
Abstract
Through considerable effort in research and clinical studies, the immune system has been identified as a participant in the onset and progression of brain injury after ischaemic stroke. Due to the involvement of all types of immune cells, the roles of the immune system in stroke pathology and associated effects are complicated. Past research concentrated on the functions of monocytes and neutrophils in the pathogenesis of ischaemic stroke and tried to demonstrate the mechanisms of tissue injury and protection involving these immune cells. Within the past several years, an increasing number of studies have elucidated the vital functions of T cells in the innate and adaptive immune responses in both the acute and chronic phases of ischaemic stroke. Recently, the phenotypes of T cells with proinflammatory or anti-inflammatory function have been demonstrated in detail. T cells with distinctive phenotypes can also influence cerebral inflammation through various pathways, such as regulating the immune response, interacting with brain-resident immune cells and modulating neurogenesis and angiogenesis during different phases following stroke. In view of the limited treatment options available following stroke other than tissue plasminogen activator therapy, understanding the function of immune responses, especially T cell responses, in the post-stroke recovery period can provide a new therapeutic direction. Here, we discuss the different functions and temporal evolution of T cells with different phenotypes during the acute and chronic phases of ischaemic stroke. We suggest that modulating the balance between the proinflammatory and anti-inflammatory functions of T cells with distinct phenotypes may become a potential therapeutic approach that reduces the mortality and improves the functional outcomes and prognosis of patients suffering from ischaemic stroke.
Similar content being viewed by others


Introduction
Stroke is not only one of the main causes of death but also the primary cause of long-term disability worldwide; however, extensive therapeutic options are lacking, which creates a dominating economic and medical burden [1]. Ischaemic stroke results from the blockade of the blood vessels supplying the brain, accounting for 87% of all strokes in the USA [1] and is currently the main focus of stroke research.
Stroke can occur at any age but mostly occurs at an older age (beyond 65 years old) [2]. Elderly patients have an elevated risk of complications and worse outcomes after treatment compared with younger patients, partially due to alterations in the immunological response to stroke [3]. Women are more vulnerable to stroke after menopause than before menopause due to the lack of female gonadal hormone protection, which may regulate T cells [4].
Despite numerous factors affecting the onset and progression of brain injury after stroke, the consistent, basic process is intimately connected with the immune response, including T cell responses. In the brain of healthy people, only a few T cells enter the central nervous system (CNS) and are found in the parenchyma, perivascular space and cerebrospinal fluid (CSF) due to the intact blood-brain barrier (BBB). These cells perform immune surveillance to maintain CNS homeostasis in cooperation with CNS-resident immune cells [5]. After stroke onset, the acute cessation of the blood supply induces primary irreversible tissue injury and results in neural cell death, the site of which constitutes the ischaemia core; neural cell death results in a subsequent release of damage-associated molecular patterns (DAMPs). The ensuing brain injury that damages the peri-infarct area (the penumbra) is caused by a rapid cascade of events such as excitotoxicity, oxidative stress and mitochondrial disturbance [6]. In the process of neural cell death, different cellular signalling pathways that regulate autophagy and apoptotic cell death (Mst1, ULK1, Bax, Caspase-3 and Bcl-2), necroptotic cell death (TRAF2 and RIPK1/RIPK3/MLKL) [7], the cellular metabolic state (TSC1/TSC2, p-mTOR, and mTORC1), the oxidative defence system (FoxO1, β-catenin/Wnt, and Yap1) and inflammatory reactions (jak2/stat3 and Adamts-1) are changed [8,9,10]. However, the cellular signalling pathways related to jak2/stat3 and Adamts-1 involved in regulating inflammatory reactions are found to be predominantly localized in macrophages/microglia [9] in the post-ischaemic brain, which may account for the fact that these pathways first trigger inflammation in brain-resident immune cells, including microglia and macrophages [11], after ischaemic stroke onset. With the release of inflammatory factors, cytokines, chemokines and DAMPs, a large number of peripheral immune cells infiltrating the injured site participate in innate and adaptive immune responses. Additionally, neutrophils, monocytes and CD8+ cells are regarded as the first peripheral immune cells to invade the injured brain within hours after stroke onset [11]. Subsequently, CD4+ cells are reported to infiltrate the brain approximately 24 h after ischaemia [11]. Regulatory T (Treg) cells remain in the injured brain for more than 30 days after ischaemia to control the aggravation of inflammation by regulating the levels of inflammatory factors [12]. Nonetheless, the roles of T cells in tissue damage and repair have not been completely elucidated.
Over the past 20 years, a number of studies have considered the immune system to have vital effects on the process and development of brain injury following ischaemia, especially the recruitment and function of macrophages and neutrophils rather than T cells in the injured brain. However, recently, due to the variety of T cell types, an increasing number of researchers have found T cells to be vitally involved in the onset and progression of ischaemic stroke because these cells not only promote the occurrence of inflammation by infiltrating the injured brain in the early stages after ischaemic stroke [13] but also exert effects on repair and functional improvement in the late stage after ischaemic stroke [14]. T helper (Th) 1 and Th2 cells were the first subsets of Th cells identified [15]. Then, Th17 cells, which differentiate in the presence of both TGF-β and IL-6, were recognized to generate proinflammatory cytokines [16]. The expression of Foxp3 is a characteristic of Treg cells that exert immunomodulatory functions [17]. Follicular T helper (Tfh) cells in the follicles were found to facilitate B cell responses [18]. Some subsets of T cells, such as Th9, Th22 and Th25 cells, are recognized by the production of different cytokines (expressing IL-9, IL-22 and IL-25, respectively) [19,20,21]. CD4loCD40+ T cells, recognized as Th40 cells, were found to greatly expand under autoimmune conditions and play a vital role in type 1 diabetes [22]. The diverse subsets of T cells are simply presented in Fig. 1.

Functions of Th cell subsets
Here, we described the different subsets of T cells and identified several vital T cell subsets related to acute and chronic processes during ischaemic stroke, especially Th1, Th2, Th17, γδ T, Treg and even Th40 cells. We also reported several very promising therapeutic targets related to the modulation of T cell responses to improve the strengths of immune responses in the injured brain. Further research towards understanding the mechanisms modulating the emergence of cerebral inflammation mediated by T cell subsets may facilitate the development of a novel or adjunctive way to treat stroke.
Subsets and plasticity of Th cells
CD4+ cells (Th cells) exert distinct effects during the immune response. Remarkably, Th cells have the capability of differentiating into subsets with functionally diverse phenotypes, such as Th1, Th2, Th9, Th17, Th22, Th25, Th40, Tfh and Treg cells, in response to different cytokine milieus and antigen stimulation [23, 24]. It is widely known that the differentiation of Th1 cells is induced by successive responses to interferon (IFN)-g and interleukin (IL)-12, which are initiated by coordinated signalling through the communication between STAT1-associated cytokine receptors and T cell receptor (TCR) [25]. Th1 cells have been shown to enhance and promote the elimination of certain intracellular pathogens [15]. Through the interaction between TCR signalling and IL-4 receptor signalling mediated by STAT6, which may react synergistically to upregulate low expression of GATA-3, a subset-determining transcription factor of Th2 cells, Th2 differentiation is induced [26], and Th2 cells function to enhance the clearance of parasites [15]. Th17 cells generate proinflammatory effector cytokines, including IL-17A, IL-17F, IL-22 and IL-26; of these cytokines, IL-17A and IL-17F modulate tissue inflammation by inducing the secretion of proinflammatory cytokines and chemokines, such as IL-1β and IL-6 [27, 28]. Compared with their proinflammatory role, the regulatory role of Th17 cells, which involves suppressing Th17-induced inflammation, may be linked with IL-10 and IL-21 by Foxp3+ Treg cells and type 1 regulatory T (Tr1) cells [29]. It has been reported that exposing effector cells to TGF-β and IL-6 sustainably drives the production of IL-17 and IL-10, which is important in modulating the level of the potentially detrimental Th17 cell-mediated immune response in an experimental autoimmune encephalomyelitis (EAE) model [30]. Th9 cells are a newly discovered CD4+ Th cell subset that mainly secrete the lineage-specific cytokine IL-9 [19, 31]. Based on the discovery study, Th9 cells have indispensable roles in the initiation and development of immune responses [32]. Generally, Th9 cells are proinflammatory and participate in various immune-related diseases, highlighting their pathological roles in inflammation and the immune system [33]. However, Th9 cells are also reported to have a protective effect on parasitic infections, indicating that these cells have various functions [33]. Th22 cells are localized in the skin by their high expression of CCR4, CCR6 and CCR10 [20, 34], and IL-22 has been shown to activate innate immune responses against pathogenic bacteria, especially in respiratory and gut epithelial cells [35], which indicates their essential roles in skin homeostasis and skin disease pathogenesis. Importantly, it has been reported that activation of AHR, a subset-determining transcription factor of Th22 cells, after acute ischaemic stroke plays a vital role in brain ischaemic injury by modulating astrogliosis and neurogenesis [36]. In addition, Th22 cells are closely related to multiple types of diseases, such as infections, autoimmune diseases, hepatitis, pancreatitis, rheumatoid arthritis (RA) and tumours, because of the wide distribution of IL-22R [37]. The Th25 cell-secreted cytokine IL-25 was reported to be related to Th2 responses [21]. Furthermore, IL-4, IL-5 and IL-13 gene expression induced by IL-25 results in a Th2-like response characterized by increased serum levels of IgE, IgG1 and IgA; blood eosinophilia; and a series of pathological changes, including infiltration of eosinophils, increased production of mucus and hyperplasia/hypertrophy of epithelial cells in the lungs and digestive tract and participates in the clinical evolution of leprosy in cooperation with the Th2 cytokine profile [35, 38, 39].
