
Abstract
The human T cell leukemia virus type 1 (HTVL-1), first reported in 1980 by Robert Gallo’s group, is the etiologic agent of both cancer and inflammatory diseases. Despite approximately 40 years of investigation, the prognosis for afflicted patients remains poor with no effective treatments. The virus persists in the infected host by evading the host immune response and inducing proliferation of infected CD4+ T-cells. Here, we will review the role that viral orf-I protein products play in altering intracellular signaling, protein expression and cell–cell communication in order to escape immune recognition and promote T-cell proliferation. We will also review studies of orf-I mutations found in infected patients and their potential impact on viral load, transmission and persistence. Finally, we will compare the orf-I gene in HTLV-1 subtypes as well as related STLV-1.
Similar content being viewed by others


Background
Human T-cell leukemia virus type-1 (HTLV-1) was discovered in 1980 in T-cells in a patient with cutaneous T-cell lymphoma [1, 2]. It is a member of the Delta retrovirus genus, alongside the closely related HTLV-2, -3, and -4 viruses, simian T-cell leukemia viruses (STLV) 1–4, and bovine leukemia virus (BLV) [3,4,5]. HTLV-1 infects approximately 5 to 10 million individuals worldwide with the highest endemic rates of infection in southern Japan, the Caribbean, Central and South America, Africa, Northeast Iran, Romania, Australia, and Melanesia [6]. HTLV-1 has seven reported subtypes (subtypes A to G), which are primarily contained to their respective geographic regions [6,7,8,9,10,11,12,13,14].
While the majority of infected individuals remain asymptomatic, a low percentage (2–5%) develop one of two major diseases after a long period of clinical latency: Adult T-cell leukemia/lymphoma (ATLL), a disease characterized by malignant proliferation of CD4+ T-lymphocytes, or HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP), a neurodegenerative condition [15,16,17,18]. Additionally, HTLV-1 is associated with other clinical disorders including HTLV-1-associated arthropathy, HTLV-1-associated uveitis, infective dermatitis, polymyositis, and chronic pulmonary disorders [18,19,20,21,22,23,24,25,26].
The manner in which HTLV-1 maintains persistent infection is likely associated with its ability to evade the host immune response. Immune evasion may also be associated with the proliferation of infected cells, leading to high proviral loads that correlate with disease progression. A high viral DNA burden in peripheral blood mononuclear cells has been associated with ATLL development [27, 28] and is considered a risk factor for HAM/TSP development [28, 29], particularly when there is a higher virus level in the cerebrospinal fluid than in peripheral blood [30]. In addition, HTLV-1-infected individuals have been shown to have diverse immunological alterations, such as high levels of inflammatory cytokines, spontaneous T-cell proliferation, and cellular maturation [31,32,33,34,35,36].
Several lines of evidence indicate that the HTLV-1 open reading frame-I (orf-I) is linked to immune evasion and viral replication and persistence. Unlike Tax and Rex, the HTLV-1 regulatory orf-I gene products are not required for virus replication and for the immortalization of human primary T-cells in vitro [37,38,39]. It has been shown, however, that human T-cell lines immortalized with HTLV-1 molecular clones lacking orf-I grow less efficiently than their wild-type counterpart clones and are more dependent upon the concentration of interleukin-2 (IL-2) in the media [40,41,42]. In addition, orf-I was found to be essential for HTLV-1 infection and replication in non-human primates, though not in rabbits [43]. In this review, we discuss the role of orf-I in immune regulation and in the context of the various HTLV subtypes.
HTLV-1A orf-I
Protein structure
The most studied orf-I gene is that of HTLV-1A, located at the 3′ end of the viral genome. It encodes the 99 amino acid p12 protein that can be proteolytically cleaved at the amino terminus to give rise to the p8 protein (Fig. 1) [44]. Amino acid sequence analysis of p12 predicts a noncanonical endoplasmic reticulum (ER) retention/retrieval signal between amino acids 1–5, two putative leucine zipper (LZ) motifs, two putative transmembrane domains between amino acids 12–30 and 48–67, a calcineurin-binding motif between amino acids 70–86, four putative proline-rich (PxxP) Src homology 3 (SH3)-binding domains, and a putative adaptin motif [45,46,47]. These structural features are thought to contribute to protein localization, dimerization, and protein–protein interactions. The naturally occurring p12 variant K88 is commonly found in HTLV-1 strains from HAM/TSP patients, while a second variant, R88, is found in virus strains from ATLL patients and healthy carriers [48]. R88 has much greater stability compared to K88, which is ubiquitinated and rapidly degraded by the proteasome [48]. Studies have found that p12 dimerization occurs through a disulfide bond at the conserved cysteine 39 residue of p12 and, when C39 is palmitoylated, the protein remains monomeric [49]. HTLV-1 strains containing either a serine (S39) or an arginine (R39) residue at this location have also been identified [50]. The actual importance of this cysteine residue to p12 function and regulation remains undetermined.

Structure of orf-I proteins p12 and p8. Amino acid sequence and putative functional domains of full length orf-I protein. The p12 protein is highly hydrophobic and contains an amino terminus noncanonical ER retention/retrieval motif (in bold), four putative proline-rich (PxxP) Src homology 3 (SH3)-binding domains, two putative leucine zipper (LZ) motifs, and an IL-2R β and γ chain binding motif (in blue boxes). The calcineurin-binding motif [70PSLP(I/L)T75] is indicated by a green box, and two transmembrane helices TM-1 and TM-2 domains are designated by black bars above the sequence. The black triangles indicate the two cleavage sites between amino acid positions 9 and 10, and 29 and 30, respectively. The asterisk denotes the position of cysteine 39. The proteolytic cleavage site G29/L30 leading to the production of p8 is indicated with a red arrow. The lysine-to-arginine variant is highlighted at position 88 by a red box. Arginine at this position increases the stability of the protein
Computer program analysis of the p12 protein sequence predicted two cleavage sites which were verified by mutagenesis studies showing that p12 undergoes a stepwise posttranslational proteolytic cleavage [46]. The first cleavage occurs between amino acids 9 and 10 and is followed by a second cleavage between amino acids 29 and 30 [46]. While the first cleavage between amino acids 9 and 10 removes the endoplasmic reticulum (ER) retention/retrieval signal of p12, cleavage between amino acids 29 and 30 generates the p8 protein (Fig. 1) [46]. Interestingly, while mutation analysis shows that cleavage first occurs at L9/10S, cleavage at the second site (G29/30L) follows so quickly afterward that detection of the first cleavage product is not seen or rarely seen in expression systems. In addition, later studies show that variation in the amino acid sequence influences protein cleavage and the abundance of p12 compared to p8. The p12 protein localizes to cellular endomembranes, particularly within the ER and Golgi apparatus, while p8 traffics to lipid rafts at the cell surface and is recruited to the immunological synapse upon T-cell receptor (TCR) ligation [46, 51,52,53]. The only protein so far identified to have amino acid similarity to p12 is the bovine papillomavirus (BPV)-transforming E5 protein, but E5 does not carry putative SH3 binding motifs [54, 55].
Indirect evidence suggests that infected individuals express orf-I-encoded proteins. The singly spliced mRNA encoding p12/p8 has been detected in ex vivo HTLV-1-infected T-cells and macrophages [44]. Moreover, sera from humans and rabbits infected with HTLV-1 contain antibodies capable of detecting overexpressed or recombinant p12 [56]. Cytotoxic T-lymphocyte (CTL) responses to orf-I products have also been detected in HTLV-1-infected individuals [57]. While the p12/p8 proteins are highly conserved, several variants have been identified [58]. Of these, G29S, P34L, S63P, R88K and S91P were the most frequent non-synonymous mutations observed. When present, G29S, P34L, and S63P were found to express a non-cleavable p12, whereas the rare D26N and D26E mutations predominantly expressed p8 [58]. Interestingly, the authors found that the pattern of p12 and p8 expression correlated with proviral load [58]. In a second study using a computational approach to examine p12/p8 sequence variants (D26N, G29S, P34L, L40F, P45L, S63P, L66P, S69G, R83C) in healthy carriers of HTLV-1 and HAM/TSP patients, P45L, S69G, and R88K were found more frequently in patients positive for HAM/TSP, and D26N, P34L, C39R, F61L, and R83C were found to be associated with low proviral load [59].
p12/p8 in T-cell proliferation
IL-2 receptor activation and STAT5 signaling
HTLV-1 persists primarily through expansion of infected cells, and while IL-2 promotes T-cell proliferation and controls immune responses [60], T-cells infected with HTLV-1 proliferate in the absence of IL-2. This IL-2 independence correlates with constitutive activation of the Janus-associated kinase and signal transducer and activator of transcription (JAK-STAT) pathway, a transcription factor cascade that affects cell proliferation, differentiation, and apoptosis [61]. Initially, p12 and p8 were not thought to have a role in IL-2 independence since they did not affect the expression of the interleukin-2 receptor (IL-2R) or the phosphorylation of JAK-STAT proteins [62]. However, later studies demonstrated that these proteins bind the β and γc chains of the immature IL-2R, stabilizing them in a pre-Golgi compartment and preventing their trafficking to the plasma membrane, leading to decreased IL-2R on the cell surface [63]. Co-immunoprecipitation experiments demonstrated that p12/p8 bind to a 20 amino acid region proximal to amino acid 350 of the IL-2R β chain that is critical for JAK1 and JAK3 recruitment [41]. The interaction of p12/p8 with the immature IL-2R leads to an increase in signal transducer and activator of transcription 5 (STAT5) phosphorylation and DNA binding activity in the absence of IL-2 [41]. Thus, the binding of p12/p8 to IL-2R allows T-cells to reproduce in the absence of IL-2 and with suboptimal antigen stimulation, providing HTLV-1-infected cells with a noteworthy proliferative advantage (Fig. 2) [41].

