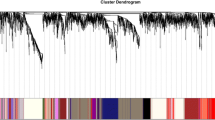
The sample cluster tree and sample dendrogram and trait heatmap are shown in Additional file 2: Figures S1 and S2. The gene expression profiles of 42 samples in cluster 1 were selected to build the weighted gene co-expression network. After the soft threshold (β = 9) was determined (Fig. 1), the weighted gene co-expression network was constructed by selecting the genes whose variance was greater than all the quartiles of variance. The adjacency matrix and correlation matrix of the gene expression profile were calculated and then transformed into a topological overlap matrix (TOM), and a clustering tree of genes based on the gene–gene non-ω similarity was obtained (Fig. 2). Combined with the TOM, the gene modules of each gene network were identified by the hierarchical average linkage clustering method, and twenty gene modules were identified by the dynamic tree cut algorithm (cut height = 0.25) (Fig. 3). The grey module contains all the genes that do not belong to the other modules and were excluded from subsequent analysis.

Analysis of network topology for various soft-thresholding powers. The left panel shows the scale-free fit index (y-axis) as a function of the soft-thresholding power (x-axis). The right panel displays the mean connectivity (degree, y-axis) as a function of the soft-thresholding power (x-axis)

Heatmap plot of the topological overlap in the gene network. In the heatmap, each row and column correspond to a gene, light colour denotes low topological overlap, and progressively darker red denotes higher topological overlap. Darker squares along the diagonal correspond to modules. The gene dendrogram and module assignment are shown along the left and top

Clustering dendrogram of genes. Gene clustering tree (dendrogram) obtained by hierarchical clustering of adjacency-based dissimilarity. The coloured row below the dendrogram indicates module membership identified by the dynamic tree cut method, together with assigned merged module colours and the original module colours
Identification of the modules of interest and functional annotation
The identification of modules that were significantly related to clinical phenotype was of high biological significance. In this study, we noticed that the royal blue module was associated with TC (r 2 = 0.38, P = 0.01), TG (r 2 = 0.41, P = 0.007) and non-HDL-C (r 2 = 0.32, P = 0.04), and the genes in the royal blue module were studied in the subsequent analyses (Fig. 4). GO and KEGG pathway enrichment analyses were used to further explore the biological functions of the genes in the royal blue module. Furthermore, we noticed that a total of 101 genes (Additional file 1: Tables S3) in the royal blue module were significantly correlated with the following pathways: hsa01100: metabolic pathways, hsa01130: biosynthesis of antibiotics, hsa00100: steroid biosynthesis, hsa01212: fatty acid metabolism, and hsa01040: biosynthesis of unsaturated fatty acids. The cell components, biological processes, molecular functions and KEGG pathway analysis of the royal blue module are also shown in Fig. 5, and more detailed information is presented in Additional file 1: Tables S4 and S5.

Module-feature associations. Each row corresponds to a module Eigengene, and the column corresponds to the clinical phenotype. Each cell contains the corresponding correlation in the first line and the P-value in the second line. The table is colour-coded by correlation according to the colour legend

GO functional and KEGG pathway enrichment analyses for genes in the object module. The x-axis shows the number of genes, and the y-axis shows the GO and KEGG pathway terms. The -log10 (P-value) of each term is coloured according to the legend. (A): GO functional enrichment analysis. (B): KEGG pathway enrichment analysis
PPI network construction and module analysis of DEGs
A PPI network including 93 notes and 333 edges was constructed by the STRING online tool. As shown in Fig. 6, the hub genes SQLE (degree = 17) and SCD (degree = 5) were identified by cytoHubba plug-ins in Molecular-1 and Molecular-2, respectively. Thus, we speculate that the genes mentioned above may be significantly correlated with blood lipid metabolism.

PPI network construction and identification of hub genes. (A) PPI network of genes in the royal blue module. The edge shows the interaction between two genes. Significant modules identified from the PPI network using MCODE with a score > 4.0. (A-1) Molecular-1 with MCODE score = 17.29. (A-2) Molecular-2 with MCODE score = 4.4
Validation analysis by RT-qPCR
As shown in Fig. 7a, the RT-qPCR results revealed that the expression of SQLE in the HCH group and SCD in the HTG group was higher than that in healthy subjects. At the same time, we also noticed that SQLE was positively correlated with TC (Fig. 7c) levels in the HCH group and that SCD was positively correlated with TG levels in the HTG group (Fig. 7d).