When CD40 is uniquely expressed on T cells, distinct from the receptor expressed on antigen-presenting cells (APCs), it serves as a functional receptor on T cells, representing a new T cell subset [40]. Studies have found that Th40 cells (CD4loCD40+ T cells) produce IFN-γ (signal of Th1 cells) and IL-17A (signal of Th17 cells) [41], are proinflammatory and balance Treg cells to maintain a homeostatic state and that Th40 cells from healthy bodies are able to produce regulatory Th2 cytokines to control autoimmunity, while pathogenic Th40 cells expand quickly throughout the progression of diabetes [42, 43]. Th40 cells not only have vital effects in autoimmunity [44] but also importantly play a pivotal role in the injured brain after cardiac arrest and cardiopulmonary resuscitation (CA/CPR) in mouse models [45]. Tfh cells, which are characterized by their expression of CXCR5 [18, 46], a B cell homing chemokine CXCL13 receptor, are a novel subset of T cells that play essential roles in facilitating B cell responses, including B cell affinity maturation, class switch recombination and plasma and memory B cell maintenance for humoral memory [47,48,49]. Studies have also verified that both Tfh cells and CD8+ T cells share key features of a memory cell precursor gene expression programme containing Bcl6 and IL-17Rα, indicating that Tfh cells have the capacity to form memory early [50]. However, they have not been researched in stroke.
Treg cells are divided into two populations, Foxp3+ Treg cells and Foxp3− Treg cells, the latter of which includes Th3 and Tr1 cells [51]. Foxp3+ Treg cells exert vital functions involved in downregulating the pathological T cell response through secretion of the anti-inflammatory cytokines IL-10 and TGF-β [17, 52], while Tr1 cells play their regulatory role by affecting different target cells, including effector CD4+ and CD8+ T cells, myeloid APCs and B cells [53]. There are also interactions between the two types of Treg cells, and it has been demonstrated that Foxp3+ Treg cells are required for the initial stage of inflammatory target organ tolerance induction, while Tr1 cells have an important effect on the maintenance of long-lasting tolerance [54, 55]. Recently, numerous studies have revealed the mechanisms underlying Treg cell participation in tissue regeneration, which occurs not only through tissue-specific anti-inflammatory effects but also through direct regenerative mechanisms [56, 57]. Th3 cells primarily secrete TGF-β, which is distinct from TGF-β secreted by Th2 cells, to provide help for IgA and suppress functions of Th1 and other immune cells in the gut [58]. Th3 cells are mainly induced by oral antigens, and enhanced differentiation from Th0 precursors can be achieved by culture with TGF-β, IL-4, IL-10 and anti-IL-12 antibodies [59]. Furthermore, Th3 cells secreting TGF-β are capable of suppressing systemic autoimmune and inflammatory responses to treat related diseases [60]. Tr1 cells generate immunosuppressive cytokines, including IL-10 and TGF-β, to inhibit the effects of effector immune cells and are reported to be induced upon antigen exposure [53]. Additional studies have tested the protective role of Tr1 cells in various mouse models, including models of multiple sclerosis, intestinal inflammation, EAE and even NOD mice with diabetes [61]. Nasal tolerance in models of atherosclerosis, cardiac ischaemia, lupus and stroke has also been identified [62]. Mucosal antigens have also been found to effectively treat animal models of stroke, but the underlying mechanisms remain to be elaborated [63]. In conclusion, recent reports indicate that endogenous Treg cells exert a neuroprotective function by secreting TGF-β and IL-10 and have a protective effect against ischaemic brain injury by increasing the number of Treg cells in the circulation [64]. Therefore, the function of Treg cells after ischaemia needs to be explored in depth.
Although there are many diverse subsets of CD4+ T cells, the plasticity of Th cells has been extensively reported and is contrary to previous views stating that the functions of distinct T cell subsets are irreversible. Initially, Th1, Th17, Th22 and Th40 cells were classified as a group that predominantly protects against extracellular pathogens by secreting cytokines with the same function, while Th2, Th9 and Th25 cells were thought to mainly exert functions in autoimmune disease and allergic inflammation [39, 65, 66]. Notably, it appears that induced Treg (iTreg) cells (induced in the periphery) and Th17 cells may be relatively unstable and have flexibility in their differentiation and function because of the unstable expression of Foxp3 by iTreg cells and that of IL-17 by Th17 cells [67, 68]. Th17 cells have been shown to transdifferentiate into Treg cells by altering their characteristic transcriptional profile and acquiring potent regulatory properties through the effects of TGF-β signalling and AHR, indicating that Th17 cells may secrete anti-inflammatory cytokines to attenuate inflammation [69]. Likewise, Th17 cells have been reported to shift into nonclassical Th1 cells and Th2 cells in the presence of IL-12 or IL-4, respectively [70, 71]. Treg cells have been demonstrated to be capable of becoming Th1 cells in the presence of IL-12 in vitro and attaining Treg-Th17 plasticity in the presence of IL-6 and TGF-β [72, 73]. There are other transitions, such as Th2 cells transforming into Th9 cells and Th0 cells in response to TGF-β or IL-12/IFNs, respectively, and Th9 cells converting into the Th1 phenotype and subsequently producing IFN-γ in vivo [74,75,76]. That means that some cell subsets can transform each other to maintain homeostasis. These findings provide novel insight into the immune response of T cells after ischaemic stroke and a more comprehensive understanding of the functions of T cell subsets.
T cells and ischaemic stroke (Fig. 2)
Spatial and temporal features of T cell responses after ischaemia
Several studies using recombination activation gene (Rag)-deficient mice and severe combined immunodeficiency (SCID) mice, which both lack T cells and B cells, have shown significantly reduced infarct volumes and lower neurological deficits in these mice compared with the corresponding wild-type mice, regardless of the stroke induction model studied. By comparing Rag1−/− mice reconstituted with B cells to Rag1−/− mice reconstituted with CD3+ T cells, studies have indicated that T cells likely have a vital effect on early stroke evolution and exert a detrimental effect as early as 24 h after stroke through an antigen-independent mechanism [77,78,79]. Our study showed that the protective roles of T cell deficiency in brain injury after ischaemic stroke were relevant to transient middle cerebral artery occlusion (tMCAO) in a rodent model, indicating that reperfusion after ischaemic stroke might be closely related to T cell responses [80]. Additionally, a study found that depletion of CD4+ T cells or CD8+ T cells reduced infarct volume in late stages after tMCAO [81]. Different studies and results indicate that distinct T cell subsets invade the brain dynamically and play various roles in the different stages after experimental ischaemic stroke (Table 1). Studies have shown that T cell infiltration occurs from hours to 30 days after stroke. Researchers have not arrived at a consistent or definite conclusion on the time of peak T cell infiltration into the injured brain after stroke.

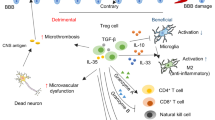
Functions of T cell subsets in the acute and chronic phases after ischaemic stroke. In the acute phase, necrotic cells release DAMPs to activate brain-resident microglia, and then activated microglia secrete cytokines, such as IL-23, to recruit γδ T cells via an antigen-independent pathway. IL-17 released by γδ T cells, other inflammatory cytokines and antigens from the injured brain induce antigen-dependent immune responses by Th1, Th2, Th17 and Th40 cells. Th2 cells exert a protective role by promoting microglial M2 polarization. Other cells interact with M1 microglia to play detrimental roles in the injured brain or secrete proinflammatory cytokines. In the chronic phase, Treg cells may exist until 1 month after stroke onset and perform roles in recovery via several pathways, including scar formation, neuronal repair and revascularization
Gelderblom et al. found that T lymphocyte numbers were increased in the infarcted hemisphere on day 3 after experimental ischaemia reperfusion (I/R) and reported that significant infiltration of CD4−/CD8− lymphocytes occurred on days 1 and 3, while the infiltration of CD4+ cells increased on day 3 after experimental I/R [91]. Additional studies demonstrated that T lymphocytes (CD3+ cells) significantly increased in number within the cerebral infarct and in peri-infarct areas, with significantly fewer T lymphocytes in the spleen at 24 h after experimental I/R, while the number of T lymphocytes in the circulation was unchanged at the same time [77, 92, 93]. Our studies also found that stroke could induce lymphopenia and reduce splenocyte T cell numbers due to the roles of T cells at 48 or 72 h after stroke [94, 95]. Saino et al. found that CD4+ cells infiltrated the infarct cortex within 3 h after stroke [96]. Another study demonstrated that Th40 lymphocyte numbers were elevated 3 h after cardiac arrest, a cause of ischaemic stroke, and CA/CPR, decreased greatly in 24 h, and increased again on day 2 and day 3 [45].
Liesz and co-workers used spectratype/immunoscope analysis to demonstrate that the number of CD3+ T cells peaked at 7 days after permanent middle cerebral artery occlusion (pMCAO) and that clonal T cell expansion occurred in T cells isolated from the ipsilateral brain at 7 days after pMCAO and in T cells isolated from the spleen at 14 days after pMCAO [82]. Moreover, Xie et al. showed that CD4+ and CD8+ T cells exhibited prolonged activation after experimental ischaemic stroke, indicating that these cells had a greater role in neuronal repair than Treg cells [85]. Vindegaard and Munoz-Briones found an obvious increase in CD3+ T cell numbers within the ipsilateral brain, not only within the infarct core but also within the corpus callosum on days 14 and 28 after pMCAO [97]. Stubbe et al. demonstrated that Treg cells accumulated and proliferated in the ipsilateral brain on days 14 and 30 post-stroke and were accompanied by increases in the number and activation of microglia after tMCAO [12]. In two previous studies, the authors demonstrated an obvious increase in T cell infiltration in the injured brain on day 7 through day 30 or 60 of the post-stroke inflammatory response in rodents after cerebrocortical photothrombosis or pMCAO, respectively [98, 99]. These results show that T cells may exert a detrimental function in the early stage while playing a protective role in the late stage of the inflammatory response after ischaemic stroke. However, the mechanisms and functions of T cells in the CNS remain to be clarified (Table 2). Although we know that Th cells exert different functions at different stages after stroke, Th cells are divided into several subsets, and we have to clarify which of these subsets play a dominant role during the onset and development of stroke.