p12/p8 protein trafficking and function. (1) In the endoplasmic reticulum (ER), p12 binds to and retains the immature forms of interleukin-2 receptor (IL-2R) β and γ subunits, decreasing their trafficking to the plasma membrane. However, accumulation of the p12/IL-2R γ and β chains in the ER results in signal transducer and activator of transcription 5 (STAT5) phosphorylation in the absence of IL-2, allowing STAT5 translocation to the nucleus to promote cellular proliferation. (2) In the ER, p12 interacts with the immature heavy chains of MHC-I (MHC-I-Hc), binding to its α chain and preventing their interaction with β2 microglobulin (β2m). This leads to its degradation by the proteasome and decreased MHC-I expression at the cell surface. (3) HTLV-1A p12 also mediates the release of calcium ions (Ca2+) from the ER by binding calreticulin and calnexin. The release of Ca2+ inhibits the binding of calcineurin to the nuclear factor of T-cells (NFAT), preventing its dephosphorylation, nuclear translocation, induction of IL-2 expression and T-cell activation. In addition, p12 can inhibit the NFAT pathway by binding to calcineurin. (4) The p12 protein is proteolytically cleaved in the ER, leading to the formation of p8 that traffics to the cell surface. There, p8 increases T-cell adhesion through lymphocyte function-associated antigen-1 (LFA-1) clustering and promotes the formation of cell-to-cell contacts. (5) Further, p8 enhances the number and length of cellular conduits between T-cells, thereby enhancing signal transduction and HTLV-1 transmission
Calcium signaling and NFAT activation
The p12 protein localizes to the endoplasmic reticulum [53], where it is able to mediate an increase in cytosolic calcium (Ca2+) through its interaction with the calcium-binding proteins calreticulin and calnexin [51]. The presence of p12 in T-cells increases calcium release from the ER through inositol trisphosphate receptors and the facilitation of capacitive calcium entry through Ca2+ channels at the plasma membrane in response to the lowered ER calcium content (Fig. 2) [64, 65]. By depleting ER calcium stores and increasing cytosolic calcium, p12 is able to modulate a range of processes including T-cell proliferation, viral replication, and viral spread.
Early studies demonstrated that p12 can increase T-cell proliferation by activating the nuclear factor of activated T-cells (NFAT), which depends on calcium-binding proteins for dephosphorylation and nuclear import (Fig. 2) [64,65,66]. NFAT proteins play several important roles in regulating T-cell activity and are involved in their regulation, differentiation, self-tolerance, and in controlling thymocyte development (reviewed in [67]). NFAT can be activated through a complex TCR signaling cascade: following TCR engagement at the cell surface, the Lck and Fyn protein tyrosine kinases phosphorylate TCRζ and the CD3 subunits, allowing ZAP70 to dock to these phosphorylated domains. Activated ZAP70 phosphorylates the linker for activation of T-cells (LAT), which in turn binds and activates phospholipase C-γ-1 (PLCγ1) and leads to the production of inositol-1,4,5-trisphosphate and the release of Ca2+ from ER calcium stores. With the increase in cytosolic calcium, calmodulin and calcineurin are activated to dephosphorylate NFAT and permit its nuclear import. By modulating the regulation of cytosolic calcium levels, p12 mediates NFAT activation independent of the proximal TCR signaling molecules LAT and PLCγ1 [64]. Since NFAT binds the IL-2 promoter to increase transcription, the expression of p12 in T-cells thus supports IL-2 production in a calcium dependent manner [65].
However, p12 has also been shown to bind calcineurin, and its calcineurin-binding motif [70PSLP(I/L)T75] is homologous to that of NFAT [PXIXIT] [66]. The p12 protein might, therefore, act as a negative regulator of NFAT activation by competing with NFAT for calcineurin binding. The calcineurin binding motif is present in both p12 and p8, but it is currently not known whether p12/p8 homodimers or p12/p8 heterodimers bind calcineurin. Additional studies have revealed that p8, which localizes at the cell surface, is also capable of downregulating NFAT activity, though in a LAT-dependent manner [68]. Besides NFAT, p12 expression influences other calcium-regulated proteins, like the transcriptional coactivator p300 [69], which can in turn modulate the transcription of viral genes from the HTLV-1 LTR [70]. Moreover, p12 can promote intercellular viral spread by inducing lymphocyte function-associated antigen 1 (LFA-1) clustering on T-cells through a calcium-dependent mechanism (Fig. 2) [71].
p12/p8 and vacuolar ATPase
As suggested by their interaction with the H+ vacuolar ATPase, p12/p8 may affect signaling. The amino acids of HTLV-1 p12/p8 were found to be similar to those of the Bovine Papilloma Virus (BPV) E5 proteins. Like the BPV E5 oncoprotein [72, 73], p12/p8 interact with the 16 kDa subunit of the V-ATPase [54, 55]. While the transmembrane domains of p12/p8 appear to be unnecessary in V-ATPase binding, the proline-rich domain between amino acids 36 and 48 strengthens the bond [54, 55]. V-ATPase is found in clathrin-coated vesicles, lysosomes, endosomes, Golgi vesicles, endoplasmic reticulum, and synaptic vesicles, where it regulates the acidification of these intracellular vesicles [74]. By binding with viral proteins such as HTLV-1 p12/p8 and BPV E5, the proton pump may potentially interfere with functions such as receptor-ligand dissociation and protein trafficking within the endosomal/lysosomal compartment, but acidification remains essential for the formation of endosome carrier vesicles, which are intermediates between early and late endosomes [75, 76]. HTLV-1 is known to infect dendritic cells, and the acidification of lysosomes could play an important role in regulating virus entry or egress [77,78,79]. Indeed, the ablation of orf-I expression impairs HTLV-1 replication in dendritic cells [43].
p12/p8 in host immunity
orf-I and MHC class I degradation
The major histocompatibility complex class I (MHC-I) antigen presentation pathway plays a central role in developing host immunity against pathogens. MHC-I molecules are expressed on the surface of all nucleated cells and present peptides to the TCRs of cytotoxic T lymphocytes. Effector CD8+ T-cells specifically recognize viral peptides via the TCR to destroy infected cells. Consequently, many viruses have evolved proteins whose main function is to interfere with this pathway [80]. In MHC-I molecules, the heavy chain (Hc) is non-covalently bound to a non-glycosylated β2 microglobulin (β2M) protein, with MHC-I-Hc binding affinity to β2M enhanced when in the presence of peptide. In this case, MHC-I-Hc folds and assembles into the peptide-MHC-I-Hc-β2M complex in the lumen of the ER [81]. The p12 proteins bind to newly synthesized MHC-I-Hc prior to their association with the β2-microglobulin necessary to form a mature MHC-I complex (Fig. 2) [52]. Improperly assembled proteins are removed from the ER for degradation [82], and p12/MHC-I-Hc complexes are thus ubiquitinated and retrotranslocated to the cytosol for degradation by the proteasome, resulting in decreased MHC-I cell surface expression. Notably, p8 also co-precipitates with MHC-I, but the biological significance of this interaction is unclear.
Orf-I mRNA is expressed early after virus entry and is critical for establishing and maintaining viral infection in vivo [40, 43, 83, 84]. In a recent report comparing the expression of MHC-I on primary CD4+ T-cells infected with HTLV-1 molecular clones expressing neither p12 or p8 (p12KO), both p12 and p8 (WT), predominantly p8 (N26), or predominantly p12 (G29S), a decrease in surface MHC-I was only observed in the CD4+ T-cells infected with the G29S virus [58]. However, it must be noted that expression of both p8 and p12 was necessary to fully protect the infected cells from CTL killing (Fig. 3) [58]. Thus, the suppression of MHC-I antigen presentation by p12/p8 may allow HTLV-1 to evade adaptive immune surveillance in vivo and contribute to the expansion and accumulation of infected CD4+ T-cell clones over time.