Validation with RT-qPCR (a) and the relationship between genes and lipid parameters in the control (b), HCH (c) and HTG (d) groups. *P < 0.001
Common and biochemical characteristics
As mentioned in Table 2, the sex ratio, age and height were similar between the controls and patients with HCH/HTG. Serum HDL-C and ApoA1 levels and the ApoA1/ApoB ratio were significantly higher, and the proportion of smokers, proportion of drinkers, systolic blood pressure, waist circumference, weight, diastolic blood pressure, glucose level, pulse pressure, body mass index (BMI), and serum LDL-C, ApoB, TG and TC levels were significantly lower in controls than in patients with hyperlipidaemia.
Discussion
Several recent studies have shown that hypertension, smoking, obesity, age, dyslipidaemia, lack of exercise, sex and diabetes mellitus are common risk factors for cardiovascular disease [36, 37]. A comprehensive understanding of the potential molecular mechanisms involved in the pathogenesis of HLP is helpful for its prevention and treatment. As a novel and practical approach to the identification of HLP susceptibility genes, a microarray analysis using WGCNA may be helpful for the diagnosis of hyperlipidaemia [20]. WGCNA could be used to build a scale-free co-expression network of lipid-associated genes by detecting gene-to-gene interactions rather than simply focusing on the differentially expressed genes (DEGs). Co-expressed genes were enriched in different modules by hierarchical average linkage cluster analysis. In the present research, we analysed a dataset from HLP patients (GSE66676) by using WGCNA and identified that the royal blue module was significantly associated with TC, TG and non-HDL. Furthermore, KEGG enrichment analyses of the genes in the royal blue module indicated that the enriched genes in this module might have significant potential biological functions that are closely related to metabolic pathways, steroid biosynthesis, fatty acid metabolism and biosynthesis of unsaturated fatty acids. Two hub genes (SQLE and SCD) were identified in the royal blue module that were detected by MCODE analysis. Moreover, the verification results were highly consistent with the above findings, and we found that the expression of the SQLE gene in patients with HCH and the SCD gene in patients with HTG was higher than that in healthy controls. Therefore, the identified SQLE gene was associated with the onset of HCH, the SCD gene was associated with the onset of HTG, and the underlying molecular mechanisms of these genes might be slightly different. In addition, SQLE and SCD were previously reported to be statin responsive, and they are known to be involved in sterol metabolism and transport; at the same time, there were significant changes in expression levels in the B-cells in response to statin treatment [38], and therefore, SQLE and SCD may be new targets for lipid-lowering therapy.
Fatty acids and cholesterol are essential lipids involved in many crucial biological processes; however, excessive free fatty acids and free cholesterol are major risk factors for type 2 diabetes and atherosclerosis [39]. Previous studies on intermediate metabolites in cholesterol biosynthesis have shown that the first oxidative step in cholesterol biosynthesis is catalysed by squalene monooxygenase (SQLE), a crucial regulator downstream of HMG-CoA reductase (HMGCR) in cholesterol synthesis [40]. Meanwhile, SQLE is suggested as the second rate-limiting enzyme in cholesterol synthesis [41, 42]. Inhibition of SQLE expression could effectively reduce cholesterol synthesis [43, 44], and the cholesterol-lowering effect is caused by the combination of multiple levels. First, SQLE and HMGCR act as direct targets of the sterol regulatory element binding protein 2 (SREBP2) transcription factor and play a crucial regulatory role in most cholesterol biosensor genes [45, 46]. Second, the N-terminus of the SQLE protein may contain a cholesterol-sensitive region that mediates the protease degradation of SQLE in a cholesterol-dependent manner by relying on an E3 ubiquitin ligase such as MARCH [47]. Interestingly, oleate acts as an unsaturated fatty acid and can stabilize SQLE by blocking MARCH6-mediated degradation [48]. In addition, Masanori Honsho et al. also noticed that inhibition of SQLE expression through elevating plasmalogen levels may be a novel and alternative potential method to reduce cholesterol synthesis [40]. Similarly, the KEGG analyses in the current study indicated that SQLE was mainly involved in metabolic pathways and steroid biosynthesis.