Different T cell subsets infiltrating the injured brain (Fig. 3)
Th1 and Th2 cells related to brain injury
We identified that Th1 and Th2 cells have vital effects on the early phase of the post-stroke inflammatory response by evaluating the infarct sizes and neurological scores 2 days after stroke in a Th1- and Th2 cell-deficient mouse model [84]. Th1 cells and Th2 cells have different impacts on ischaemic brain injury. Th1 cells secrete proinflammatory cytokines, such as IFN-γ and chemokines, and produce numerous reactive oxygen species (ROS) and nitric oxide to destroy the BBB. Th2 cells secrete anti-inflammatory cytokines, such as IL-4, IL-10 and IL-13, to promote nerve growth factor (NGF) production, debris removal, tissue remodelling and repair, and angiogenesis after brain ischaemia [83, 86]. Interestingly, Th1/Th17 cells and Th2 cells separately perform crosstalk with M1 and M2 microglia [100]. Initially, microglia are derived from macrophages that undergo migration and differentiation in the process of original haematopoiesis in the foetal yolk sac, and then they are localized in the brain with the ability to proliferate in the process of neonatal growth, while granulocyte-monocyte progenitors are the precursors of macrophages in the periods of development and adulthood [87, 101]. Both microglia-derived macrophages (MiDM) and monocyte-derived macrophages (MoDM) in the injured brain show the capabilities of polarizing into a proinflammatory or anti-inflammatory (M1 or M2, respectively) phenotype and performing phagocytosis function, as well as exhibiting a high degree of morphological plasticity [101]. However, MiDM perform more vital roles due to their abilities to facilitate neuronal viability and modulate neuronal excitability as well as secrete NGF [88]. At the same time, activation of MiDM relies on ATP/ADP signalling, which may account for the number of MiDM being influenced by energy deficiency and alterations in local blood perfusion [89, 90]. Th1 cells can promote M1 polarization through the induction of proinflammatory cytokines, including TNF-α and IFN-γ [102]. M1 microglia also induce and recruit Th1 cells by secreting IL-12 and TNF-α and expressing chemokines, such as CXCL9 and CXCL10 [103]. Th2 cells promote M2 polarization by secreting anti-inflammatory cytokines (IL-4, IL-10 and IL-13) [102, 104, 105] and increase the levels of insulin-like growth factors, neurotrophic factors secreted by microglia, to augment the neuroprotective role of microglia [83, 106, 107]. M2 cells can induce and recruit Th2 cells by secreting IL-4, CCL17, CCL22 and CCL24 [102, 103]. Th17 cells have also been shown to cause brain injury through crosstalk with M1 microglia via secreted IL-17 [100]. A recent study found that Th2/Th17 cells could enhance blood perfusion in ischaemic injury by regulating angiogenesis and inducing endothelial sprouting [108]. However, there are studies implying that M1/M2 cells have complicated roles in view of the complexity and diversity of M2 subtypes [74] and the phenotypic transformation between M2 and M1; these studies have identified that both M1 cells and M2 cells have proinflammatory and anti-inflammatory functions [101] rather than oversimplified single functions and that the functions of these cells are more complex in vivo than in vitro and are harder to study in humans than in mice. Whether and how these functions occur after ischaemic stroke remain to be elucidated.

Different T cell subsets infiltrate the injured brain. Th1 cells secrete proinflammatory cytokines, such as IFN-γ and TNF-α, to promote M1 polarization. M1 microglia also induce and recruit Th1 cells by secreting IL-12 and TNF-α and expressing chemokines, such as CXCL9 and CXCL10. Moreover, Th2 cells secrete anti-inflammatory cytokines, such as IL-4, IL-10 and IL-13, to promote M2 polarization, and M2 cells can induce and recruit Th2 cells by secreting IL-4, CCL17, CCL22 and CCL24. Th40 cells, which are proinflammatory, secrete both IFN-γ and IL-17A and infiltrate the injured brain in the early stage after brain injury. γδ T cells secrete proinflammatory IL-17 to aggravate brain injury. Moreover, γδ T cells and Th17 cells activate proinflammatory microglia by modulating the FasL/PTPN2/TNF-α signalling pathway, which aggravates ischaemic brain injury. Treg cells interact with microglia and modulate microglial polarization from the M1 phenotype into the M2 phenotype via IL-10, and they also regulate astrogliosis by producing the cytokine amphiregulin (Areg). CD8+ T cells can recruit CD4+ mononuclear cells via the cytokine IL-16 after femoral artery ligation, and CD4+ T cells contribute to the imbalance in M1 and M2 polarization
Th40 cells related to brain injury
Deng et al. identified a new T cell subset, the Th40 cell subset, that is proinflammatory, secretes both IFN-γ and IL-17A and infiltrates the injured brain in the early stage (within 3 h) after CA/CPR or global cerebral ischaemia to contribute to neuronal injury [45]. Additionally, Th40 cell numbers increase again at 72 h, indicating a role in the sustained immune response [45]. Many studies have identified roles for these cells in autoimmune diseases, such as type 1 diabetes. Nonetheless, little is known about the roles of Th40 cells in the MCAO mouse model of ischaemia. More studies on Th40 cells in the injured brain after ischaemic stroke are needed.
Γδ T and Th17 cells related to brain injury
Shichita et al. proposed that γδ T cells, not Th17 cells, secrete proinflammatory IL-17 to aggravate I/R brain injury in the delayed phase (day 3) [109], although a previous study identified that IL-17-producing cell numbers peaked 3–5 days after injury in the ipsilateral cerebral hemispheres of patients following ischaemic stroke [110]. CD3+CD4−CD8− T cells, also recognized as double-negative T cells (DNTs) and including γδ T cells, have been shown to coordinate immune and inflammatory homeostasis to exert functions in peripheral immune-related diseases [111, 112]. Subsequently, Meng et al. discovered that the number of DNTs was significantly elevated in a time-dependent manner, in both the injured brain and the peripheral blood, in both stroke patients and an MCAO mouse model. The experimental model showed that DNTs prominently infiltrated the injured brain from day 1 to day 3 after MCAO. The infiltrating DNTs activated proinflammatory microglia by orchestrating the FasL/PTPN2/TNF-α signalling pathway and then augmented cerebral immune and inflammatory responses to aggravate ischaemic brain injury [113]. Nonetheless, there are studies supporting the conclusion that Th17 cells play a detrimental role in the chronic phase of ischaemic stroke. These cells were found to exist in the injured brain at 1 week after traumatic brain injury (TBI) and drove the cytotoxicity of CD8+ T cells at the later stage, which potentiated the detrimental effects of CD8+ T cells seen in TBI [114, 115]. Likewise, increasing levels of IL-17 associated with worse neurological outcomes were found in the peripheral blood through 3 days after stroke onset in stroke patients [116, 117]. It remains to be clarified which types of immune cells are responsible for producing IL-17.
Studies have also found that γδ T cells link the innate immune response with the adaptive immune response. Because studies have shown that the acute adverse effects of T cells following acute ischaemic stroke are not associated with adaptive immune mechanisms, such as antigen recognition or costimulatory pathways [78], γδ T cells and natural killer T (NKT) cells may play detrimental roles in brain injury through the innate immune response.
Treg cells related to brain injury
Liesz et al. demonstrated that Treg cells were activated 5 days after MCAO and restrained to the peri-infarct zone, playing a protective role after brain injury [14]. Furthermore, Treg cells prevented secondary infarct growth by suppressing excessive production of proinflammatory cytokines and by orchestrating infiltration of lymphocytes and microglia, mainly via IL-10 signalling [14], in the ischaemic brain. Treg cells interact with microglia [100] and modulate microglial polarization from the M1 phenotype to the M2 phenotype via IL-10 [118]. Interestingly, a study showed that Treg cells gathered in the ipsilateral cerebral hemisphere during the chronic phase of ischaemic brain injury and regulated astrogliosis by producing the cytokine amphiregulin (Areg) [57]. Studies have also shown that Treg cells may adapt to different tissue environments by expressing distinct genes related to the tissue site and augment neurological recovery by suppressing neurotoxic astrogliosis by producing Areg in the context of the massive amplification and infiltration occurring in the chronic phase of stroke [119]. The 5-HT7 expression represents a specific way, one of hundreds, to amplify brain Treg cell functions [119]. Conversely, the function of Treg cells is controversial due to Treg cell depletion leading to a better outcome within 24 h and no progression until 1 week by ameliorating microvascular thrombus formation [120]. Additionally, Kleinschnitz et al. verified that Treg cells exerted a detrimental effect on mice with acute ischaemic stroke by inducing dysfunction in the cerebral microvasculature in the early phase [120]. In addition, the phenomenon of stroke-induced immunosuppression characterized by lymphopenia presents with a reduction in natural killer (NK) cell, B cell and T cell numbers in the peripheral blood and spleen and is thus vulnerable to bacterial infection, especially urinary tract infection and pneumonia in the subacute and chronic stages of ischaemic stroke [86]. This immunosuppression poses the question of whether adoptive transfer of Treg cells will create a more unbalanced immune state leading to an increase in the incidence of infection. Luckily, Li et al. revealed that Treg cells not only contribute to brain tissue protection and modulate CNS damage from a peripheral location but also regulate the homeostatic equilibrium of peripheral immune responses, such as simultaneously correcting immunosuppression and attenuating peripheral inflammation [121]. Previous literature has shown that Th cells possess plasticity, indicating that Th17 cells can transform into another T cell subset (such as Treg cells) within the injured brain during the process of neuroinflammation [69].
Nonetheless, neutrophils are still recognized as one of the first cells to infiltrate the injured brain, causing BBB disruption, cerebral oedema and brain injury, which indirectly prompt the infiltration of T cells [122]. In conclusion, more research is required to elucidate the biological roles of Treg cells to better understand how to modulate the immune system and facilitate healing after stroke.
