
Abstract
Background
Perioperative neurocognitive disorders (PNDs) occur frequently after surgery and worsen patient outcome. How C-X-C motif chemokine (CXCL) 13 and its sole receptor CXCR5 contribute to PNDs remains poorly understood.
Methods
A PND model was created in adult male C57BL/6J and CXCR5−/− mice by exploratory laparotomy. Mice were pretreated via intracerebroventricular injection with recombinant CXCL13, short hairpin RNA against CXCL13 or a scrambled control RNA, or ERK inhibitor PD98059. Then surgery was performed to induce PNDs, and animals were assessed in the Barnes maze trial followed by a fear-conditioning test. Expression of CXCL13, CXCR5, and ERK in hippocampus was examined using Western blot, quantitative PCR, and immunohistochemistry. Levels of interleukin-1 beta (IL-1β) and tumor necrosis factor alpha (TNF-α) in hippocampus were assessed by Western blot.
Results
Surgery impaired learning and memory, and it increased expression of CXCL13 and CXCR5 in the hippocampus. CXCL13 knockdown partially reversed the effects of surgery on CXCR5 and cognitive dysfunction. CXCR5 knockout led to similar cognitive outcomes as CXCL13 knockdown, and it repressed surgery-induced activation of ERK and production of IL-1β and TNF-α in hippocampus. Recombinant CXCL13 induced cognitive deficits and increased the expression of phospho-ERK as well as IL-1β and TNF-α in hippocampus of wild-type mice, but not CXCR5−/− mice. PD98059 partially blocked CXCL13-induced cognitive dysfunction as well as production of IL-1β and TNF-α.
Conclusions
CXCL13-induced activation of CXCR5 may contribute to PNDs by triggering ERK-mediated production of pro-inflammatory cytokines in hippocampus.
Similar content being viewed by others


Background
Perioperative neurocognitive disorders (PNDs) refer to pre- and post-operative reduction in hippocampus-dependent cognitive functions involving disturbance of memory, consciousness and attention, as well as changes in personality [1, 2]. PNDs are a well-recognized clinical complication and patients of any age undergoing surgical procedure are susceptible. Among patients undergoing major noncardiac surgery, PNDs occur before discharge in 37% of patients aged 18–39 years, 30% of patients aged 40–59, and 41% of patients 60 years or older [3]. PNDs not only increase the burden on the healthcare system but also lead to postoperative long-term disability and even mortality. The use of volatile anesthetics during surgery has long been suspected of increasing risk of PNDs [4, 5]. As the number of surgeries continues to increase, PNDs are expected to become an epidemic [1]. Increasing evidence suggests that PNDs involve neuroinflammation in the hippocampus, at least in their early stages [2, 4]. Under certain conditions, secreted chemokines contribute to neuroinflammation in the central nervous system (CNS) [6,7,8]. Whether and how chemokines may contribute to PNDs is unclear, particularly in the hippocampus.
C-X-C motif chemokine (CXCL) 13 recruits B cells to the CNS in different neuroinflammatory diseases [6]. C-X-C motif chemokine 13 (CXCL13) helps mediate the inflammatory response to infection and injury, and it exerts local, concentration-dependent effects on hippocampal memory function [9]. Elevated levels of CXCL13 have been reported in the cerebrospinal fluid of patients with multiple sclerosis or neuromyelitis optica, and it has even been proposed as a biomarker for these neurodegenerative conditions [7]. The only known CXCL13 receptor is C-X-C motif chemokine receptor 5 (CXCR5), which is highly expressed on B lymphocytes and certain T cells in cerebrospinal fluid, and which helps guide lymphocyte tracking within secondary lymphoid tissues [10,11,12,13]. Our previous work showed that CXCR5 contributes to learning and memory impairment in an animal model of sepsis [14]. CXCR5 is expressed in neuronal precursor cells of the brain, which migrate across brain endothelial cells upon exposure to CXCL13 [11]. CXCL13/CXCR5 signaling in the spinal cord mediates neuroinflammation induced by neuropathic pain [13]. Expression of CXCL13 and CXCR5 is altered in intractable temporal lobe epilepsy patients and epileptic rats that show neurodegeneration and cognitive impairment [9]. These considerations led us to ask whether CXCL13 and CXCR5 are involved in PNDs.
We began to investigate this question by focusing on extracellular signal-regulated kinase (ERK). The CXCL13/CXCR5 axis acts via ERK to trigger production of pro-inflammatory cytokines induced by orofacial neuropathic pain [13], and CXCL13 appears to activate ERK by triggering its phosphorylation to p-ERK [15, 16]. ERK is highly expressed in the neurons of the developing and adult brain, functioning as an important mediator of synaptic plasticity and cell survival [17]. Changes in hippocampal levels of p-ERK have been linked to cognitive impairment in a mouse model of Alzheimer’s disease [18], and p-ERK appears to induce production of pro-inflammatory cytokines, such as tumor necrosis factor alpha (TNF-α) and interleukin-1 beta (IL-1β), in animal models of cognitive deficits [19].
The purpose of the present study was to examine whether CXCL13 may contribute to PNDs in an animal model, and more concretely whether it does so via ERK.
Methods
Animals
The animal protocol was approved by the Institutional Animal Ethics Committee of Nanjing Medical University. All experiments described here were conducted in strict accordance with institutional guidelines.