Metabolic risk factors such as insulin resistance, obesity, hypertension and dyslipidaemia are correlated with each other, so their combination is generally referred to as “metabolic syndrome” (MetS). Abnormal stearoyl-coenzyme A desaturase (SCD) expression/activity has been noticed in subjects with metabolic syndrome, indicating that SCD may be related to the pathogenesis of metabolic syndrome. By querying the GENE database in NCBI, we noticed that SCD (also known as SCD1; FADS5; SCDOS; hSCD1; MSTP008; gene ID: 6319, HGNC: 10571, OMIM: 604031) is positioned on chromosome 10q24.31 (exon count: 6) and encodes a biological synthase, which is mainly involved in the metabolism of fatty acids, especially oleic acid. This protein is an intact membrane protein located in the endoplasmic reticulum and is a member of the fatty acid desaturase family. Herman-Edelstein M et al. proved that SREBPs are transcription factors that activate the synthesis of fatty acids (FAs), triglycerides (TGs), and cholesterol, and SREBP2 activates cholesterol production, whereas SREBP1 primarily activates FA and TG synthesis [49]. ATP-citrate lyase (ACLY), a cytosolic enzyme that generates acetyl-CoA for cholesterol and de novo fatty acid synthesis, is a potential target for hyperlipidaemic intervention [50]. ACLY acts as a critical enzyme involved in de novo fatty acid synthesis and catalyses the conversion of citrate to cytosolic acetyl-CoA. Acetyl-CoA is converted to malonyl CoA via acetyl-CoA carboxylase (ACC), which plays a key role in the first committed step in the synthesis of fatty acids [51]. SCD is another key rate-limiting enzyme in fatty acid metabolism downstream of ACLY; it can convert different saturated fatty acids into monounsaturated fatty acids, and its expression is directly regulated by SREBP1 [52,53,54,55]. Both animal and human studies have shown that SCD is associated with obesity and insulin resistance [56, 57]. Mice with the SCD gene exhibited reduced diet-induced weight gain and improved insulin resistance compared to wild-type controls [58]. Deletion of the SCD1 gene product in mice could effectively improve insulin sensitivity, reduce plasma non-HDL cholesterol and triglyceride levels and liver lipid accumulation and increase beneficial HDL cholesterol levels [59]. Daniel Castellano-Castillo et al. also found a negative relationship between SCD DNA methylation and BMI and the MetS index [60]. In the current study, we also noticed that SCD was mainly involved in fatty acid metabolism and the biosynthesis pathways of unsaturated fatty acids.
Several recent studies have indicated that smoking [61, 62] and excessive drinking [63] were associated directly to HLP development and progression along with its complications. In recent years, the influence of smoking on HLP has attracted increasing attention. A compelling research has indicated the existence of lower HDL-C and higher TC, LDL-C and TG levels in smokers than in non-smokers [61]. In addition, atherosclerosis formation has also been shown to be influenced by different alcohol doses [64]. Moderate alcohol consumption may be protective against cardiovascular events, a phenomenon that has been attributed to elevated levels of ApoA1 and HDL-C [65]. Nevertheless, frequent heavy drinking leading to dyslipidaemia, alcoholic fatty liver and abnormal liver function is known to increase risk of CAD mortality [66]. In the present study, we found that the percentage of participants who smoking and excessive drinking was greater in the hyperlipidaemic group than in the normal group. Therefore, the preventive effect of a healthy lifestyle on hyperlipidaemia should not be ignored when exploring new therapeutic targets for hyperlipidaemia.
This research had several limitations. First, this is a single-centre study comprising a small patient number, and large multicentre studies are necessary to validate our findings. Second, the molecular mechanisms of SQLE and SCD involved in HLP are still not fully defined and require further cytology and animal experiments to further outline their respective roles in vivo and in vitro.