Functions of CD4+ and CD8+ T cells related to stroke
Feng et al. found that chronic colitis exacerbated brain injury after stroke by inducing gut-derived CD4+ T cells to create an imbalance in M1 and M2 microglia/macrophages and increase the numbers of non-gut-derived CD4+ T cells infiltrating the brain [123]. CD4+ T cells have been identified to aggravate brain inflammation and induce neuronal death [100, 123]. Removal of the CD4+ T cell population attenuates apoptosis and enhances neurogenesis, while the elimination of CD25+ T cells, which include Treg cells, impairs functional recovery partly through the inhibition of neurogenesis after permanent experimental stroke [96]. Nonetheless, researchers have demonstrated that reducing astrogliosis and/or preserving neurogenesis play a vital role in protecting the injured brain during the repair stage after stroke [36, 119]. It is not clear which subsets of T cells are primarily responsible. Other researchers have identified that CD8+ T cells not only infiltrate the site of collateral vessel growth but also recruit CD4+ mononuclear cells to this site via the cytokine IL-16 after femoral artery ligation [124]. Interestingly, studies have found that stroke-induced immunodepression may be protective by reducing naive T cell and CD8+ CD45RA+ effector memory T cell (TEMRA) numbers to attenuate detrimental long-term antigen-specific immune responses in the CNS [125].
Therapy related to T cells and stroke (Table 3)
Cytokines, small molecules, neutralizing antibodies and cell epitopes as targets
We identified that IL-4 knockout (KO) mice have worse neurological outcomes than wild-type mice due to the increases in the Th1/Th2 ratio and Th1 polarization associated with greater injury [126]. Another study proposed that the source of IL-4 is neurons rather than T cells in tissue in the context of ischaemic brain injury, and the authors proposed that administering recombinant mouse IL-4 (rIL-4) subcutaneously could have a delayed role in protecting ischaemic brain tissue and improving outcomes, probably by polarizing the microglia into the healing M2 phenotype [148]. The specific mechanism remains to be clarified. Li and co-workers showed that astrocytic IL-15 could increase the severity of post-ischaemic brain injury by activating NK-, CD8+ T- and CD4+ T cell-mediated immunity [127]. Lee et al. identified that ablation of IL-15 using an anti-IL-15 neutralizing antibody decreased brain damage after ischaemic stroke by decreasing NK, CD8+ T and CD4+ T cell infiltration into the brain [128]. However, IL-15 was also reported to defend astrocytes against oxygen-glucose deprivation (OGD)-induced damage and death, and astrocytes could protect neurons from ischaemic injury and sustain BBB integrity [129], which are contradictory to IL-15 deficiency exerting a protective role in brain injury after ischaemic stroke. Clarkson et al. illustrated that treatments blocking T cell-derived IL-21 might improve neurological outcomes by reducing lymphocytic brain infiltration and attenuating neuronal autophagy [149]. Xiao et al. found that pretreatment with IL-33, a new member of the IL-1 cytokine family, improved neurological outcomes by suppressing the Th1 cell response and improving the Treg cell response in mice, indicating that IL-33 might play a long-term protective role by modulating peripheral immune responses after ischaemia [130, 150].
Bodhankar et al. identified that PD-1 and CTLA-4 had inhibitory effects on the activation of T cells in a rodent stroke model [151] and that blockade of the PD-L1 checkpoint significantly limited the CNS inflammatory response and improved neurological outcomes by partially reversing splenic atrophy and increasing the accumulation of CD8+ Treg cells in the lesioned brain hemisphere [131]. This suggests the application potential of a novel therapy using accessible humanized anti-PD-L1 antibodies to treat human stroke subjects and confirms that PD-1 is inversely correlated with the absolute amount of CD4+ T central memory (TCM) cells in ischaemic patients [135]. However, these conclusions remain to be validated in clinical trials.
Systemic administration of docosahexaenoic acid (DHA), a major form of omega-3 polyunsaturated fatty acids (n-3 PUFAs) in the CNS, may reduce post-stroke brain injury by attenuating T cell infiltration, thereby decreasing the immune response in injured brain tissue and promoting the polarization of macrophages into the healing M2 phenotype [133].
Granulocyte colony-stimulating factor (G-CSF) was reported to have immunomodulatory effects and suppress the migration and maturation of dendritic cells (DCs) to exert neuroprotective effects [136]. The administration of a single dose of G-CSF can attenuate the recruitment of T cells to the injured brain following stroke, which has positive effects [140].
Toll-like receptors (TLRs), especially TLR2 and TLR4, have been broadly reported to play detrimental roles following ischaemic stroke, and TLR2-deficient and TLR4-deficient mice have been shown to have neurological function protection after ischaemic stroke mediated by attenuation of the activation of T cells. TLR8 may produce the same effect [141]. Inhibition of acetyl coenzyme A carboxylase 1 (ACC1), which has been achieved by either conditional knockout or pre-treatment with caloric restriction, is a novel approach to balance Treg cells and Th17 cells and has a protective effect on brain injury after ischaemic stroke [142].
Recombinant T cell receptor ligands (RTLs) have been studied to find a novel target to improve neurological outcomes after ischaemic stroke [152]. Zhu et al. demonstrated that in addition to RTL551, RTL1000 could improve long-term neurological outcomes following ischaemic stroke by inhibiting the activation or infiltration of CD3+ T cells and other proinflammatory cells [152].
Treg cells as a therapeutic target
Treg cells may improve stroke outcomes by suppressing IL-17+ γδ T cell proliferation by altering the intestinal flora rather than being present in the brain [153]. Directly augmenting Treg cell numbers through adoptive transfer has been shown to be an efficacious way to protect the injured brain and promote long-term recovery after stroke [144]. However, there are many issues with this approach, such as the requirement for ex vivo-expanded Treg cells and the aggravation of stroke-induced immunosuppression [144]. Numerous studies have demonstrated that inducing a Treg cell response to a brain antigen, such as myelin basic protein (MBP) [154], E-selectin [143] or myelin oligodendrocyte glycoprotein (MOG) [132, 155], through intranarial instillation can improve neurological outcomes via the “bystander suppression” approach. “Bystander suppression” is defined as an immune response in which Treg cells are stimulated in an antigen-specific manner but secrete cytokines modulating immune responses in an antigen-nonspecific manner, implying that a therapeutic immunomodulatory response can be induced regardless of whether the pathogenic antigen is known [154]. The immunomodulatory response is thought to occur through enhancement of a Th3 (TGF-β)-type response or other Treg response and suppression of the Th1 response or other immune responses [134, 154, 155] and may promote adult neurogenesis after ischaemia [137]. However, tolerization to antigens such as MBP prior to ischaemia may cause detrimental autoimmunity via the development of a Th1 response to the antigen by 3 months after ischaemia [63]. Other studies have illustrated that passive CXCL14 supplementation improves neurological deficits after ischaemic stroke by promoting immature dendritic cell (iDC) secretion of IL-2, which induces Treg cell differentiation and other positive pathways [138]. A super-agonistic anti-CD28 monoclonal antibody (CD28SA) can expand and amplify Treg cells that produce IL-10 to attenuate brain damage after ischaemic stroke [145]. However, both of the clinical trials (NCT00012454 and NCT00069069) evaluating E-selectin nasal instillation have failed.
Using drugs for therapy
Some immunomodulatory drugs have shown promise as novel therapies that decrease morbidity and mortality following ischaemic stroke. Glycyrrhizin (Gly) is thought to protect against brain damage induced by ischaemic stroke by inhibiting the activation of CD8+ and CD4+ T cells mediated by IFN and partly regulated by HMGB1 activity [147]. Administration of exogenous vitamin D3 prior to stroke may improve neurological deficits and produce an acute anti-inflammatory response by reducing the Th17/γδ T cell response and increasing the Treg cell response [146]. Levodopa/benserazide treatment after stroke onset was shown to play a protective role by reducing CD8+ cell infiltration into the injured brain [139]. Blocking α4 integrin on leukocytes with natalizumab can provide delayed protection in a mouse model [81], but there is not enough evidence that it is effective in clinical trials [156]. Therefore, the protective role of natalizumab in determining functional outcomes in ischaemic stroke requires further clinical research. Fingolimod, an oral S1P receptor modulator used to reduce peripheral lymphocyte numbers [157], was shown to enhance short-term and long-term neurological recovery in clinical trials with or without alteplase [158,159,160]. These two drugs are very promising for the future treatment of ischaemic stroke [161, 162].
Using intravenous cells for therapy
Intravenous cellular therapies have intrigued many researchers and clinicians over the past decades because of their potential advantage of affecting immune responses through multiple mechanisms and actions [163]. Both animal stroke models and a multi-arm phase 2 clinical trial have shown that intravenous injection of multipotent adult progenitor cells (MAPCs) enhances long-term neurological recovery by modulating immune responses. The underlying mechanism may involve reducing the levels of proinflammatory cells such as CD3+, CD4+ and CD8+ T cells while promoting Treg cell accumulations [163].
Despite discrepancies and heterogeneity amongst studies, new therapeutic targets that can balance the immune response of T cells to protect against the acute and chronic phases after ischaemic stroke are being discovered.
Conclusion
Despite the various types and functions of T cells, most studies have focused on common T cell subsets, including Th1, Th2, Th17, γδ T and Treg cells. These cells intricately communicate with each other and with injured brain tissue via proinflammatory cytokines and anti-inflammatory cytokines and perform immunomodulatory roles. While the inconsistent description of the roles of T cells may be partly due to the differences in stroke models and measurement methods, as well as discrepant post-stroke outcomes, the influx of different subsets of T cells at different stages after ischaemic stroke requires more study. Nevertheless, the evidence reviewed here demonstrates that the interactions of T cells with the CNS and the connections of these cells with other immune cells are complicated and need further elaboration [164]. In conclusion, although a number of studies have elucidated that T cell numbers peak in the infarct zone and peri-infarct zone within 30 days after ischaemic stroke, T cells play an indispensable long-term role after ischaemic stroke through mechanisms such as tissue remodelling and revascularization [165] and therefore are a new target for clinical stroke treatment.