Effect of p12/p8 on cytotoxic T-cells (CTLs). Cytotoxic CD8 T-cells (CTL) recognize target cells bearing an appropriate antigen-MHC I complex via the T-cell receptor (TCR). CTLs carry out target cell killing by releasing the cytotoxic proteins, granzyme B and perforin. Both p12 and p8 expression are important for HTLV-1 inhibition of CTL killing. By inducing the proteasome degradation of immature MHC I, p12 decreases MHC I surface expression, reducing antigen presentation to CTLs. In addition, the reduction of ICAM-1 expression in infected cells further reduces cell adhesion. The p8 protein enhances the number and length of cellular conduits between T-cells, allowing for the transfer of target cell proteins to other cells, including p8 itself. Transferred p8 could alter intracellular signaling and dampen TCR signaling to inhibit CTL killing. The p8 protein also promotes T-cell adhesion through lymphocyte function-associated antigen-1 (LFA-1) clustering and by enhancing the formation of cell-to-cell contacts promoting viral transmission
orf-I and NK cell recognition
While down modulation of MHC-I surface expression may allow infected cells to evade CTL recognition, it makes them susceptible to natural killer (NK) cell lysis. NK cells recognize and destroy cells that express low levels of MHC-I at their surface. Like CTLs, NK cells can kill infected cells directly by mediating cytolysis through perforin and granzyme production (reviewed in [85]). NK-target cell immune synapse is mediated by integrins such as leukocyte function antigen 1 (LFA-1) on the NK cell, and its ligand intercellular adhesion molecule 1 (ICAM-1) on the target cell. As shown in early studies, overexpression of the Tax protein increases the presence of adhesion molecules like LFA-3 and ICAM-1 [86, 87]. While high levels of ICAM-1 were found on transformed HTLV-1 cell lines that expressed Tax, the ligand was instead downregulated in several ATLL cell lines [87]. In more recent studies, a significant decrease in surface MHC-I and ICAM-1 and ICAM-2 (but not ICAM-3) was observed in primary CD4+ T-cells infected with HTLV-1 [88]. Furthermore, the infected cells in this study were shown to resist NK cell killing, which was moderately ameliorated by pretreatment of NK cells with IL-2 [88]. The majority of infected primary CD4+ T-cells did not express ligands for the NK cell activating receptors, natural cytotoxicity receptors, or NKG2D [88]. The authors did go on to show that orf-I expression was sufficient to decrease ICAM-1 and ICAM-2 expression in primary CD4+ T-cells. Treatment of Tax-expressing HTLV-1 transformed cells with pomalidomide (POM), an immunomodulatory drug used in the treatment of multiple myeloma [89], led to an increase in both surface MHC-I and ICAM-1. The effect of pomalidomide was shown to be orf-I dependent: MHC-I and ICAM-I expression increased in wild type (WT) HTLV-1 immortalized CD4+ T-cells after POM treatment, but their levels did not rise in HTLV-1 orf-I knockout immortalized CD4+ T-cells [90]. Thus, p12/p8 inhibit NK cell adhesion to T-cells and protect virus-infected cells from recognition in the presence of low levels of MHC-I (Fig. 4).

Impact of p12/p8 proteins on the host immune response. Summary of p12 and p8 influence on the host immune response to HTLV-1-infected cells. Expression of HTLV-1 p12/p8 results in decreased intercellular adhesion molecules (ICAM) and MHC-I expression, resulting in the inhibition of natural killer (NK) cell recognition and cytotoxic T-cell (CTL) killing. The p8 protein induces increased cell adhesion through increased lymphocyte function-associated antigen-1 (LFA-1) expression, increased virus transmission and cell signaling through tunneling nanotubes (TNTs) and virological synapse formation, and dampens T-cell receptor (TCR) signaling
Interestingly, additional proteins with functions similar to that of p12 have been identified in HTLV-2 and STLV-3. For instance, the corresponding p10 protein is encoded by the orf-I region of HTLV-2, and an additional protein, p11, is encoded by the orf-V region, which overlaps with orf-I. Both proteins were shown to bind the MHC heavy chain, but do not bind the IL2Rβ chain or the 16-kDa subunit of the vacuolar H+ ATPase [91]. Moreover, STLV-3 p9 shares some structural and functional features with HTLV-1 p12. Despite a very low percent sequence similarity between the two proteins, STLV-3 p9 and HTLV-1 p12 showed identical localization patterns in transfected cells, and p9 expression promoted the downregulation of calreticulin signaling [92].
orf-I in virus transmission
Surface adhesion molecules like LFA and ICAM are also important in viral transmission. Cell-free virions are not easily detected in the blood plasma of infected individuals and cell-free virus is poorly infectious for most cell types except dendritic cells [77, 93,94,95]. HTLV-1 is transmitted through cell-to-cell contact via the virological synapse, biofilm-like extracellular viral assemblies, and cellular conduits [96,97,98,99]. The transfer of a virus through the virological synapse requires polarization of adhesion and cytoskeletal proteins to the point of cellular contact [96]. Recent evidence suggests that p8 enhances polysynapse formation and modulates LFA-1 clustering to increase the formation of cell to cell contact and facilitate virus transfer [71, 98]. In addition, p8 promotes the formation of thin membranous cellular conduits that allow intracellular communication between several different cell types (Figs. 3 and 4) [98, 100, 101]. Among these, p8 induces tunneling nanotubes (TNTs), thin structures containing F-actin and lacking tubulin that allow for communication between cells at a distance. Immune cells such as NK cells, macrophages, T-cells, and B-cells are known to interact through TNTs [102, 103], and the induction of TNTs by other viruses has been reported [104,105,106,107,108]. Through these structures, the HTLV-1 proteins Tax, Gag, and Env are transferred to target T-cells [98]. When HTLV-1-infected T-cells are treated with Cytarabine, a molecule shown to reduce TNT formation [109], virus production and transmission are shown to decrease [110].
Furthermore, the p8 protein was also shown to be transferred to target cells [98, 110]. Using a quantitative flow cytometry method, p8 was transferred to approximately 5% of recipient T-cells after 5 min of co-culture, in a process dependent on actin polymerization [111]. The presence of p8 was also shown to decrease T-cell activation by inhibiting proximal TCR signaling [68]. Upon ligation of the TCR to the major histocompatibility complex class II (MHC-II) of an antigen presenting cell, p8 localizes to the immunological synapse, where it decreases phosphorylation of LAT, PLCγ1, and Vav by a LAT-dependent mechanism [46, 68]. By dampening TCR signaling, p8 downregulates NFAT activation, which is a crucial pathway in T-cell activation [64, 68]. Induction of T-cell anergy, a state in which T-cells become unresponsive to TCR stimulation, results in decreased Tax activity and HTLV-1 replication [68]. Since it has recently been shown that p8 transfers to target cells, it is possible that p8 induces T-cell anergy in cells that neighbor HTLV-1-infected cells to expand opportunities to safely transfer the virus to target cells [98].
Role of p12/p8 in HTLV-1 infectivity in vivo
To more closely examine the role of p12 in the initial stages of infection, investigators used animal models to study HTLV-1 molecular clones [43, 83]. Before identification of HBZ [112, 113], it was reported that orf-I expression was necessary for HTLV-1 infection in the rabbit model [83]. In addition to the deletion of orf-I in those studies, however, the molecular clone used had a frameshift affecting the gene encoding HBZ (Additional file 1: Figure S1). Therefore, it is unclear whether the results were due to the deletion of hbz, orf-I, or both. A more recent study using an HTLV-1 molecular clone which selectively disrupted orf-I expression revealed that orf-I is essential for infectivity in the macaque model, but not in the rabbit model [43]. Moreover, the finding that p12 is required for viral infectivity in macaques was found to be related to its role in supporting HTLV-1 infectivity of dendritic cells in vitro [43]. Additional in vivo studies in macaques have provided further support for the notion that p8 and p12 are important for viral persistence and spread [58]. When these molecular clones were used in a humanized mouse model, wild type HTLV-1 virus caused a polyclonal expansion of infected CD4+CD25+ T-cells. Notably, when the p12 knockout virus was used instead, infection only occurred after the virus reverted to wild type [84]. These studies suggest that maintaining p12/p8 expression is important for enabling viral infection and persistence. This is in line with results on HTLV-2 in the rabbit model. The authors showed that the sequences at the 3′ end of the proximal portion of HTLV-2, corresponding to the p12 region in HTLV-1, are not necessary for infection, but confer increased replicative capacity in vivo [114].
orf-I genetic variation of HTLV-1 subtypes
Genetic variation in HTLV-1A and HTLV-1C orf-I
Four major geographic subtypes of HTLV-1 have been identified: HTLV-1A, HTLV-1B, HTLV-1C and HTLV-1D [14, 115, 116], with HTLV-1C being the most divergent. To investigate the degree of divergence between the two HTLV-1 clades A and C, we compare three HTLV-1A representative sequences (NC-001436, J02029 and AF033817) and the seven HTLV-1C Australian and Melanesian complete genome sequences available online (GenBank KF242505, KF242506, JX891478, JX891479, KX905202, KX905203, L02534). Pairwise comparison at the nucleotide level shows higher conservation among structural genes env, pol, pro, gag compared to the regulatory genes p30, p27, and p12 [117]. Interestingly, the greatest nucleotide and amino acid divergence between these two clades was observed in orf-I. In all seven HTLV-1C sequences, nucleotide substitution was present within the p12 orf-I at position 6840. This mutation leads to substitution of the start codon AUG (methionine) to ACG (threonine) in all Australian HTLV-1C isolates, and to UCG (serine) in the Melanesian isolate [118,119,120]. Moreover, the multiple sequence alignment of 22 HTLV-1C-infected patients [117] from an indigenous Australian cohort reveals the presence of this T6840C nucleotide substitution in 100% of the subjects (Fig. 5). Given that serine and threonine are both small polar amino acids, the different amino acid substitutions between the two clades suggests the occurrence of an evolutionary event in the isolated endemic population. In addition to this mutation, amino acid comparison demonstrated 21 significant amino acid substitutions, with 11 observed in the first 30 amino acids of the p12 that is cleaved in the endoplasmic reticulum (ER) to process the p8 isoform (Fig. 5). The significance of p12 substitutions and deletions remain unclear, but it is likely that variations within HTLV-1C p12 are implicated in its novel disease progression. Although ATLL and HAM/TSP cases have been identified in HTLV-1C-infected individuals, the majority of patients develop bronchiectasis and lung disease [24, 121]. It is important to note that since the antisense transcribed hbz overlaps 303 nucleotides within the orf-I, any changes in the HTLV-1C p12 coding region could also potentially affect HBZ amino acid sequence, expression, and function. Since HBZ and Tax are thought to play distinct but related roles during the multi-step oncogenesis and inflammation caused by the virus, the imbalanced expression of HBZ and Tax in HTLV-1C patients may impact its novel disease progression.