Conclusions
WGCNA identified that the royal blue module was significantly associated with TC, TG and non-HDL. GO and KEGG enrichment analyses revealed that the hub genes of SQLE were associated with TC and that SCD was associated with TG metabolism. The verification results of RT-qPCR revealed that the expression of SQLE in hypercholesterolaemia and SCD in hypertriglyceridaemia was higher than that in normal controls, which further increased the credibility of the conclusion. Thus, we speculated that SQLE may be a novel target for cholesterol-lowering therapy and that SCD may be a novel target for triglyceride-lowering therapy.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- WGCNA:
-
Weighted gene co-expression network analysis
- HCH:
-
Hypercholesterolemia
- HTG:
-
Hypertriglyceridemia
- SQLE:
-
Squalene epoxidase
- SCD:
-
Stearoyl-CoA desaturase
- DAVID:
-
Database for Annotation, Visualization and Integrated Discovery
- T2DM:
-
Type 2 diabetes mellitus
- GO:
-
Gene Ontology
- HDL-C:
-
High-density lipoprotein cholesterol
- IS:
-
Ischemic stroke
- KEGG:
-
Kyoto Encyclopedia of Genes and genomes
- LDL-C:
-
Low-density lipoprotein cholesterol
- MCODE:
-
Molecular Complex Detection
- Apo:
-
Apolipoprotein
- PPI:
-
Protein–protein interaction
- GEO:
-
Gene Expression Omnibus
- BMI:
-
Body mass index
- TG:
-
Triglyceride
- RT-qPCR:
-
Quantitative real time polymerase chain reaction
- TC:
-
Total cholesterol
- ACC:
-
Acetyl CoA carboxylase
- ACLY:
-
ATP—Citrate Lyase
- FAs:
-
Fatty acids
- MetS:
-
Metabolic syndrome
- HMGCR:
-
HMG-CoA reductase
- SREBP2:
-
Sterol regulatory element binding protein 2
- CAD:
-
Coronary artery disease
- HLP:
-
Hyperlipidaemia
- ACC:
-
American College of Cardiology
- AHA:
-
American Heart Association
- ACS:
-
Acute coronary syndrome
- TOM:
-
Topological overlap matrix
- MM:
-
Module membership
- Mes:
-
Module eigengenes
- Mes:
-
Module eigengenes
- DNA:
-
Deoxyribonucleic acid
- PBMCs:
-
Peripheral blood monocytes
- DEGs:
-
Differentially expressed genes
References
Houston M. The role of noninvasive cardiovascular testing, applied clinical nutrition and nutritional supplements in the prevention and treatment of coronary heart disease. Ther Adv Cardiovasc Dis. 2018;12(3):85–108.
Yokokawa H, Yasumura S, Tanno K, Ohsawa M, Onoda T, Itai K, et al. Serum low-density lipoprotein to high-density lipoprotein ratio as a predictor of future acute myocardial infarction among men in a 2.7-year cohort study of a Japanese northern rural population. J Atheroscler Thromb. 2011;18(2):89–98.
Finegold JA, Asaria P, Francis DP. Mortality from ischaemic heart disease by country, region, and age: statistics from World Health Organisation and United Nations. Int J Cardiol. 2013;168(2):934–45.
Chow CK, Jolly S, Rao-Melacini P, Fox KA, Anand SS, Yusuf S. Association of diet, exercise, and smoking modification with risk of early cardiovascular events after acute coronary syndromes. Circulation. 2010;121(6):750–8.
Li Y, Tang K, Zhou K, Wei Z, Zeng Z, He L, et al. Quantitative assessment of the effect of ABCA1 R219K polymorphism on the risk of coronary heart disease. Mol Biol Rep. 2012;39(2):1809–13.
Libby P, Theroux P. Pathophysiology of coronary artery disease. Circulation. 2005;111(25):3481–8.
Bosomworth NJ. Approach to identifying and managing atherogenic dyslipidemia: a metabolic consequence of obesity and diabetes. Can Fam Physician. 2013;59(11):1169–80.
Robins SJ, Lyass A, Zachariah JP, Massaro JM, Vasan RS. Insulin resistance and the relationship of a dyslipidemia to coronary heart disease: the Framingham Heart Study. Arterioscler Thromb Vasc Biol. 2011;31(5):1208–14.
Gotto AM Jr, Brinton EA. Assessing low levels of high-density lipoprotein cholesterol as a risk factor in coronary heart disease: a working group report and update. J Am Coll Cardiol. 2004;43(5):717–24.
Grundy SM, Cleeman JI, Merz CN, Brewer HB Jr, Clark LT, Hunninghake DB, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III Guidelines. J Am Coll Cardiol. 2004;44(3):720–32.
Chapman MJ, Ginsberg HN, Amarenco P, Andreotti F, Borén J, Catapano AL, et al. Triglyceride-rich lipoproteins and high-density lipoprotein cholesterol in patients at high risk of cardiovascular disease: evidence and guidance for management. Eur Heart J. 2011;32(11):1345–61.
Can M, Acikgoz S, Mungan G, Ugurbas E, Ankarali H, Sumbuloglu V, et al. Is direct method of low density lipoprotein cholesterol measurement appropriate for targeting lipid lowering therapy? Int J Cardiol. 2010;142(1):105–7.