Future research must not only examine how the immune response mediated by T cells is initiated and maintained but also differentiate the various roles of T cell subsets in the onset and process of post-stroke tissue injury and repair. These studies could inform approaches for designing immunoregulatory therapies that regulate T cells in the acute stage following stroke to improve the functional outcome and long-term sequelae of patients suffering from ischaemic stroke. Although current studies have identified a large number of targets, such as cytokines, small molecules, neutralizing antibodies, cell epitopes and injectable cellular products, to regulate the immune response and inflammation related to T cells in the acute or chronic phase following stroke, the most important issue is whether a further understanding of T cell inflammation will provide more comprehensive therapeutic targets and lead to successful clinical translation of immune modulators for stroke. The results of previous studies that manipulated T cell responses have not been completely clarified. The protective effect of suppressing T cells in the acute phase may be based on attenuating neuroinflammation, and long-term protection may refer to the role of Treg cells in tissue repair. However, no comprehensive clinical trial has demonstrated the clinical efficacy or safety of these treatments. Nonetheless, great effort is being put into exploring the underlying mechanisms of these therapies. We still have many challenges to overcome in the pursuit of understanding the pathogeneses and therapies of ischaemic stroke.
We need to perform more studies to understand the roles and mechanisms of T cells in the onset and evolution of ischaemic stroke and to further explore the modulation of both local and peripheral T cell responses, with the goal of attenuating acute neuroinflammation and improving long-term neurological function following ischaemic stroke.
Availability of data and materials
Not applicable
Abbreviations
- DAMPs:
-
Damage-associated molecular patterns
- CD:
-
Cluster of differentiation
- TCR:
-
T cell receptor
- Th cells:
-
T helper cells
- STAT:
-
Signal-transducing activator of transcription
- IFN:
-
Interferon
- IL:
-
Interleukin
- Treg cells:
-
Regulatory T cells
- NKT cells:
-
Natural killer T cells
- IRF:
-
Interferon-regulatory factor
- TGF-β:
-
Transforming growth factor-β
- GM-CSF:
-
Granulocyte macrophage colony-stimulating factor
- EAE:
-
Experimental autoimmune encephalomyelitis
- TNF:
-
Tumour necrosis factor
- AHR:
-
Aryl hydrocarbon receptor
- CCR:
-
C-C chemokine receptor
- APCs:
-
Antigen-presenting cells
- RA:
-
Rheumatoid arthritis
- NBNT:
-
Non-B/non-T
- NF-κB:
-
Nuclear factor kappa-light-chain-enhancer of activated B cells
- Tnfrsf:
-
TNF receptor super family
- TRAFs:
-
TNF receptor-associated factors
- CA/CPR:
-
Cardiac arrest and cardiopulmonary resuscitation
- Tfh:
-
Follicular helper T cells
- CXCR:
-
C-X-C chemokine receptor
- CXCL:
-
Chemokine (C-X-C motif) ligand
- Bcl6:
-
B cell lymphoma 6 protein
- c-Maf:
-
Cellular muscular aponeurotic fibrosarcoma
- Batf:
-
B cell-activating transcription factor
- Blimp1:
-
B lymphocyte-induced maturation protein-1
- S1PR2:
-
Sphingosine-1-phosphate receptor-2
- GCs:
-
Germinal centres
- LPS:
-
Lipopolysaccharide
- ICOSL:
-
Inducible co-stimulator ligand
- Rag:
-
Recombination activation gene
- SCID:
-
Severe combined immunodeficiency
- tMCAO:
-
Transient middle cerebral artery occlusion
- pMCAO:
-
Permanent middle cerebral artery occlusion
- CNS:
-
Central nervous system
- ROS:
-
Reactive oxygen species
- BBB:
-
Blood-brain barrier
- CCL:
-
Chemokine (C-C motif) ligand
- I/R:
-
Ischaemia-reperfusion
- DNTs:
-
Double-negative T cells
- Fas ligand:
-
FasL
- Areg:
-
Amphiregulin
- TEMRA:
-
CD45RA+ effector memory T cell
- IL-4 KO:
-
IL-4 knockout
- rIL-4:
-
Recombinant mouse IL-4
- NK:
-
Natural killer
- OGD:
-
Oxygen-glucose deprivation
- PD-1:
-
Programmed cell death-1
- CTLA:
-
Cytotoxic T lymphocyte antigen
- PD-L1:
-
PD-ligand1
- TCM:
-
T central memory
- DHA:
-
Docosahexaenoic acid
- n-3 PUFAs:
-
Omega-3 polyunsaturated fatty acids
- G-CSF:
-
Granulocyte-colony stimulating factor
- DCs:
-
Dendritic cells
- TLRs:
-
Toll-like receptors
- ACC1:
-
Acetyl coenzyme A carboxylase 1
- RTLs:
-
Recombinant T cell receptor ligands
- MBP:
-
Myelin basic protein
- MOG:
-
Myelin oligodendrocyte glycoprotein
- iDC:
-
Immature dendritic cell
- CD28SA:
-
Super-agonistic anti-CD28 monoclonal antibody
- Gly:
-
Glycyrrhizin
- HMGB1:
-
High-mobility group box 1
- MAPCs:
-
Multipotent adult progenitor cells
References
Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart Disease and Stroke Statistics-2019 update: a report from the American Heart Association. Circulation. 2019;139(10):e56–e528.
Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, et al. Heart Disease and Stroke Statistics--2012 update: a report from the American Heart Association. Circulation. 2012;125(1):e2–e220.
Ritzel RM, Lai YJ, Crapser JD, Patel AR, Schrecengost A, Grenier JM, et al. Aging alters the immunological response to ischemic stroke. Acta Neuropathol. 2018;136(1):89–110.
Bravo-Alegria J, McCullough LD, Liu F. Sex differences in stroke across the lifespan: the role of T lymphocytes. Neurochemistry Int. 2017;107:127–37.
Evans FL, Dittmer M, de la Fuente AG, Fitzgerald DC. Protective and regenerative toles of T cells in central nervous system disorders. Front Immunol. 2019;10:2171.
Shi K, Tian DC, Li ZG, Ducruet AF, Lawton MT, Shi FD. Global brain inflammation in stroke. Lancet Neurol. 2019;18(11):1058–66.
Jun-Long H, Yi L, Bao-Lian Z, Jia-Si L, Ning Z, Zhou-Heng Y, et al. Necroptosis signaling pathways in stroke: from mechanisms to therapies. Curr Neuropharmacol. 2018;16(9):1327–39.
Amani H, Mostafavi E, Alebouyeh MR, Arzaghi H, Akbarzadeh A, Pazoki-Toroudi H, et al. Would colloidal gold nanocarriers present an effective diagnosis or treatment for ischemic stroke? Int J Nanomedicine. 2019;14:8013–31.
Amani H, Habibey R, Shokri F, Hajmiresmail SJ, Akhavan O, Mashaghi A, et al. Selenium nanoparticles for targeted stroke therapy through modulation of inflammatory and metabolic signaling. Sci Rep. 2019;9(1):6044.
Li J, Zhang J, Zhang Y, Wang Z, Song Y, Wei S, et al. TRAF2 protects against cerebral ischemia-induced brain injury by suppressing necroptosis. Cell Death Dis. 2019;10(5):328.
Chu HX, Kim HA, Lee S, et al. Immune cell infiltration in malignant middle cerebral artery infarction: comparison with transient cerebral ischemia. J Cereb Blood Flow Metab. 2014;34(3):450–9.
Stubbe T, Ebner F, Richter D, Engel O, Klehmet J, Royl G, et al. Regulatory T cells accumulate and proliferate in the ischemic hemisphere for up to 30 days after MCAO. Journal of cerebral blood flow and metabolism : official journal of the International Society of Cerebral Blood Flow and Metabolism. 2013;33(1):37–47.
Gill D, Veltkamp R. Dynamics of T cell responses after stroke. Curr Opin Pharmacol. 2016;26:26–32.
Liesz A, Suri-Payer E, Veltkamp C, Doerr H, Sommer C, Rivest S, et al. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat Med. 2009;15(2):192–9.
Zhang Y, Zhang Y, Gu W, He L, Sun B. Th1/Th2 cell’s function in immune system. In: Sun B, editor. T helper cell differentiation and their function. Dordrecht: Springer Netherlands; 2014. p. 45–65.
Weaver CT, Harrington LE, Mangan PR, Gavrieli M, Murphy KM. Th17: an effector CD4 T cell lineage with regulatory T cell ties. Immunity. 2006;24(6):677–88.
Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nature immunology. 2003;4(4):330–6.
Breitfeld D, Ohl L, Kremmer E, Ellwart J, Sallusto F, Lipp M, et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med. 2000;192(11):1545–52.
Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, et al. Transforming growth factor-beta ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9-producing subset. Nat Immunol. 2008;9(12):1341–6.
Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol. 2009;10(8):857–63.
Fallon PG, Ballantyne SJ, Mangan NE, et al. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J Exp Med. 2006;203(4):1105–16.
Waid DM, Vaitaitis GM, Wagner DH Jr. Peripheral CD4loCD40+ auto-aggressive T cell expansion during insulin-dependent diabetes mellitus. Eur J Immunol. 2004;34(5):1488–97.
Tian Y, Zajac AJ. IL-21 and T cell differentiation: consider the context. Trends Immunol. 2016;37(8):557–68.
Wagner DH Jr, Vaitaitis G, Sanderson R, Poulin M, Dobbs C, Haskins K. Expression of CD40 identifies a unique pathogenic T cell population in type 1 diabetes. Proc Natl Acad Sci USA. 2002;99(6):3782–7.
Hsieh CS, Macatonia SE, Tripp CS, Wolf SF, O'Garra A, Murphy KM. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science (New York, NY). 1993;260(5107):547–9.
Ouyang W, Ranganath SH, Weindel K, Bhattacharya D, Murphy TL, Sha WC, et al. Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity. 1998;9(5):745–55.
Akdis M, Palomares O, van de Veen W, van Splunter M, Akdis CA. TH17 and TH22 cells: a confusion of antimicrobial response with tissue inflammation versus protection. J Allergy Clin Immunol. 2012;129(6):1438–49; quiz50-1.