Amino acid sequence analysis of HTLV and STLV orf-I proteins. Alignment of amino acid sequences of p12 from HTLV-1A prototype (NC-001436; J02029; AF033817), and other previously described p12 sequences for HTLV-1A, HTLV-1B, HTLV-1C and STLV-1 available on Genbank. A dash (–) indicates a gap in the amino acid alignment, an asterisk (*) represents a stop codon, and a period (.) represents amino acid similarity. Functional elements are indicated as follows: the proteolytic cleavage sites between positions 9 and 10, and between 29 and 30 are highlighted in blue. The calcineurin binding motif is highlighted in gray, the ubiquitylation site is highlighted in yellow, the four SH3 binding domains are outlined in red, and IL-2Rβ and γ binding domain is outlined in magenta. The multi-alignment was performed with the Mega7 program using default parameters
HTLV-1 subtype orf-I expression by alternative splicing
Alternatively, the virus may compensate for the mutation at the p12 initiating methionine to produce a protein of similar function by using an alternative splice acceptor or start site from a different gene region. The existence of different, alternative acceptor splice sites at the 3′ end of HTLV-1A have previously been described. Splice acceptor sites at position 6383, 6478, and 6875 were defined in infected cell lines and patient samples [44, 122,123,124,125,126,127]. One bicistronic mRNA, rex-orf-I, uses an initiation codon in exon 2 and the acceptor splice site at position 6383 to encode a protein of 152 amino acid referred to as the Rex-orf-I protein of 19 kDa. In this mRNA, the first coding exon of the Rex protein is joined in frame to p12/p8. Interestingly, ectopic expression of the of rex-orf-I cDNA yielded a 12-kDa protein that has the same relative migration as the p12 protein expressed by the singly spliced orf-I RNA. It has been demonstrated that this mRNA could encode p12 since the internal orf-I AUG is present in this mRNA, or p12 could be generated by posttranslational cleavage of the larger precursor protein [53, 124, 128]. Therefore, it is likely that both the doubly and singly spliced mRNAs encode the p12 protein. Moreover, the putative protein products from rex-orf-I show conservation of the amino acids that constitute the hallmark motifs implicated in p12 function. Thus, the protein products of rex-orf-I could possibly compensate for the role of p12 in viruses that do not contain the canonical initiation codon of p12 (Table 1).
orf-I genetic variation in STLV-1 subtypes
Analyses of HTLV-1 and STLV-1 viral strains from throughout the world have shown that they are closely related genetically, and they have been grouped together under the label, primate T-cell lymphotropic virus type 1 (PTLV-1). Moreover, it has been suggested that HTLV-1 has a simian origin and was originally acquired by humans through interspecies transmission from STLV-1-infected Old World monkeys. This hypothesis was supported by the high percentage of identity between STLV-1 strains from chimpanzees and mandrills with some HTLV-1 strains present in inhabitants of West and Central Africa. In these specific areas, zoonotic transmission from non-human primates (NHP) infected with STLV-1-to humans is still ongoing today [120, 130,131,132].
Despite its wide geographic distribution and identification in more than 20 Old World primate species in both Asia and Africa, only a few complete STLV-1 genome sequences are available [131, 133,134,135,136]. An early in vitro transcription-translation analysis of STLV-1 strains from naturally infected feral monkeys from Central and West Africa showed that STLV-1 p12 sequences exhibit interstrain genetic variability at both the nucleotide and amino acid level. Interestingly, this high variability seems to be specific to the STLV-1 p12 region since low genetic variability has been described in other genomic regions of STLV-1 [137,138,139]. Moreover, STLV-1 was shown to encode a 91 amino acid p12 protein in contrast to the 99 amino acid p12 protein found among HTLV-1A strains around the globe [129, 140]. The truncated STLV-1 p12 protein is the result of a change from glutamine to a stop codon, leading to a premature termination codon at amino acid residue 92, except in the STLV-1 Tan90 isolate (AF074966), where this UAG codon was found at residue 87 (Fig. 5) [129]. This feature had not been reported in HTLV-1 and was thought to be an important genetic difference between STLV-1 and HTLV-1.
Is orf-I expression dispensable in humans?
This review has highlighted the important functions of orf-I in promoting infected cell proliferation and their evasion from host immune recognition. Further, expression of p12/p8 is necessary in both the macaque and humanized mouse models. One would therefore expect that the viral protein is important for establishing a lifelong infection in humans as well. This hypothesis was supported by a study that looked at 160 HTLV-1-infected individuals (HAM/TSP or carriers) from various geographical regions, in which none of the approximately 1600 orf-I cloned sequences analyzed had a premature stop codon [58]. However, an earlier study identified truncated p12 proteins at position 82, 87, and 91, in HAM/TSP and ATLL patients [141]. In a study analyzing p12 sequences from 144 HAM/TSP patients, 41 ATLL patients, and 46 carriers from the Kagoshima region of Japan, the authors found 8 HAM/TSP patients and 2 ATLL patients with truncated (82 aa or 87 aa) p12 proteins, for a total frequency of 4% (Fig. 5) [141].
Sequence analysis, however, shows that these truncated proteins retain the leucine zipper sites, the dileucine motif, the calcineurin binding sites, and the receptor binding sites for IL-2 beta and gamma chains and both cleavage sites (Fig. 5). As mentioned earlier, p12 contains four SH3 binding motifs. While SH3-2 and SH3-4 were demonstrated to positively regulate NFAT, SH3-1 and SH3-3 were found to exert a negative effect on NFAT activation. Thus, the premature stop codon of p12 in HTLV-1 and STLV-1 generating 82 and 87 aa sequences may negatively impact the activation of NFAT, as shown in vitro in studies by Ding et al. [142]. However, the functional consequence of these truncations is not yet known.
In the Japanese cohort analysis, the authors also found that the premature stop codon in the orf-I gene was stably maintained over years in these individuals. Moreover, they found one HAM/TSP patient in which a nucleotide substitution from G to A at position 6836 resulted in the destruction of the initiation codon of p12. This virus was also found in two of the patient’s sisters, one carrier and one with HAM/TSP, indicating maternal transmission [141] similar to the mutation previously described in STLV-1 [140]. This study did not exclude the possibility that orf-I could be expressed by an alternative doubly spliced mRNA in these patients (Table 1). It is important to note that studies that examine the p12 sequence from patient PBMCs in vivo have relied on PCR amplification and cloning of viral DNA regions, resulting in amplification of the most dominant clones [58, 59, 141] that may not be infectious. Therefore, it is possible that where premature termination of p12 was found, minor intact clones are also present that contribute to infection and/or viral persistence.
Conclusions
The lifetime risk of developing ATLL or TSP/HAM has been estimated to be 2–5% depending on the ethnic origin of the infected individual, with a latency period between 40 and 60 years (reviewed in [143, 144]). Therefore, infected T-cells must be able to evade the host immune response to establish a persistent infection. The role of p12 and p8 in HTLV-1A pathogenesis is beginning to be uncovered, and evidence points to a central role for the orf-I protein products not only in viral transmission, but also in virus immune evasion and persistence. Sparing HTLV-1-infected cells may account for clonal expansion and contribute to disease development. Although the genomic organization of HTLV-1C closely resembles that of the cosmopolitan HTLV-1A, several differences at the nucleotide and amino acid level appear to be unique to the Australo-Melanesian HTLV-1C. Among these differences is the absence of the orf-I initiation codon in 100% of sequences from a remote Indigenous Australian cohort and in the complete HTLV-1C genome sequences now available online.
We don’t believe this observation suggests that orf-I expression is dispensable for HTLV-1C transmission and infectivity. Instead, we favor the hypothesis that an alternatively spliced mRNA may be used to provide the AUG initiation codon for the orf-I encoded protein(s). Interestingly, proteins that show p12 like functions have been identified in HTLV-2 and STLV-3 [91, 92]. Further studies are still needed to determine the role of rex-orf-I in infection, transmission, and pathogenesis of different HTLV-1 subtypes. Despite having had the complete genomic sequence for HTLV-1 for over 30 years, the mechanisms that govern disease status and host immune response are still unclear. We believe that developing a greater understanding of the complex kinetics, expression level, and function of the genes encoded in the 3′ end of the virus will allow us to develop novel therapeutic approaches for the treatment of HTLV-1 infection.
Availability of data and materials
Not applicable.