Ferrieres J, Amber V, Crisan O, Chazelle F, Junger C, Wood D. Total lipid management and cardiovascular disease in the dyslipidemia international study. Cardiology. 2013;125(3):154–63.
Gencer B, Koskinas KC, Räber L, Karagiannis A, Nanchen D, Auer R, et al. Eligibility for PCSK9 inhibitors according to American College of Cardiology (ACC) and European Society of Cardiology/European Atherosclerosis Society (ESC/EAS) guidelines after acute coronary syndromes. J Am Heart Assoc. 2017;6(11):e006537.
Ma J, Dempsey AA, Stamatiou D, Marshall KW, Liew CC. Identifying leukocyte gene expression patterns associated with plasma lipid levels in human subjects. Atherosclerosis. 2007;191(1):63–72.
Ntzani EE, Ioannidis JP. Predictive ability of DNA microarrays for cancer outcomes and correlates: an empirical assessment. Lancet. 2003;362(9394):1439–44.
Ein-Dor L, Kela I, Getz G, Givol D, Domany E. Outcome signature genes in breast cancer: is there a unique set? Bioinformatics. 2005;21(2):171–8.
Horvath S, Dong J. Geometric interpretation of gene coexpression network analysis. PLoS Comput Biol. 2008;4(8):e1000117.
Ruan J, Dean AK, Zhang W. A general co-expression network-based approach to gene expression analysis: comparison and applications. BMC Syst Biol. 2010;4(1):8.
Miao L, Yin RX, Pan SL, Yang S, Yang DZ, Lin WX. Weighted gene co-expression network analysis identifies specific modules and hub genes related to hyperlipidemia. Cell Physiol Biochem. 2018;48(3):1151–63.
Gautier L, Cope L, Bolstad BM, Irizarry RA. affy–analysis of Affymetrix GeneChip data at the probe level. Bioinformatics. 2004;20(3):307–15.
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 2008;9(1):559.
Fuller TF, Ghazalpour A, Aten JE, Drake TA, Lusis AJ, Horvath S. Weighted gene coexpression network analysis strategies applied to mouse weight. Mamm Genome. 2007;18(6–7):463–72.
Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(Database issue):D447–52.
Smoot ME, Ono K, Ruscheinski J, Wang PL, Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27(3):431–2.
Bader GD, Hogue CWV. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003;4(1):2.
Chin C-H, Chen S-H, Wu H-H, Ho C-W, Ko M-T, Lin C-Y. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. 2014;8(4):S11.
An epidemiological study of cardiovascular and cardiopulmonary disease risk factors in four populations in the People's Republic of China. Baseline report from the P.R.C.-U.S.A. Collaborative Study. People's Republic of China--United States Cardiovascular and Cardiopulmonary Epidemiology Research Group. Circulation, 1992;85(3):1083–96.
Guo T, Yin RX, Li H, Wang YM, Wu JZ, Yang DZ. Association of the Trp316Ser variant (rs1801690) near the apolipoprotein H (beta2-glycoprotein-I) gene and serum lipid levels. Int J Clin Exp Pathol. 2015;8(6):7291–304.
Sun JQ, Yin RX, Shi GY, Shen SW, Chen X, Bin Y, et al. Association of the ARL15 rs6450176 SNP and serum lipid levels in the Jing and Han populations. Int J Clin Exp Pathol. 2015;8(10):12977–94.
Zheng P-F, Yin R-X, Liu C-X, Deng G-X, Guan Y-Z, Wei B-L. SYNE1-QK1 SNPs, G × G and G × E interactions on the risk of hyperlipidaemia. J Cell Mol Med. 2020;24(10):5772–85.
Alberti KG, Zimmet PZ. Definition, diagnosis and classification of diabetes mellitus and its complications. Part 1: diagnosis and classification of diabetes mellitus provisional report of a WHO consultation. Diabet Med. 1998;15(7):539–53.
Whitworth JA. 2003 World Health Organization (WHO)/International Society of Hypertension (ISH) statement on management of hypertension. J Hypertens. 2003;21(11):1983–92.
Khounphinith E, Yin R-X, Cao X-L, Huang F, Wu J-Z, Li H. TIMD4 rs6882076 SNP is associated with decreased levels of triglycerides and the risk of coronary heart disease and ischemic stroke. Int J Med Sci. 2019;16(6):864–71.