Burgler S, Ouaked N, Bassin C, Basinski TM, Mantel P-Y, Siegmund K, et al. Differentiation and functional analysis of human TH17 cells. J Allergy Clin Immunol. 2009;123(3):588–95.e7.
Peters A, Lee Y, Kuchroo VK. The many faces of Th17 cells. Curr Opin Immunol. 2011;23(6):702–6.
McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, et al. TGF-β and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain TH-17 cell-mediated pathology. Nat Immunol. 2007;8(12):1390–7.
Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, et al. IL-4 inhibits TGF-β-induced Foxp3+ T cells and, together with TGF-β, generates IL-9+ IL-10+ Foxp3− effector T cells. Nat Immunol. 2008;9(12):1347–55.
Pan H-F, Leng R-X, Li X-P, Zheng SG, Ye D-Q. Targeting T-helper 9 cells and interleukin-9 in autoimmune diseases. Cytokine Growth Factor Rev. 2013;24(6):515–22.
Yazdani R, Shapoori S, Rezaeepoor M, Sanaei R, Ganjalikhani-Hakemi M, Azizi G, et al. Features and roles of T helper 9 cells and interleukin 9 in immunological diseases. Allergologia et immunopathologia. 2019;47(1):90–104.
Trifari S, Kaplan CD, Tran EH, Crellin NK, Spits H. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from TH-17, TH1 and TH2 cells. Nature immunology. 2009;10(8):864–71.
Farahani R, Sherkat R, Hakemi MG, Eskandari N, Yazdani R. Cytokines (interleukin-9, IL-17, IL-22, IL-25 and IL-33) and asthma. Adv Biomed Res. 2014;3:127.
Chen WC, Chang LH, Huang SS, Huang YJ, Chih CL, Kuo HC, et al. Aryl hydrocarbon receptor modulates stroke-induced astrogliosis and neurogenesis in the adult mouse brain. J Neuroinflammation. 2019;16(1):187.
Jia L, Wu C. The biology and functions of Th22 cells. Adv Exp Med Biol. 2014;841:209–30.
Fort MM, Cheung J, Yen D, Li J, Zurawski SM, Lo S, et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 2001;15(6):985–95.
de Sousa JR, Quaresma JAS. The role of T helper 25 cells in the immune response to Mycobacterium leprae. J Am Acad Dermatol. 2018;78(5):1009–11.
Vaitaitis G, Dan W, Jr DHW. The expanding role of TNF-receptor super family member CD40 (tnfrsf5) in autoimmune disease: focus on Th40 cells Curr Immunol Rev. 2010;6(2):130–137.
Vaitaitis GM, Wagner DH Jr. High distribution of CD40 and TRAF2 in Th40 T cell rafts leads to preferential survival of this auto-aggressive population in autoimmunity. Plos One. 2008;3(4):e2076.
Waid DM, Wagner RJ, Putnam A, Vaitaitis GM, Pennock ND, Calverley DC, et al. A unique T cell subset described as CD4loCD40+ T cells (TCD40) in human type 1 diabetes. Clin Immunol. 2007;124(2):138–48.
Vaitaitis GM, Rihanek M, Alkanani AK, Waid DM, Gottlieb PA, Wagner DH, et al. Biomarker discovery in pre-type 1 diabetes; Th40 cells as a predictive risk factor. J Clin Endocrinol Metab. 2019;104(9):4127–42.
Horwitz MS, Vaitaitis GM, Yussman MG, Waid DM, Wagner DH. Th40 cells (CD4+CD40+ Tcells) drive a more severe form of experimental autoimmune encephalomyelitis than conventional CD4 T cells. Plos One. 2017;12(2):e0172037.
Deng G, Carter J, Traystman RJ, Wagner DH, Herson PS. Pro-inflammatory T-lymphocytes rapidly infiltrate into the brain and contribute to neuronal injury following cardiac arrest and cardiopulmonary resuscitation. J Neuroimmunol. 2014;274(1-2):132–40.
Schaerli P, Willimann K, Lang AB, Lipp M, Loetscher P, Moser B. CXC chemokine receptor 5 expression defines follicular homing T cells with B cell helper function. J Exp Med. 2000;192(11):1553–62.
Kim CH, Rott LS, Clark-Lewis I, Campbell DJ, Wu L, Butcher EC. Subspecialization of CXCR5+ T cells: B helper activity is focused in a germinal center-localized subset of CXCR5+ T cells. J Exp Med. 2001;193(12):1373–81.
Yu D, Vinuesa CG. The elusive identity of T follicular helper cells. Trends Immunol. 2010;31(10):377–83.
Powell MD, Read KA, Sreekumar BK, Jones DM, Oestreich KJ. IL-12 signaling drives the differentiation and function of a TH1-derived TFH1-like cell population. Sci Rep. 2019;9(1):13991.
Choi YS, Yang JA, Yusuf I, Johnston RJ, Greenbaum J, Peters B, et al. Bcl6 expressing follicular helper CD4 T cells are fate committed early and have the capacity to form memory. J Immunol. 2013;190(8):4014–26.
Curotto de Lafaille MA, Lafaille JJ. Natural and adaptive foxp3+ regulatory T cells: more of the same or a division of labor? Immunity. 2009;30(5):626–35.
Wing JB, Sakaguchi S. Multiple Treg suppressive modules and their adaptability. Frontiers in Immunology. 2012;3:178.
Roncarolo MG, Gregori S, Bacchetta R, Battaglia M, Gagliani N. The biology of T regulatory type 1 cells and their therapeutic application in immune-mediated diseases. Immunity. 2018;49(6):1004–19.
Gagliani N, Gregori S, Jofra T, Valle A, Stabilini A, Rothstein DM, et al. Rapamycin combined with anti-CD45RB mAb and IL-10 or with G-CSF induces tolerance in a stringent mouse model of islet transplantation. Plos One. 2011;6(12):e28434.
Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, Roncarolo MG. Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol. 2006;177(12):8338–47.
de la Vega GN, Dittmer M, Dombrowski Y, Fitzgerald DC. Regenerating CNS myelin: emerging roles of regulatory T cells and CCN proteins. Neurochem Int. 2019;130:104349.
Ito M, Komai K, Nakamura T, Srirat T, Yoshimura A. Tissue regulatory T cells and neural repair. Int Immunol. 2019;31(6):361–9.
Chen Y, Kuchroo VK, Inobe J, Hafler DA, Weiner HL. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science. 1994;265(5176):1237.
Weiner HL. Oral tolerance: immune mechanisms and the generation of Th3-type TGF-beta-secreting regulatory cells. Microbes Infect. 2001;3(11):947–54.
Weiner HL, da Cunha AP, Quintana F, Wu H. Oral tolerance. Immunolo Rev. 2011;241(1):241–59.
Clemente-Casares X, Blanco J, Ambalavanan P, Yamanouchi J, Singha S, Fandos C, et al. Expanding antigen-specific regulatory networks to treat autoimmunity. Nature. 2016;530(7591):434–40.
Faria AM, Weiner HL. Oral tolerance. Immunol Rev. 2005;206:232–59.
Gee JM, Zierath D, Hadwin J, Savos A, Kalil A, Thullbery M, et al. Long term immunologic consequences of experimental stroke and mucosal tolerance. Exp Transl Stroke Med. 2009;1:3.
Mao L, Li P, Zhu W, Cai W, Liu Z, Wang Y, et al. Regulatory T cells ameliorate tissue plasminogen activator-induced brain haemorrhage after stroke. Brain. 2017;140(7):1914–31.
Cosmi L, Maggi L, Santarlasci V, Liotta F, Annunziato F. T helper cells plasticity in inflammation. Cytometry A. 2014;85(1):36–42.
Ivanova EA, Orekhov AN. T Helper lymphocyte subsets and plasticity in autoimmunity and cancer: an overview. Biomed Res Int. 2015;2015:327470.
Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity. 2009;30(5):646–55.
Jiang C, Wang H, Xue M, Lin L, Wang J, Cai G, et al. Reprograming of peripheral Foxp3(+) regulatory T cell towards Th17-like cell in patients with active systemic lupus erythematosus. Clin Immunol (Orlando). 2019;209:108267.
Gagliani N, Vesely MCA, Iseppon A, Brockmann L, Xu H, Palm NW, et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature. 2015;523(7559):221–5.
Maggi L, Capone M, Giudici F, Santarlasci V, Querci V, Liotta F, et al. CD4+CD161+ T lymphocytes infiltrate Crohn’s disease-associated perianal fistulas and are reduced by anti-TNF-α local therapy. Int Arch Allergy Immunol. 2013;161(1):81–6.
Cosmi L, Maggi L, Santarlasci V, Capone M, Cardilicchia E, Frosali F, et al. Identification of a novel subset of human circulating memory CD4(+) T cells that produce both IL-17A and IL-4. J Allergy Clin Immunol. 2010;125(1):222–30.e1-4.
Xu L, Kitani A, Fuss I, Strober W. Cutting edge: regulatory T cells induce CD4+CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J Immunol. 2007;178(11):6725–9.
Voo KS, Wang YH, Santori FR, Boggiano C, Wang YH, Arima K, et al. Identification of IL-17-producing FOXP3+ regulatory T cells in humans. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(12):4793–8.
Franco R, Fernández-Suárez D. Alternatively activated microglia and macrophages in the central nervous system. Progress in neurobiology. 2015;131:65–86.
Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, et al. Transforming growth factor-β ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9–producing subset. Nat Immunol. 2008;9(12):1341–6.
Annunziato F, Cosmi L, Manetti R, Brugnolo F, Parronchi P, Maggi E, et al. Reversal of human allergen-specific CRTH2+ T(H)2 cells by IL-12 or the PS-DSP30 oligodeoxynucleotide. J Allergy Clin Immunol. 2001;108(5):815–21.
Yilmaz G, Arumugam TV, Stokes KY, Granger DN. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation. 2006;113(17):2105–12.