Abbreviations
- ATLL:
-
adult T-cell leukemia/lymphoma
- β2M:
-
β2 microglobulin
- BLV:
-
bovine leukemia virus
- BPV:
-
bovine papillomavirus
- Ca2+ :
-
cytosolic calcium
- CTL:
-
cytotoxic T-lymphocyte
- ER:
-
endoplasmic reticulum
- HAM/TSP:
-
HTLV-1-associated myelopathy/tropical spastic paraparesis
- Hc:
-
heavy chain
- HTLV:
-
human T-cell leukemia virus
- ICAM:
-
intercellular adhesion molecule
- IL-2:
-
interleukin-2
- IL-2R:
-
interleukin-2 receptor
- JAK-STAT:
-
Janus-associated kinase and signal transducer and activator of transcription
- LAT:
-
linker for activation of T-cells
- LFA:
-
lymphocyte function-associated antigen
- LZ:
-
leucine zipper
- MHC:
-
major histocompatibility complex
- NFAT:
-
nuclear factor of activated T-cells
- NHP:
-
non-human primate
- NK:
-
natural killer
- orf:
-
open reading frame
- PLCγ1:
-
phospholipase C-γ-1
- POM:
-
pomalidomide
- PTLV:
-
primate T-cell lymphotropic virus
- PxxP:
-
proline-rich
- SH3:
-
Src homology 3
- STAT5:
-
signal transducer and activator of transcription 5
- STLV:
-
simian T-cell leukemia virus
- TCR:
-
T-cell receptor
- TM:
-
transmembrane
- TNT:
-
tunneling nanotube
- V-ATPase:
-
vacuolar ATPase
- WT:
-
wild type
References
Poiesz BJ, Ruscetti FW, Gazdar AF, Bunn PA, Minna JD, Gallo RC. Detection and isolation of type C retrovirus particles from fresh and cultured lymphocytes of a patient with cutaneous T-cell lymphoma. Proc Natl Acad Sci USA. 1980;77(12):7415–9.
Poiesz BJ, Ruscetti FW, Mier JW, Woods AM, Gallo RC. T-cell lines established from human T-lymphocytic neoplasias by direct response to T-cell growth factor. Proc Natl Acad Sci USA. 1980;77(11):6815–9.
Verdonck K, Gonzalez E, Van Dooren S, Vandamme AM, Vanham G, Gotuzzo E. Human T-lymphotropic virus 1: recent knowledge about an ancient infection. Lancet Infect Dis. 2007;7(4):266–81.
Guo HG, Wong-Stall F, Gallo RC. Novel viral sequences related to human T-cell leukemia virus in T cells of a seropositive baboon. Science. 1984;223(4641):1195–7.
Hunsmann G, Schneider J, Schmitt J, Yamamoto N. Detection of serum antibodies to adult T-cell leukemia virus in non-human primates and in people from Africa. Int J Cancer. 1983;32(3):329–32.
Gessain A, Cassar O. Epidemiological aspects and world distribution of HTLV-1 infection. Front Microbiol. 2012;3:388.
Hahn BH, Shaw GM, Popovic M, Lo Monico A, Gallo RC, Wong-Staal F. Molecular cloning and analysis of a new variant of human T-cell leukemia virus (HTLV-ib) from an African patient with adult T-cell leukemia-lymphoma. Int J Cancer. 1984;34(5):613–8.
Gessian A, Yanagihara R, Franchini G, Garruto RM, Jenkins CL, Ajdukiewicz AB, et al. Highly divergent molecular variants of human T-lymphotropic virus type I from isolated populations in Papua New Guinea and the Solomon Islands. Proc Natl Acad Sci USA. 1991;88(17):7694–8.
Salemi M, Van Dooren S, Audenaert E, Delaporte E, Goubau P, Desmyter J, et al. Two new human T-lymphotropic virus type I phylogenetic subtypes in seroindeterminates, a Mbuti pygmy and a Gabonese, have closest relatives among African STLV-I strains. Virology. 1998;246(2):277–87.
Miura T, Fukunaga T, Igarashi T, Yamashita M, Ido E, Funahashi S, et al. Phylogenetic subtypes of human T-lymphotropic virus type I and their relations to the anthropological background. Proc Natl Acad Sci USA. 1994;91(3):1124–7.
Chen J, Zekeng L, Yamashita M, Takehisa J, Miura T, Ido E, et al. HTLV type I isolated from a Pygmy in Cameroon is related to but distinct from the known central African type. AIDS Res Hum Retrovir. 1995;11(12):1529–31.
Wolfe ND, Heneine W, Carr JK, Garcia AD, Shanmugam V, Tamoufe U, et al. Emergence of unique primate T-lymphotropic viruses among central African bushmeat hunters. Proc Natl Acad Sci USA. 2005;102(22):7994–9.
Ehrlich GD, Andrews J, Sherman MP, Greenberg SJ, Poiesz BJ. DNA sequence analysis of the gene encoding the HTLV-I p21e transmembrane protein reveals inter- and intraisolate genetic heterogeneity. Virology. 1992;186(2):619–27.
Gessain A, Gallo RC, Franchini G. Low degree of human T-cell leukemia/lymphoma virus type I genetic drift in vivo as a means of monitoring viral transmission and movement of ancient human populations. J Virol. 1992;66(4):2288–95.
Gessain A, Barin F, Vernant JC, Gout O, Maurs L, Calender A, et al. Antibodies to human T-lymphotropic virus type-I in patients with tropical spastic paraparesis. Lancet. 1985;2(8452):407–10.
Gallo RC. HTLV: the family of human T-lymphotropic retroviruses and their role in leukemia and AIDS. Med Oncol Tumor Pharmacother. 1986;3(3–4):265–7.
Yoshida M, Miyoshi I, Hinuma Y. Isolation and characterization of retrovirus from cell lines of human adult T-cell leukemia and its implication in the disease. Proc Natl Acad Sci USA. 1982;79(6):2031–5.
Nishioka K, Maruyama I, Sato K, Kitajima I, Nakajima Y, Osame M. Chronic inflammatory arthropathy associated with HTLV-I. Lancet. 1989;1(8635):441.
Morgan OS, Rodgers-Johnson P, Mora C, Char G. HTLV-1 and polymyositis in Jamaica. Lancet. 1989;2(8673):1184–7.
Eguchi K, Matsuoka N, Ida H, Nakashima M, Sakai M, Sakito S, et al. Primary Sjogren’s syndrome with antibodies to HTLV-I: clinical and laboratory features. Ann Rheum Dis. 1992;51(6):769–76.
Nakagawa M, Izumo S, Ijichi S, Kubota H, Arimura K, Kawabata M, et al. HTLV-I-associated myelopathy: analysis of 213 patients based on clinical features and laboratory findings. J Neurovirol. 1995;1(1):50–61.
Kamoi K, Mochizuki M. HTLV-1 uveitis. Front Microbiol. 2012;3:270.
Araya N, Sato T, Yagishita N, Ando H, Utsunomiya A, Jacobson S, et al. Human T-lymphotropic virus type 1 (HTLV-1) and regulatory T cells in HTLV-1-associated neuroinflammatory disease. Viruses. 2011;3(9):1532–48.
Einsiedel L, Pham H, Wilson K, Walley R, Turpin J, Bangham C, et al. Human T-Lymphotropic Virus type 1c subtype proviral loads, chronic lung disease and survival in a prospective cohort of Indigenous Australians. PLoS Negl Trop Dis. 2018;12(3):e0006281.
Osame M, Usuku K, Izumo S, Ijichi N, Amitani H, Igata A, et al. HTLV-I associated myelopathy, a new clinical entity. Lancet. 1986;1(8488):1031–2.
Mochizuki M, Watanabe T, Yamaguchi K, Takatsuki K, Yoshimura K, Shirao M, et al. HTLV-I uveitis: a distinct clinical entity caused by HTLV-I. Jpn J Cancer Res. 1992;83(3):236–9.
Iwanaga M, Watanabe T, Utsunomiya A, Okayama A, Uchimaru K, Koh KR, et al. Human T-cell leukemia virus type I (HTLV-1) proviral load and disease progression in asymptomatic HTLV-1 carriers: a nationwide prospective study in Japan. Blood. 2010;116(8):1211–9.
Nagai M, Usuku K, Matsumoto W, Kodama D, Takenouchi N, Moritoyo T, et al. Analysis of HTLV-I proviral load in 202 HAM/TSP patients and 243 asymptomatic HTLV-I carriers: high proviral load strongly predisposes to HAM/TSP. J Neurovirol. 1998;4(6):586–93.
Furtado Mdos S, Andrade RG, Romanelli LC, Ribeiro MA, Ribas JG, Torres EB, et al. Monitoring the HTLV-1 proviral load in the peripheral blood of asymptomatic carriers and patients with HTLV-associated myelopathy/tropical spastic paraparesis from a Brazilian cohort: ROC curve analysis to establish the threshold for risk disease. J Med Virol. 2012;84(4):664–71.
Demontis MA, Hilburn S, Taylor GP. Human T cell lymphotropic virus type 1 viral load variability and long-term trends in asymptomatic carriers and in patients with human T cell lymphotropic virus type 1-related diseases. AIDS Res Hum Retrovir. 2013;29(2):359–64.
Futsch N, Prates G, Mahieux R, Casseb J, Dutartre H. Cytokine networks dysregulation during HTLV-1 infection and associated diseases. Viruses. 2018;10(12):691.
Hanon E, Goon P, Taylor GP, Hasegawa H, Tanaka Y, Weber JN, et al. High production of interferon gamma but not interleukin-2 by human T-lymphotropic virus type I-infected peripheral blood mononuclear cells. Blood. 2001;98(3):721–6.
Pinto LA, Galvao Castro B, Soares MB, Grassi MF. An evaluation of the spontaneous proliferation of peripheral blood mononuclear cells in HTLV-1-infected individuals using flow cytometry. ISRN Oncol. 2011;2011:326719.
Sakai JA, Nagai M, Brennan MB, Mora CA, Jacobson S. In vitro spontaneous lymphoproliferation in patients with human T-cell lymphotropic virus type I-associated neurologic disease: predominant expansion of CD8+ T cells. Blood. 2001;98(5):1506–11.