Greenland P, Knoll MD, Stamler J, Neaton JD, Dyer AR, Garside DB, et al. Major risk factors as antecedents of fatal and nonfatal coronary heart disease events. JAMA. 2003;290(7):891–7.
Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, Lanas F, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case–control study. Lancet. 2004;364(9438):937–52.
Bolotin E, Armendariz A, Kim K, Heo SJ, Boffelli D, Tantisira K, et al. Statin-induced changes in gene expression in EBV-transformed and native B-cells. Hum Mol Genet. 2014;23(5):1202–10.
Wang Y-J, Bian Y, Luo J, Lu M, Xiong Y, Guo S-Y, et al. Cholesterol and fatty acids regulate cysteine ubiquitylation of ACAT2 through competitive oxidation. Nat Cell Biol. 2017;19(7):808–19.
Honsho M, Abe Y, Fujiki Y. Dysregulation of plasmalogen homeostasis impairs cholesterol biosynthesis. J Biol Chem. 2015;290(48):28822–33.
Hidaka Y, Satoh T, Kamei T. Regulation of squalene epoxidase in HepG2 cells. J Lipid Res. 1990;31(11):2087–94.
Gonzalez R, Carlson JP, Dempsey ME. Two major regulatory steps in cholesterol synthesis by human renal cancer cells. Arch Biochem Biophys. 1979;196(2):574–80.
Hidaka Y, Hotta H, Nagata Y, Iwasawa Y, Horie M, Kamei T. Effect of a novel squalene epoxidase inhibitor, NB-598, on the regulation of cholesterol metabolism in Hep G2 cells. J Biol Chem. 1991;266(20):13171–7.
Sawada M, Matsuo M, Hagihara H, Tenda N, Nagayoshi A, Okumura H, et al. Effect of FR194738, a potent inhibitor of squalene epoxidase, on cholesterol metabolism in HepG2 cells. Eur J Pharmacol. 2001;431(1):11–6.
Sharpe LJ, Brown AJ. Controlling cholesterol synthesis beyond 3-hydroxy-3-methylglutaryl-CoA reductase (HMGCR). J Biol Chem. 2013;288(26):18707–15.
Cai D, Wang J, Gao B, Li J, Wu F, Zou JX, et al. RORγ is a targetable master regulator of cholesterol biosynthesis in a cancer subtype. Nat Commun. 2019;10(1):4621.
Zelcer N, Sharpe LJ, Loregger A, Kristiana I, Cook EC, Phan L, et al. The E3 ubiquitin ligase MARCH6 degrades squalene monooxygenase and affects 3-hydroxy-3-methyl-glutaryl coenzyme A reductase and the cholesterol synthesis pathway. Mol Cell Biol. 2014;34(7):1262–70.
Stevenson J, Luu W, Kristiana I, Brown AJ. Squalene mono-oxygenase, a key enzyme in cholesterol synthesis, is stabilized by unsaturated fatty acids. Biochem J. 2014;461(3):435–42.
Herman-Edelstein M, Thomas MC, Thallas-Bonke V, Saleem M, Cooper ME, Kantharidis P. Dedifferentiation of immortalized human podocytes in response to transforming growth factor-β: a model for diabetic podocytopathy. Diabetes. 2011;60(6):1779–88.
Pearce NJ, Yates JW, Berkhout TA, Jackson B, Tew D, Boyd H, et al. The role of ATP citrate-lyase in the metabolic regulation of plasma lipids. Biochem J. 1998;334(1):113–9.
Scaglia N, Chisholm JW, Igal RA. Inhibition of stearoylCoA desaturase-1 inactivates acetyl-CoA carboxylase and impairs proliferation in cancer cells: role of AMPK. PLoS ONE. 2009;4(8):e6812.
Jones DT, Valli A, Haider S, Zhang Q, Smethurst EA, Schug ZT, et al. 3D growth of cancer cells elicits sensitivity to kinase inhibitors but not lipid metabolism modifiers. Mol Cancer Ther. 2019;18(2):376–88.
Ntambi JM, Miyazaki M. Regulation of stearoyl-CoA desaturases and role in metabolism. Prog Lipid Res. 2004;43(2):91–104.
Forrest LM, Lough CM, Chung S, Boudyguina EY, Gebre AK, Smith TL, et al. Echium oil reduces plasma triglycerides by increasing intravascular lipolysis in apoB100-only low density lipoprotein (LDL) receptor knockout mice. Nutrients. 2013;5(7):2629–45.