Kleinschnitz C, Schwab N, Kraft P, Hagedorn I, Dreykluft A, Schwarz T, et al. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood. 2010;115(18):3835–42.
Gelderblom M, Weymar A, Bernreuther C, Velden J, Arunachalam P, Steinbach K, et al. Neutralization of the IL-17 axis diminishes neutrophil invasion and protects from ischemic stroke. Blood. 2012;120(18):3793.
Xiong X, Gu L, Zhang H, Xu B, Zhu S, Zhao H. The protective effects of T cell deficiency against brain injury are ischemic model-dependent in rats. Neurochem Int. 2013;62(3):265–70.
Liesz A, Zhou W, Mracskó É, Karcher S, Bauer H, Schwarting S, et al. Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain. 2011;134(3):704–20.
Liesz A, Karcher S, Veltkamp R. Spectratype analysis of clonal T cell expansion in murine experimental stroke. J Neuroimmunol. 2013;257(1-2):46–52.
Arumugam TV, Granger DN, Mattson MP. Stroke and T-cells. Neuro Mol Med. 2005;7(3):229–42.
Gu L, Xiong X, Zhang H, Xu B, Steinberg GK, Zhao H. Distinctive effects of T cell subsets in neuronal injury induced by cocultured splenocytes in vitro and by in vivo stroke in mice. Stroke. 2012;43(7):1941–6.
Xie L, Li W, Hersh J, Liu R, Yang SH. Experimental ischemic stroke induces long-term T cell activation in the brain. J Cereb Blood Flow Metab. 2019;39(11):2268–76.
Gu L, Jian Z, Stary C, Xiong X. T cells and cerebral ischemic stroke. Neurochem Res. 2015;40(9):1786–91.
Kim E, Cho S. Microglia and monocyte-derived macrophages in stroke. Neurotherapeutics. 2016;13(4):702–18.
Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol. 2009;9(4):259–70.
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, et al. ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci. 2005;8(6):752–8.
Atkinson SJ, Hosford MA, Molitoris BA. Mechanism of actin polymerization in cellular ATP depletion. J Biol Chem. 2004;279(7):5194–9.
Gelderblom M, Leypoldt F, Steinbach K, Behrens D, Choe CU, Siler DA, et al. Temporal and spatial dynamics of cerebral immune cell accumulation in stroke. Stroke. 2009;40(5):1849–57.
Brait VH, Jackman KA, Walduck AK, Selemidis S, Diep H, Mast AE, et al. Mechanisms contributing to cerebral infarct size after stroke: gender, reperfusion, T lymphocytes, and Nox2-derived superoxide. J Cereb Blood Flow Metab. 2010;30(7):1306–17.
Hurn PD, Subramanian S, Parker SM, Afentoulis ME, Kaler LJ, Vandenbark AA, et al. T- and B-cell-deficient mice with experimental stroke have reduced lesion size and inflammation. J Cereb Blood Flow Metab. 2007;27(11):1798–805.
Gu L, Xiong X, Wei D, Gao X, Krams S, Zhao H. T cells contribute to stroke-induced lymphopenia in rats. Plos One. 2013;8(3):e59602.
Gu LJ, Xiong XX, Ito T, Lee J, Xu BH, Krams S, et al. Moderate hypothermia inhibits brain inflammation and attenuates stroke-induced immunodepression in rats. CNS Neurosci Ther. 2014;20(1):67–75.
Saino O, Taguchi A, Nakagomi T, Nakano-Doi A, Kashiwamura S, Doe N, et al. Immunodeficiency reduces neural stem/progenitor cell apoptosis and enhances neurogenesis in the cerebral cortex after stroke. J Neurosci Res. 2010;88(11):2385–97.
Vindegaard N, Muñoz-Briones C, El Ali HH, Kristensen LK, Rasmussen RS, Johansen FF, et al. T-cells and macrophages peak weeks after experimental stroke: spatial and temporal characteristics. Neuropathology: official journal of the Japanese Society of Neuropathology. 2017;37(5):407–14.
Schroeter M, Jander S, Witte OW, Stoll G. Local immune responses in the rat cerebral cortex after middle cerebral artery occlusion. J Neuroimmunol. 1994;55(2):195–203.
Jander S, Kraemer M, Schroeter M, Witte OW, Stoll G. Lymphocytic infiltration and expression of intercellular adhesion molecule-1 in photochemically induced ischemia of the rat cortex. J Cereb Blood Flow Metab. 1995;15(1):42–51.
Wang S, Zhang H, Xu Y. Crosstalk between microglia and T cells contributes to brain damage and recovery after ischemic stroke. Neurol Res. 2016;38(6):495–503.
Jian Z, Liu R, Zhu X, Smerin D, Zhong Y, Gu L, et al. The involvement and therapy target of immune cells after ischemic stroke. Front Immunol. 2019;10:2167.
Klebe D, McBride D, Flores JJ, Zhang JH, Tang J. Modulating the immune response towards a neuroregenerative peri-injury milieu after cerebral hemorrhage. J Neuroimmune Pharmacol. 2015;10(4):576–86.
Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11(10):889–96.
Korhonen P, Kanninen KM, Lehtonen Š, Lemarchant S, Puttonen KA, Oksanen M, et al. Immunomodulation by interleukin-33 is protective in stroke through modulation of inflammation. Brain Behav Immun. 2015;49:322–36.
Xiong X, Xu L, Wei L, White RE, Ouyang YB, Giffard RG. IL-4 is required for sex differences in vulnerability to focal ischemia in mice. Stroke. 2015;46(8):2271–6.
Appel SH. CD4+ T cells mediate cytotoxicity in neurodegenerative diseases. J Clin Invest. 2009;119(1):13–5.
Butovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S, et al. Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesis from adult stem/progenitor cells. Mol Cell Neurosci. 2006;31(1):149–60.
Kwee BJ, Budina E, Najibi AJ, Mooney DJ. CD4 T-cells regulate angiogenesis and myogenesis. Biomaterials. 2018;178:109–21.
Shichita T, Sugiyama Y, Ooboshi H, Sugimori H, Nakagawa R, Takada I, et al. Pivotal role of cerebral interleukin-17-producing γδT cells in the delayed phase of ischemic brain injury. Nat Med. 2009;15(8):946–50.
Li GZ, Zhong D, Yang LM, Sun B, Zhong ZH, Yin YH, et al. Expression of interleukin-17 in ischemic brain tissue. Scand J Immunol. 2005;62(5):481–6.
Hillhouse EE, Lesage S. A comprehensive review of the phenotype and function of antigen-specific immunoregulatory double negative T cells. J Autoimmun. 2013;40:58–65.
D’Acquisto F, Crompton T. CD3+CD4−CD8− (double negative) T cells: Saviours or villains of the immune response? Biochem Pharmacol. 2011;82(4):333–40.
Meng H, Zhao H, Cao X, Hao J, Zhang H, Liu Y, et al. Double-negative T cells remarkably promote neuroinflammation after ischemic stroke. Proc Natl Acad Sci U S A. 2019;116(12):5558–63.
Braun M, Vaibhav K, Saad N, Fatima S, Brann DW, Vender JR, et al. Activation of myeloid TLR4 mediates T lymphocyte polarization after traumatic brain injury. J Immunol. 2017;198(9):3615–26.
Daglas M, Draxler DF, Ho H, McCutcheon F, Galle A, Au AE, et al. Activated CD8+ T cells cause long-term neurological impairment after traumatic brain injury in mice. Cell Rep. 2019;29(5):1178–91 e6.
Kostulas N, Pelidou SH, Kivisäkk P, Kostulas V, Link H. Increased IL-1beta, IL-8, and IL-17 mRNA expression in blood mononuclear cells observed in a prospective ischemic stroke study. Stroke. 1999;30(10):2174–9.
Selvaraj UM, Stowe AM. Long-term T cell responses in the brain after an ischemic stroke. Discov Med. 2017;24(134):323–33.
Zhou K, Zhong Q, Wang YC, Xiong XY, Meng ZY, Zhao T, et al. Regulatory T cells ameliorate intracerebral hemorrhage-induced inflammatory injury by modulating microglia/macrophage polarization through the IL-10/GSK3beta/PTEN axis. J Cereb Blood Flow Metab. 2017;37(3):967–79.
Ito M, Komai K, Mise-Omata S, Iizuka-Koga M, Noguchi Y, Kondo T, et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature. 2019;565(7738):246–50.
Kleinschnitz C, Kraft P, Dreykluft A, Hagedorn I, Göbel K, Schuhmann MK, et al. Regulatory T cells are strong promoters of acute ischemic stroke in mice by inducing dysfunction of the cerebral microvasculature. Blood. 2013;121(4):679–91.
Li P, Mao L, Zhou G, Leak RK, Sun BL, Chen J, et al. Adoptive regulatory T-cell therapy preserves systemic immune homeostasis after cerebral ischemia. Stroke. 2013;44(12):3509–15.
Jickling GC, Liu D, Ander BP, Stamova B, Zhan X, Sharp FR. Targeting neutrophils in ischemic stroke: translational insights from experimental studies. J Cereb Blood Flow Metab. 2015;35(6):888–901.
Feng Y, He X, Luo S, Chen X, Long S, Liang F, et al. Chronic colitis induces meninges traffic of gut-derived T cells, unbalances M1 and M2 microglia/macrophage and increases ischemic brain injury in mice. Brain Res. 2019;1707:8–17.
Stabile E, Kinnaird T, la Sala A, Hanson SK, Watkins C, Campia U, et al. CD8+ T lymphocytes regulate the arteriogenic response to ischemia by infiltrating the site of collateral vessel development and recruiting CD4+ mononuclear cells through the expression of interleukin-16. Circulation. 2006;113(1):118–24.
Klehmet J, Hoffmann S, Walter G, Meisel C, Meisel A. Stroke induces specific alteration of T memory compartment controlling auto-reactive CNS antigen-specific T cell responses. J Neurol Sci. 2016;368:77–83.