Nascimento CR, Lima MA, de Andrada Serpa MJ, Espindola O, Leite AC, Echevarria-Lima J. Monocytes from HTLV-1-infected patients are unable to fully mature into dendritic cells. Blood. 2011;117(2):489–99.
Santos SB, Porto AF, Muniz AL, de Jesus AR, Magalhaes E, Melo A, et al. Exacerbated inflammatory cellular immune response characteristics of HAM/TSP is observed in a large proportion of HTLV-I asymptomatic carriers. BMC Infect Dis. 2004;4:7.
Derse D, Mikovits J, Ruscetti F. X-I and X-II open reading frames of HTLV-I are not required for virus replication or for immortalization of primary T-cells in vitro. Virology. 1997;237(1):123–8.
Lairmore MD, Albrecht B, D’Souza C, Nisbet JW, Ding W, Bartoe JT, et al. In vitro and in vivo functional analysis of human T cell lymphotropic virus type 1 pX open reading frames I and II. AIDS Res Hum Retrovir. 2000;16(16):1757–64.
Robek MD, Wong FH, Ratner L. Human T-cell leukemia virus type 1 pX-I and pX-II open reading frames are dispensable for the immortalization of primary lymphocytes. J Virol. 1998;72(5):4458–62.
Albrecht B, Collins ND, Burniston MT, Nisbet JW, Ratner L, Green PL, et al. Human T-lymphotropic virus type 1 open reading frame I p12(I) is required for efficient viral infectivity in primary lymphocytes. J Virol. 2000;74(21):9828–35.
Nicot C, Mulloy JC, Ferrari MG, Johnson JM, Fu K, Fukumoto R, et al. HTLV-1 p12(I) protein enhances STAT5 activation and decreases the interleukin-2 requirement for proliferation of primary human peripheral blood mononuclear cells. Blood. 2001;98(3):823–9.
Taylor JM, Brown M, Nejmeddine M, Kim KJ, Ratner L, Lairmore M, et al. Novel role for interleukin-2 receptor-Jak signaling in retrovirus transmission. J Virol. 2009;83(22):11467–76.
Valeri VW, Hryniewicz A, Andresen V, Jones K, Fenizia C, Bialuk I, et al. Requirement of the human T-cell leukemia virus p12 and p30 products for infectivity of human dendritic cells and macaques but not rabbits. Blood. 2010;116(19):3809–17.
Koralnik IJ, Gessain A, Klotman ME, Lo Monico A, Berneman ZN, Franchini G. Protein isoforms encoded by the pX region of human T-cell leukemia/lymphotropic virus type I. Proc Natl Acad Sci USA. 1992;89(18):8813–7.
Albrecht B, Lairmore MD. Critical role of human T-lymphotropic virus type 1 accessory proteins in viral replication and pathogenesis. Microbiol Mol Biol Rev. 2002;66(3):396–406 (Table of contents).
Fukumoto R, Andresen V, Bialuk I, Cecchinato V, Walser JC, Valeri VW, et al. In vivo genetic mutations define predominant functions of the human T-cell leukemia/lymphoma virus p12I protein. Blood. 2009;113(16):3726–34.
Edwards D, Fenizia C, Gold H, de Castro-Amarante MF, Buchmann C, Pise-Masison CA, et al. Orf-I and orf-II-encoded proteins in HTLV-1 infection and persistence. Viruses. 2011;3(6):861–85.
Trovato R, Mulloy JC, Johnson JM, Takemoto S, de Oliveira MP, Franchini G. A lysine-to-arginine change found in natural alleles of the human T-cell lymphotropic/leukemia virus type 1 p12(I) protein greatly influences its stability. J Virol. 1999;73(8):6460–7.
Edwards D, Fukumoto R, de Castro-Amarante MF, Alcantara LC, Galvao-Castro B, Washington Parks R, et al. Palmitoylation and p8-mediated human T-cell leukemia virus type 1 transmission. J Virol. 2014;88(4):2319–22.
Georgieva ER. Non-Structural proteins from human T-cell leukemia virus type 1 in cellular membranes-mechanisms for viral survivability and proliferation. Int J Mol Sci. 2018;19(11):3508.
Ding W, Albrecht B, Luo R, Zhang W, Stanley JR, Newbound GC, et al. Endoplasmic reticulum and cis-Golgi localization of human T-lymphotropic virus type 1 p12(I): association with calreticulin and calnexin. J Virol. 2001;75(16):7672–82.
Johnson JM, Nicot C, Fullen J, Ciminale V, Casareto L, Mulloy JC, et al. Free major histocompatibility complex class I heavy chain is preferentially targeted for degradation by human T-cell leukemia/lymphotropic virus type 1 p12(I) protein. J Virol. 2001;75(13):6086–94.
Koralnik IJ, Fullen J, Franchini G. The p12I, p13II, and p30II proteins encoded by human T-cell leukemia/lymphotropic virus type I open reading frames I and II are localized in three different cellular compartments. J Virol. 1993;67(4):2360–6.
Koralnik IJ, Mulloy JC, Andresson T, Fullen J, Franchini G. Mapping of the intermolecular association of human T cell leukaemia/lymphotropic virus type I p12I and the vacuolar H+-ATPase 16 kDa subunit protein. J Gen Virol. 1995;76(Pt 8):1909–16.
Franchini G, Mulloy JC, Koralnik IJ, Lo Monico A, Sparkowski JJ, Andresson T, et al. The human T-cell leukemia/lymphotropic virus type I p12I protein cooperates with the E5 oncoprotein of bovine papillomavirus in cell transformation and binds the 16-kilodalton subunit of the vacuolar H+ ATPase. J Virol. 1993;67(12):7701–4.
Dekaban GA, Peters AA, Mulloy JC, Johnson JM, Trovato R, Rivadeneira E, et al. The HTLV-I orfI protein is recognized by serum antibodies from naturally infected humans and experimentally infected rabbits. Virology. 2000;274(1):86–93.
Pique C, Dokhelar MC. In vivo production of Rof and Tof proteins of HTLV type 1: evidence from cytotoxic T lymphocytes. AIDS Res Hum Retrovir. 2000;16(16):1783–6.
Pise-Masison CA, de Castro-Amarante MF, Enose-Akahata Y, Buchmann RC, Fenizia C, Washington Parks R, et al. Co-dependence of HTLV-1 p12 and p8 functions in virus persistence. PLoS Pathog. 2014;10(11):e1004454.
Barreto FK, Khouri R, Rego FFA, Santos LA, Castro-Amarante MF, Bialuk I, et al. Analyses of HTLV-1 sequences suggest interaction between ORF-I mutations and HAM/TSP outcome. Infect Genet Evol. 2016;45:420–5.
Ross SH, Cantrell DA. Signaling and function of interleukin-2 in T lymphocytes. Annu Rev Immunol. 2018;36:411–33.
Migone TS, Lin JX, Cereseto A, Mulloy JC, O’Shea JJ, Franchini G, et al. Constitutively activated Jak-STAT pathway in T cells transformed with HTLV-I. Science. 1995;269(5220):79–81.
Collins ND, D’Souza C, Albrecht B, Robek MD, Ratner L, Ding W, et al. Proliferation response to interleukin-2 and Jak/Stat activation of T cells immortalized by human T-cell lymphotropic virus type 1 is independent of open reading frame I expression. J Virol. 1999;73(11):9642–9.
Mulloy JC, Crownley RW, Fullen J, Leonard WJ, Franchini G. The human T-cell leukemia/lymphotropic virus type 1 p12I proteins bind the interleukin-2 receptor beta and gammac chains and affects their expression on the cell surface. J Virol. 1996;70(6):3599–605.
Albrecht B, D’Souza CD, Ding W, Tridandapani S, Coggeshall KM, Lairmore MD. Activation of nuclear factor of activated T cells by human T-lymphotropic virus type 1 accessory protein p12(I). J Virol. 2002;76(7):3493–501.
Ding W, Albrecht B, Kelley RE, Muthusamy N, Kim SJ, Altschuld RA, et al. Human T-cell lymphotropic virus type 1 p12(I) expression increases cytoplasmic calcium to enhance the activation of nuclear factor of activated T cells. J Virol. 2002;76(20):10374–82.
Kim SJ, Ding W, Albrecht B, Green PL, Lairmore MD. A conserved calcineurin-binding motif in human T lymphotropic virus type 1 p12I functions to modulate nuclear factor of activated T cell activation. J Biol Chem. 2003;278(18):15550–7.
Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5(6):472–84.
Fukumoto R, Dundr M, Nicot C, Adams A, Valeri VW, Samelson LE, et al. Inhibition of T-cell receptor signal transduction and viral expression by the linker for activation of T cells-interacting p12(I) protein of human T-cell leukemia/lymphoma virus type 1. J Virol. 2007;81(17):9088–99.
Nair AM, Michael B, Datta A, Fernandez S, Lairmore MD. Calcium-dependent enhancement of transcription of p300 by human T-lymphotropic type 1 p12I. Virology. 2006;353(2):247–57.
Bex F, Yin MJ, Burny A, Gaynor RB. Differential transcriptional activation by human T-cell leukemia virus type 1 Tax mutants is mediated by distinct interactions with CREB binding protein and p300. Mol Cell Biol. 1998;18(4):2392–405.
Kim SJ, Nair AM, Fernandez S, Mathes L, Lairmore MD. Enhancement of LFA-1-mediated T cell adhesion by human T lymphotropic virus type 1 p12I1. J Immunol. 2006;176(9):5463–70.