Herman-Edelstein M, Scherzer P, Tobar A, Levi M, Gafter U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J Lipid Res. 2014;55(3):561–72.
Rahman SM, Dobrzyn A, Lee SH, Dobrzyn P, Miyazaki M, Ntambi JM. Stearoyl-CoA desaturase 1 deficiency increases insulin signaling and glycogen accumulation in brown adipose tissue. Am J Physiol Endocrinol Metab. 2005;288(2):E381–7.
García-Serrano S, Moreno-Santos I, Garrido-Sánchez L, Gutierrez-Repiso C, García-Almeida JM, García-Arnés J, et al. Stearoyl-CoA desaturase-1 is associated with insulin resistance in morbidly obese subjects. Mol Med. 2011;17(3–4):273–80.
Ntambi JM, Miyazaki M, Stoehr JP, Lan H, Kendziorski CM, Yandell BS, et al. Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity. Proc Natl Acad Sci U S A. 2002;99(17):11482–6.
MacDonald MLE, Singaraja RR, Bissada N, Ruddle P, Watts R, Karasinska JM, et al. Absence of stearoyl-CoA desaturase-1 ameliorates features of the metabolic syndrome in LDLR-deficient mice. J Lipid Res. 2008;49(1):217–29.
Castellano-Castillo D, Moreno-Indias I, Sanchez-Alcoholado L, Ramos-Molina B, Alcaide-Torres J, Morcillo S, et al. Altered adipose tissue DNA methylation status in metabolic syndrome: relationships between global DNA methylation and specific methylation at adipogenic, lipid metabolism and inflammatory candidate genes and metabolic variables. J Clin Med. 2019;8(1):87.
Rao ChS, Subash YE. The effect of chronic tobacco smoking and chewing on the lipid profile. J Clin Diagn Res. 2013;7(1):31–4.
Maeda K, Noguchi Y, Fukui T. The effects of cessation from cigarette smoking on the lipid and lipoprotein profiles: a meta-analysis. Prev Med. 2003;37(4):283–90.
Ruixing Y, Jinzhen W, Yaoheng H, Jing T, Hai W, Muyan L, et al. Associations of diet and lifestyle with hyperlipidemia for middle-aged and elderly persons among the Guangxi Bai Ku Yao and Han populations. J Am Diet Assoc. 2008;108(6):970–6.
Rimm EB, Williams P, Fosher K, Criqui M, Stampfer MJ. Moderate alcohol intake and lower risk of coronary heart disease: meta-analysis of effects on lipids and haemostatic factors. BMJ. 1999;319(7224):1523–8.
Ruixing Y, Shangling P, Hong C, Hanjun Y, Hai W, Yuming C, et al. Diet, alcohol consumption, and serum lipid levels of the middle-aged and elderly in the Guangxi Bai Ku Yao and Han populations. Alcohol. 2008;42(3):219–29.
Pai JK, Mukamal KJ, Rimm EB. Long-term alcohol consumption in relation to all-cause and cardiovascular mortality among survivors of myocardial infarction: the Health Professionals Follow-up Study. Eur Heart J. 2012;33(13):1598–605.
Acknowledgements
We thank all the participants of this study.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81960047).
Author information
Authors and Affiliations
Contributions
F-JL and P-FZ conceived the study, carried out the epidemiological survey and collected the samples, participated in the design, and drafted the manuscript. YZG performed the statistical analyses. HWP helped to modify the manuscript. WL conceived the study, participated in the design, carried out the epidemiological survey, and helped to draft the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The study design was approved by the Ethics Committee of the Affiliated Hospital of Guizhou Medical University. Informed consent was obtained from all participants.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
More details in Gene expression profile (Additional Table S1), Clinical phenotype (Additional Table S2), Module Genes (Additional Table S3), GO functional enrichment (Additional Table S4) and KEGG pathway enrichment analyses (Additional Table S5).
Additional file 2.
The Figures of Clustering dendrogram of samples, and Sample dendrogram and trait heatmap of selected samples in cluster 1.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Liao, FJ., Zheng, PF., Guan, YZ. et al. Weighted gene co-expression network analysis to identify key modules and hub genes related to hyperlipidaemia. Nutr Metab (Lond) 18, 24 (2021). https://doi.org/10.1186/s12986-021-00555-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12986-021-00555-2