Xiong X, Barreto GE, Xu L, Ouyang YB, Xie X, Giffard RG. Increased brain injury and worsened neurological outcome in interleukin-4 knockout mice after transient focal cerebral ischemia. Stroke. 2011;42(7):2026–32.
Li M, Li Z, Yao Y, Jin WN, Wood K, Liu Q, et al. Astrocyte-derived interleukin-15 exacerbates ischemic brain injury via propagation of cellular immunity. Proc Natl Acad Sci U S A. 2017;114(3):E396–405.
Lee GA, Lin TN, Chen CY, Mau SY, Huang WZ, Kao YC, et al. Interleukin 15 blockade protects the brain from cerebral ischemia-reperfusion injury. Brain Behav Immun. 2018;73:562–70.
Lee GA, Lai YG, Chen RJ, Liao NS. Interleukin 15 activates Akt to protect astrocytes from oxygen glucose deprivation-induced cell death. Cytokine. 2017;92:68–74.
Guo S, Luo Y. Brain Foxp3(+) regulatory T cells can be expanded by interleukin-33 in mouse ischemic stroke. Int Immunopharmacol. 2019;81:106027.
Bodhankar S, Chen Y, Lapato A, Dotson AL, Wang J, Vandenbark AA, et al. PD-L1 monoclonal antibody treats ischemic stroke by controlling central nervous system inflammation. Stroke. 2015;46(10):2926–34.
Vandenbark AA, Meza-Romero R, Benedek G, Offner H. A novel neurotherapeutic for multiple sclerosis, ischemic injury, methamphetamine addiction, and traumatic brain injury. J Neuroinflammation. 2019;16(1):14.
Cai W, Liu S, Hu M, Sun X, Qiu W, Zheng S, et al. Post-stroke DHA treatment protects against acute ischemic brain injury by skewing macrophage polarity toward the M2 phenotype. Transl Stroke Res. 2018;9(6):669–80.
Chen Y, Ruetzler C, Pandipati S, Spatz M, McCarron RM, Becker K, et al. Mucosal tolerance to E-selectin provides cell-mediated protection against ischemic brain injury. Proc Natl Acad Sci U S A. 2003;100(25):15107–12.
Zhang Y, Wei L, Du Y, Xie Y, Wu W, Yuan Y. Association between programed cell death-1 and CD4+ T cell alterations in different phases of ischemic stroke patients. Front Cell Neurosci. 2018;12:170.
Dietel B, Cicha I, Kallmünzer B, Tauchi M, Yilmaz A, Daniel WG, et al. Suppression of dendritic cell functions contributes to the anti-inflammatory action of granulocyte-colony stimulating factor in experimental stroke. Exp Neurol. 2012;237(2):379–87.
Ishibashi S. Immunomodulation by inducing tolerance to E-selectin and adult neurogenesis after stroke. Rinsho shinkeigaku. 2010;50(11):882–5.
Lee HT, Liu SP, Lin CH, Lee SW, Hsu CY, Sytwu HK, et al. A crucial role of CXCL14 for promoting regulatory T cells activation in stroke. Theranostics. 2017;7(4):855–75.
Kuric E, Ruscher K. Reduction of rat brain CD8+ T-cells by levodopa/benserazide treatment after experimental stroke. The European journal of neuroscience. 2014;40(2):2463–70.
Dietel B, Cicha I, Achenbach S, Kollmar R, Garlichs C, Tauchi M. Different treatment settings of granulocyte-colony stimulating factor and their impact on T cell-specific immune response in experimental stroke. Immunology letters. 2014;158(1-2):95–100.
Tang SC, Yeh SJ, Li YI, Wang YC, Baik SH, Santro T, et al. Evidence for a detrimental role of TLR8 in ischemic stroke. Exp Neurol. 2013;250:341–7.
Wang X, Zhou Y, Tang D, Zhu Z, Li Y, Huang T, et al. ACC1 (acetyl coenzyme A carboxylase 1) is a potential immune modulatory target of cerebral ischemic stroke. Stroke. 2019;50(7):1869–78.
Yun W. qing-cheng L, Lei Y, Jia-yin M. Mucosal tolerance to E-selectin provides protection against cerebral ischemia–reperfusion injury in rats. J Neuroimmunol. 2008;205(1):73–9.
Xia Y, Cai W, Thomson AW, Hu X. Regulatory T cell therapy for ischemic stroke: how far from clinical translation? Transl Stroke Res. 2016;7(5):415–9.
Na SY, Mracsko E, Liesz A, Hunig T, Veltkamp R. Amplification of regulatory T cells using a CD28 superagonist reduces brain damage after ischemic stroke in mice. Stroke. 2015;46(1):212–20.
Evans MA, Kim HA, Ling YH, Uong S, Vinh A, De Silva TM, et al. Vitamin D supplementation reduces subsequent brain injury and inflammation associated with ischemic stroke. Neuromolecular Med. 2018;20(1):147–59.
Xiong X, Gu L, Wang Y, Luo Y, Zhang H, Lee J, et al. Glycyrrhizin protects against focal cerebral ischemia via inhibition of T cell activity and HMGB1-mediated mechanisms. J Neuroinflammation. 2016;13(1):241.
Zhao X, Wang H, Sun G, Zhang J, Edwards NJ, Aronowski J. Neuronal interleukin-4 as a modulator of microglial pathways and ischemic brain damage. J Neurosci. 2015;35(32):11281–91.
Clarkson BD, Ling C, Shi Y, Harris MG, Rayasam A, Sun D, et al. T cell-derived interleukin (IL)-21 promotes brain injury following stroke in mice. J Exp Med. 2014;211(4):595–604.
Xiao W, Guo S, Chen L, Luo Y. The role of interleukin-33 in the modulation of splenic T-cell immune responses after experimental ischemic stroke. J Neuroimmunol. 2019;333:576970.
Bodhankar S, Chen Y, Lapato A, Vandenbark AA, Murphy SJ, Offner H. Targeting immune co-stimulatory effects of PD-L1 and PD-L2 might represent an effective therapeutic strategy in stroke. Front Cell Neurosci. 2014;8:228.
Zhu W, Dotson AL, Libal NL, Lapato AS, Bodhankar S, Offner H, et al. Recombinant T-cell receptor ligand RTL1000 limits inflammation and decreases infarct size after experimental ischemic stroke in middle-aged mice. Neuroscience. 2015;288:112–9.
Benakis C, Brea D, Caballero S, Faraco G, Moore J, Murphy M, et al. Commensal microbiota affects ischemic stroke outcome by regulating intestinal γδ T cells. Nat Med. 2016;22(5):516–23.
Gee JM, Kalil A, Thullbery M, Becker KJ. Induction of immunologic tolerance to myelin basic protein prevents central nervous system autoimmunity and improves outcome after stroke. Stroke. 2008;39(5):1575–82.
Frenkel D, Huang Z, Maron R, Koldzic DN, Moskowitz MA, Weiner HL. Neuroprotection by IL-10-producing MOG CD4+ T cells following ischemic stroke. J Neurol Sci. 2005;233:125–32.
Elkins J, Veltkamp R, Montaner J, Johnston SC, Singhal AB, Becker K, et al. Safety and efficacy of natalizumab in patients with acute ischaemic stroke (ACTION): a randomised, placebo-controlled, double-blind phase 2 trial. Lancet Neurol. 2017;16(3):217–26.
Han M, Sun T, Chen H, Han M, Wang D. Potential sphingosine-1-phosphate-related therapeutic targets in the treatment of cerebral ischemia reperfusion injury. Life Sci. 2020;249:117542.
Fu Y, Zhang N, Ren L, Yan Y, Sun N, Li YJ, et al. Impact of an immune modulator fingolimod on acute ischemic stroke. Proc Natl Acad Sci U S A. 2014;111(51):18315–20.
Zhu Z, Fu Y, Tian D, Sun N, Han W, Chang G, et al. Combination of the immune modulator fingolimod with alteplase in acute ischemic stroke: a pilot trial. Circulation. 2015;132(12):1104–12.
Tian DC, Shi K, Zhu Z, Yao J, Yang X, Su L, et al. Fingolimod enhances the efficacy of delayed alteplase administration in acute ischemic stroke by promoting anterograde reperfusion and retrograde collateral flow. Ann Neurol. 2018;84(5):717–28.
Fu Y, Liu Q, Anrather J, Shi F-D. Immune interventions in stroke. Nat Rev Neurol. 2015;11(9):524–35.
Veltkamp R, Gill D. Clinical trials of immunomodulation in ischemic stroke. Neurotherapeutics. 2016;13(4):791–800.
Mays RW, Savitz SI. Intravenous cellular therapies for acute ischemic stroke. Stroke. 2018;49(5):1058–65.
Rayasam A, Hsu M, Kijak JA, Kissel L, Hernandez G, Sandor M, et al. Immune responses in stroke: how the immune system contributes to damage and healing after stroke and how this knowledge could be translated to better cures? Immunology. 2018;154(3):363–76.
Stubbe T, Ebner F, Richter D, Engel OR, Klehmet J, Royl G, et al. Regulatory T cells accumulate and proliferate in the ischemic hemisphere for up to 30 days after MCAO. J Cereb Blood Flow Metab. 2012;33(1):37–47.
Acknowledgements
We thank Xueting Wu for her artistic drawing.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81870939 and 81571147 to XX Xiong; No. 82070526 to HF Zhang) and the Natural Science Foundation of Guangdong Province, Guangdong (No. 2019A1515010654 to HF Zhang).
Author information
Authors and Affiliations
Contributions
Lei TY wrote the initial draft. Zhu XQ contributed to reviewing the literature. Figures and the submission were prepared by Daniel Smerin and Ye YZ. Gu LJ and Xiong XX prepared the final version. Zhang HF and Jian ZH recommended a structure for the review and substantially advancing the draft. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lei, TY., Ye, YZ., Zhu, XQ. et al. The immune response of T cells and therapeutic targets related to regulating the levels of T helper cells after ischaemic stroke. J Neuroinflammation 18, 25 (2021). https://doi.org/10.1186/s12974-020-02057-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-020-02057-z