Goldstein DJ, Finbow ME, Andresson T, McLean P, Smith K, Bubb V, et al. Bovine papillomavirus E5 oncoprotein binds to the 16 K component of vacuolar H(+)-ATPases. Nature. 1991;352(6333):347–9.
Schapiro F, Sparkowski J, Adduci A, Suprynowicz F, Schlegel R, Grinstein S. Golgi alkalinization by the papillomavirus E5 oncoprotein. J Cell Biol. 2000;148(2):305–15.
Finbow ME, Pitts JD, Goldstein DJ, Schlegel R, Findlay JB. The E5 oncoprotein target: a 16-kDa channel-forming protein with diverse functions. Mol Carcinog. 1991;4(6):441–4.
Clague MJ, Urbe S, Aniento F, Gruenberg J. Vacuolar ATPase activity is required for endosomal carrier vesicle formation. J Biol Chem. 1994;269(1):21–4.
Nelson N. Energizing porters by proton-motive force. J Exp Biol. 1994;196:7–13.
Liu J, Yu Q, Stone GW, Yue FY, Ngai N, Jones RB, et al. CD40L expressed from the canarypox vector, ALVAC, can boost immunogenicity of HIV-1 canarypox vaccine in mice and enhance the in vitro expansion of viral specific CD8+ T cell memory responses from HIV-1-infected and HIV-1-uninfected individuals. Vaccine. 2008;26(32):4062–72.
Prchla E, Kuechler E, Blaas D, Fuchs R. Uncoating of human rhinovirus serotype 2 from late endosomes. J Virol. 1994;68(6):3713–23.
Nawa M. Japanese encephalitis virus infection in Vero cells: the involvement of intracellular acidic vesicles in the early phase of viral infection was observed with the treatment of a specific vacuolar type H+-ATPase inhibitor, bafilomycin A1. Microbiol Immunol. 1997;41(7):537–43.
Yewdell JW, Hill AB. Viral interference with antigen presentation. Nat Immunol. 2002;3(11):1019–25.
Pamer E, Cresswell P. Mechanisms of MHC class I–restricted antigen processing. Annu Rev Immunol. 1998;16:323–58.
Hegde RS, Ploegh HL. Quality and quantity control at the endoplasmic reticulum. Curr Opin Cell Biol. 2010;22(4):437–46.
Collins ND, Newbound GC, Albrecht B, Beard JL, Ratner L, Lairmore MD. Selective ablation of human T-cell lymphotropic virus type 1 p12I reduces viral infectivity in vivo. Blood. 1998;91(12):4701–7.
Galli V, Nixon CC, Strbo N, Artesi M, de Castro-Amarante MF, McKinnon K, et al. Essential role of HTLV-1 orf-I in lethal proliferation of CD4(+) cells in humanized mice. J Virol. 2019;93(19):e00565-19.
Topham NJ, Hewitt EW. Natural killer cell cytotoxicity: how do they pull the trigger? Immunology. 2009;128(1):7–15.
Tanaka Y, Fukudome K, Hayashi M, Takagi S, Yoshie O. Induction of ICAM-1 and LFA-3 by Tax1 of human T-cell leukemia virus type 1 and mechanism of down-regulation of ICAM-1 or LFA-1 in adult-T-cell-leukemia cell lines. Int J Cancer. 1995;60(4):554–61.
Fukudome K, Furuse M, Fukuhara N, Orita S, Imai T, Takagi S, et al. Strong induction of ICAM-1 in human T cells transformed by human T-cell-leukemia virus type 1 and depression of ICAM-1 or LFA-1 in adult T-cell-leukemia-derived cell lines. Int J Cancer. 1992;52(3):418–27.
Banerjee P, Feuer G, Barker E. Human T-cell leukemia virus type 1 (HTLV-1) p12I down-modulates ICAM-1 and -2 and reduces adherence of natural killer cells, thereby protecting HTLV-1-infected primary CD4+ T cells from autologous natural killer cell-mediated cytotoxicity despite the reduction of major histocompatibility complex class I molecules on infected cells. J Virol. 2007;81(18):9707–17.
Lindner S, Kronke J. The molecular mechanism of thalidomide analogs in hematologic malignancies. J Mol Med. 2016;94(12):1327–34.
Davis DA, Shrestha P, Aisabor AI, Stream A, Galli V, Pise-Masison CA, et al. Pomalidomide increases immune surface marker expression and immune recognition of oncovirus-infected cells. Oncoimmunology. 2019;8(2):e1546544.
Johnson JM, Mulloy JC, Ciminale V, Fullen J, Nicot C, Franchini G. The MHC class I heavy chain is a common target of the small proteins encoded by the 3′ end of HTLV type 1 and HTLV type 2. AIDS Res Hum Retrovir. 2000;16(16):1777–81.
Turpin J, Journo C, Ko NL, Sinet F, Carpentier A, Galioot A, et al. Discovery and characterization of auxiliary proteins encoded by type 3 simian T-cell lymphotropic viruses. J Virol. 2015;89(2):931–51.
Fan N, Gavalchin J, Paul B, Wells KH, Lane MJ, Poiesz BJ. Infection of peripheral blood mononuclear cells and cell lines by cell-free human T-cell lymphoma/leukemia virus type I. J Clin Microbiol. 1992;30(4):905–10.
Derse D, Hill SA, Lloyd PA, Chung H, Morse BA. Examining human T-lymphotropic virus type 1 infection and replication by cell-free infection with recombinant virus vectors. J Virol. 2001;75(18):8461–8.
Demontis MA, Sadiq MT, Golz S, Taylor GP. HTLV-1 viral RNA is detected rarely in plasma of HTLV-1 infected subjects. J Med Virol. 2015;87(12):2130–4.
Igakura T, Stinchcombe JC, Goon PK, Taylor GP, Weber JN, Griffiths GM, et al. Spread of HTLV-I between lymphocytes by virus-induced polarization of the cytoskeleton. Science. 2003;299(5613):1713–6.
Pais-Correia AM, Sachse M, Guadagnini S, Robbiati V, Lasserre R, Gessain A, et al. Biofilm-like extracellular viral assemblies mediate HTLV-1 cell-to-cell transmission at virological synapses. Nat Med. 2010;16(1):83–9.
Van Prooyen N, Gold H, Andresen V, Schwartz O, Jones K, Ruscetti F, et al. Human T-cell leukemia virus type 1 p8 protein increases cellular conduits and virus transmission. Proc Natl Acad Sci USA. 2010;107(48):20738–43.
Yamamoto N, Okada M, Koyanagi Y, Kannagi M, Hinuma Y. Transformation of human leukocytes by cocultivation with an adult T cell leukemia virus producer cell line. Science. 1982;217(4561):737–9.
Sowinski S, Jolly C, Berninghausen O, Purbhoo MA, Chauveau A, Kohler K, et al. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat Cell Biol. 2008;10(2):211–9.
Watkins SC, Salter RD. Functional connectivity between immune cells mediated by tunneling nanotubules. Immunity. 2005;23(3):309–18.
Chauveau A, Aucher A, Eissmann P, Vivier E, Davis DM. Membrane nanotubes facilitate long-distance interactions between natural killer cells and target cells. Proc Natl Acad Sci USA. 2010;107(12):5545–50.
Onfelt B, Nedvetzki S, Yanagi K, Davis DM. Cutting edge: membrane nanotubes connect immune cells. J Immunol. 2004;173(3):1511–3.
Eugenin EA, Gaskill PJ, Berman JW. Tunneling nanotubes (TNT) are induced by HIV-infection of macrophages: a potential mechanism for intercellular HIV trafficking. Cell Immunol. 2009;254(2):142–8.
Hashimoto M, Bhuyan F, Hiyoshi M, Noyori O, Nasser H, Miyazaki M, et al. Potential role of the formation of tunneling nanotubes in HIV-1 spread in macrophages. J Immunol. 2016;196(4):1832–41.
Okafo G, Prevedel L, Eugenin E. Tunneling nanotubes (TNT) mediate long-range gap junctional communication: implications for HIV cell to cell spread. Sci Rep. 2017;7(1):16660.
Panasiuk M, Rychlowski M, Derewonko N, Bienkowska-Szewczyk K. Tunneling nanotubes as a novel route of cell-to-cell spread of herpesviruses. J Virol. 2018;92(10):e00090-18.
Roberts KL, Manicassamy B, Lamb RA. Influenza A virus uses intercellular connections to spread to neighboring cells. J Virol. 2015;89(3):1537–49.
Omsland M, Bruserud O, Gjertsen BT, Andresen V. Tunneling nanotube (TNT) formation is downregulated by cytarabine and NF-kappaB inhibition in acute myeloid leukemia (AML). Oncotarget. 2017;8(5):7946–63.
Omsland M, Pise-Masison C, Fujikawa D, Galli V, Fenizia C, Parks RW, et al. Inhibition of tunneling nanotube (TNT) formation and human t-cell leukemia virus type 1 (HTLV-1) transmission by cytarabine. Sci Rep. 2018;8(1):11118.
Donhauser N, Heym S, Thoma-Kress AK. Quantitating the transfer of the HTLV-1 p8 protein between T-cells by flow cytometry. Front Microbiol. 2018;9:400.
Gaudray G, Gachon F, Basbous J, Biard-Piechaczyk M, Devaux C, Mesnard JM. The complementary strand of the human T-cell leukemia virus type 1 RNA genome encodes a bZIP transcription factor that down-regulates viral transcription. J Virol. 2002;76(24):12813–22.
Satou Y, Yasunaga J, Yoshida M, Matsuoka M. HTLV-I basic leucine zipper factor gene mRNA supports proliferation of adult T cell leukemia cells. Proc Natl Acad Sci USA. 2006;103(3):720–5.
Cockerell GL, Rovnak J, Green PL, Chen IS. A deletion in the proximal untranslated pX region of human T-cell leukemia virus type II decreases viral replication but not infectivity in vivo. Blood. 1996;87(3):1030–5.
Yanagihara R. Geographic-specific genotypes or topotypes of human T-cell lymphotropic virus type I as markers for early and recent migrations of human populations. Adv Virus Res. 1994;43:147–86.
Cassar O, Capuano C, Bassot S, Charavay F, Duprez R, Afonso PV, et al. Human T lymphotropic virus type 1 subtype C melanesian genetic variants of the Vanuatu Archipelago and Solomon Islands share a common ancestor. J Infect Dis. 2007;196(4):510–21.
Khoury G, Yurick D, Hirons A, Einsiedel L, Purcell DFJ. Deleted HTLV-1 subtype C proviruses that retain only the X-region persist and predominate in infected Aboriginal Central Australians. 2019. (Unpublished data)
Gessain A, Boeri E, Yanagihara R, Gallo RC, Franchini G. Complete nucleotide sequence of a highly divergent human T-cell leukemia (lymphotropic) virus type I (HTLV-I) variant from melanesia: genetic and phylogenetic relationship to HTLV-I strains from other geographical regions. J Virol. 1993;67(2):1015–23.
Cassar O, Einsiedel L, Afonso PV, Gessain A. Human T-cell lymphotropic virus type 1 subtype C molecular variants among indigenous australians: new insights into the molecular epidemiology of HTLV-1 in Australo-Melanesia. PLoS Negl Trop Dis. 2013;7(9):e2418.
Cassar O, Charavay F, Touzain F, Jeannin P, Grangeon JP, Laumond S, et al. A novel human T-lymphotropic virus type 1c molecular variant in an indigenous individual from New Caledonia, Melanesia. PLoS Negl Trop Dis. 2017;11(1):e0005278.
Einsiedel L, Cassar O, Goeman E, Spelman T, Au V, Hatami S, et al. Higher human T-lymphotropic virus type 1 subtype C proviral loads are associated with bronchiectasis in indigenous australians: results of a case-control study. Open Forum Infect Dis. 2014;1(1):ofu023.
Berneman ZN, Gartenhaus RB, Reitz MS Jr, Blattner WA, Manns A, Hanchard B, et al. Expression of alternatively spliced human T-lymphotropic virus type I pX mRNA in infected cell lines and in primary uncultured cells from patients with adult T-cell leukemia/lymphoma and healthy carriers. Proc Natl Acad Sci USA. 1992;89(7):3005–9.
Caputo A, Haseltine WA. Reexamination of the coding potential of the HTLV-1 pX region. Virology. 1992;188(2):618–27.
Ciminale V, Pavlakis GN, Derse D, Cunningham CP, Felber BK. Complex splicing in the human T-cell leukemia virus (HTLV) family of retroviruses: novel mRNAs and proteins produced by HTLV type I. J Virol. 1992;66(3):1737–45.
Cereseto A, Berneman Z, Koralnik I, Vaughn J, Franchini G, Klotman ME. Differential expression of alternatively spliced pX mRNAs in HTLV-I-infected cell lines. Leukemia. 1997;11(6):866–70.
Ciminale V, D’Agostino DM, Zotti L, Franchini G, Felber BK, Chieco-Bianchi L. Expression and characterization of proteins produced by mRNAs spliced into the X region of the human T-cell leukemia/lymphotropic virus type II. Virology. 1995;209(2):445–56.
Princler GL, Julias JG, Hughes SH, Derse D. Roles of viral and cellular proteins in the expression of alternatively spliced HTLV-1 pX mRNAs. Virology. 2003;317(1):136–45.
Koralnik IJ, Lemp JF Jr, Gallo RC, Franchini G. In vitro infection of human macrophages by human T-cell leukemia/lymphotropic virus type I (HTLV-I). AIDS Res Hum Retrovir. 1992;8(11):1845–9.
Saksena NK, Srinivasan A, Ge YC, Xiang SH, Azad A, Bolton W, et al. Simian T cell leukemia virus type I from naturally infected feral monkeys from central and west Africa encodes a 91-amino acid p12 (ORF-I) protein as opposed to a 99-amino acid protein encoded by HTLV type I from humans. AIDS Res Hum Retrovir. 1997;13(5):425–32.
Slattery JP, Franchini G, Gessain A. Genomic evolution, patterns of global dissemination, and interspecies transmission of human and simian T-cell leukemia/lymphotropic viruses. Genome Res. 1999;9(6):525–40.
Koralnik IJ, Boeri E, Saxinger WC, Monico AL, Fullen J, Gessain A, et al. Phylogenetic associations of human and simian T-cell leukemia/lymphotropic virus type I strains: evidence for interspecies transmission. J Virol. 1994;68(4):2693–707.
Watanabe T, Seiki M, Tsujimoto H, Miyoshi I, Hayami M, Yoshida M. Sequence homology of the simian retrovirus genome with human T-cell leukemia virus type I. Virology. 1985;144(1):59–65.
Watanabe T, Seiki M, Hirayama Y, Yoshida M. Human T-cell leukemia virus type I is a member of the African subtype of simian viruses (STLV). Virology. 1986;148(2):385–8.
Song KJ, Nerurkar VR, Saitou N, Lazo A, Blakeslee JR, Miyoshi I, et al. Genetic analysis and molecular phylogeny of simian T-cell lymphotropic virus type I: evidence for independent virus evolution in Asia and Africa. Virology. 1994;199(1):56–66.
Van Dooren S, Verschoor EJ, Fagrouch Z, Vandamme AM. Phylogeny of primate T lymphotropic virus type 1 (PTLV-1) including various new Asian and African non-human primate strains. Infect Genet Evol. 2007;7(3):374–81.
Sintasath DM, Wolfe ND, Zheng HQ, LeBreton M, Peeters M, Tamoufe U, et al. Genetic characterization of the complete genome of a highly divergent simian T-lymphotropic virus (STLV) type 3 from a wild Cercopithecus mona monkey. Retrovirology. 2009;6:97.
Saksena NK, Herve V, Sherman MP, Durand JP, Mathiot C, Muller M, et al. Sequence and phylogenetic analyses of a new STLV-I from a naturally infected tantalus monkey from Central Africa. Virology. 1993;192(1):312–20.
Saksena NK, Herve V, Durand JP, Leguenno B, Diop OM, Digouette JP, et al. Seroepidemiologic, molecular, and phylogenetic analyses of simian T-cell leukemia viruses (STLV-I) from various naturally infected monkey species from central and western Africa. Virology. 1994;198(1):297–310.
Ibrahim F, de The G, Gessain A. Isolation and characterization of a new simian T-cell leukemia virus type 1 from naturally infected celebes macaques (Macaca tonkeana): complete nucleotide sequence and phylogenetic relationship with the Australo-Melanesian human T-cell leukemia virus type 1. J Virol. 1995;69(11):6980–93.
Van Dooren S, Meertens L, Lemey P, Gessain A, Vandamme AM. Full-genome analysis of a highly divergent simian T-cell lymphotropic virus type 1 strain in Macaca arctoides. J Gen Virol. 2005;86(Pt 7):1953–9.
Furukawa Y, Usuku K, Izumo S, Osame M. Human T cell lymphotropic virus type I (HTLV-I) p12I is dispensable for HTLV-I transmission and maintenance of infection in vivo. AIDS Res Hum Retrovir. 2004;20(10):1092–9.
Ding W, Kim SJ, Nair AM, Michael B, Boris-Lawrie K, Tripp A, et al. Human T-cell lymphotropic virus type 1 p12I enhances interleukin-2 production during T-cell activation. J Virol. 2003;77(20):11027–39.
Goncalves DU, Proietti FA, Ribas JG, Araujo MG, Pinheiro SR, Guedes AC, et al. Epidemiology, treatment, and prevention of human T-cell leukemia virus type 1-associated diseases. Clin Microbiol Rev. 2010;23(3):577–89.
Tagaya Y, Matsuoka M, Gallo R. 40 years of the human T-cell leukemia virus: past, present, and future. F1000Res. 2019. https://doi.org/10.12688/f1000research.17479.1.
Acknowledgements
We thank D. Ahern for editorial support.
Funding
This work was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, and the National Health and Medical Research Council of Australia (APP1129320 and APP1052979).
Author information
Authors and Affiliations
Contributions
SS, CPM, and GF wrote the manuscript; SS, RM, and VGproduced the figures; GK, DY, and DFJP provided HTLV-1C sequence information. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
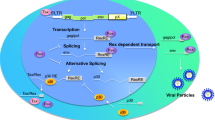
Structure of the HTLV-1 proviral genome. The proviral DNA with the LTRs, and the unspliced, singly spliced and doubly spliced mRNA transcripts are shown. The names of the gene transcripts are depicted inside each specific box. Solid lines indicate the exons and the dotted lines indicate the introns.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Sarkis, S., Galli, V., Moles, R. et al. Role of HTLV-1 orf-I encoded proteins in viral transmission and persistence. Retrovirology 16, 43 (2019). https://doi.org/10.1186/s12977-019-0502-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12977-019-0502-1