Male C57BL/6J wild-type (WT) mice (3 months old, 33–40 g) were obtained from the Experimental Animal Center of Nanjing First Hospital. CXCR5−/− mice [B6.129S2 (Cg)-CXCR5 tm1Lipp/J, stock number 006659] bred on a B6 background were kindly provided by Professor Yongjing Gao (Pain Research Laboratory, Nantong University, China) [13]. WT and CXCR5−/− mice were age-, gender-, and weight-matched. All mice were housed in a pathogen-free facility in the Experimental Animal Center at Nanjing First Hospital and maintained under a 12-h light-dark cycle with free access to food and water.
Experimental design
The overall experimental design is summarized in Fig. 1. The mouse PND model was established using exploratory laparotomy. All pharmacological treatments were given via intracerebroventricular (i.c.v.) injection into the left ventricle. Several experimental procedures with different timelines were performed in order to comprehensively assess the roles of CXCL13, CXCR5, and ERK in PNDs. The following is a brief description of the experimental groups, and their manipulations are described in detail in subsequent subsections. (1) A cohort of WT mice was divided into two groups, one of which underwent laparotomy (n = 8) and the other of which did not (n = 8; termed controls). No additional pre-treatments were administered. (2) A cohort of WT mice was divided to randomly receive short hairpin RNA (shRNA) targeting CXCL13 or scrambled control shRNA at 3 days before surgery (n = 8). Additionally, ERK inhibitor PD98059 or vehicle was injected 1 h before surgery (n = 8). (3) A cohort of WT and CXCR5−/− mice was randomly selected to undergo surgery without prior treatment (n = 8 of each strain). (4) A cohort of WT and CXCR5−/− mice that did not undergo surgery was injected i.c.v. with recombinant murine CXCL13 (to mimic surgical insult) or with vehicle (n = 8). (5) A cohort of WT mice was injected with ERK inhibitor PD98059 at 1 h before treatment with recombinant CXCL13 (n = 8).

Schematic illustration of the experimental design. CXCL13 C-X-C motif chemokine 13, CXCR5 C-X-C motif chemokine receptor 5, HE hematoxylin-eosin staining, i.c.v. intracerebroventricular, IL-1β interleukin-1 beta, TNF-α tumor necrosis factor alpha, WT wide-type
Exploratory laparotomy model
Exploratory laparotomy was performed on mice in a sterile environment as described [20], with some modifications. To ensure a similar degree of stimulation, every mouse was explored using the same set of procedures. Briefly, mice were anesthetized by 1.5% isoflurane inhalation and kept under spontaneous respiration with an inspiration of 50% O2. Temperature was monitored rectally (Techman Software, Chengdu, China) and maintained at 37 ± 0.5 °C with the aid of a heating blanket. To mimic clinical exploratory laparotomy, a longitudinal midline incision was made in the abdomen from the xiphoid process to the superior margin of the pubic symphysis. A cotton tip wetted with 0.9% sterile saline (Baxter Healthcare, Shanghai, China) explored abdominal organs in the following order: liver, spleen, left and right kidneys, bladder and bowel. The incision site was infiltrated with bupivacaine (0.25%, 3 mg/kg) for postoperative analgesia. Then, the peritoneum and skin were sutured separately. The total duration of exploration and anesthesia lasted around 2 h. After animals recovered, they were returned to their cages with food and water ad libitum.
Pharmacological treatments
All drugs were prepared under sterile conditions. The dose for i.c.v. injection was 5 μg for PD98059 (Sigma, St. Louis, MO, USA) [17] as well as recombinant murine CXCL13 (catalog no. 250-24, PeproTech, Rocky Hills, NJ, USA) [13]. The final DMSO concentration was 2.5%. Vehicle solution was 2.5% DMSO. Solutions were administered by i.c.v. injection. An shRNA targeting CXCL13 was expressed from the sequence 5′-TCG TGC CAA ATG GTT ACA A-3′ in a plasmid (GenePharma, Shanghai, China) [13], while negative control scrambled shRNA was expressed from the sequence 5′-TTC TCC GAA CGT GTC ACG T-3′. The shRNAs (5 μg) were dissolved in 5 μL RNase-free water, mixed with 5 μL transfection reagent (Entranster™-in vivo, Engreen Biosystem, Beijing, China), and injected i.c.v. at 3 days before exploratory laparotomy as described previously [14]. CXCL13 knockdown was verified using Western blot analysis.
Intracerebroventricular injections
Mice were anesthetized with 1.5% isoflurane inhalation, and placed in a stereotaxic apparatus (Stoelting, Wood Dale, IL, USA). I.c.v. injection was performed using a 32-gauge microsyringe (Hamilton, Reno, NV, USA) over a period of 10 min, and the microsyringe was left in place for 5 min after completing the infusion. The stereotaxic coordinates were determined as 1.0 mm to the left from midline, 0.5 mm posterior to the bregma, and 2.5 mm below the dural surface [21]. The animals were taken to the individual cages after the i.c.v. injection.
Barnes maze test
The Barnes maze trial was performed to assess the spatial learning and memory of mice as described [22], with modifications. Animals were placed in the middle of a round platform with 20 equal-sized peripheral holes spaced at regular intervals. One of the holes, called the “target hole,” was connected to a dark box. After the mouse was placed in the middle of the platform, aversive noise (85 dB) and bright light from a 200-W bulb were used to stimulate mice to seek and enter the target hole. Mice were trained in a spatial acquisition phase (3 min per trial, 2 trials each day at a 2-h interval for 4 days) beginning on day 7 after surgery. The spatial memory test was conducted 1 day after training ended (day 11 post-surgery) to measure short-term retention and 8 days after training (day 18 after surgery) to measure long-term retention. Mouse activity and the latency to find the target hole during each trial were recorded using the ANY-Maze video tracking system (SD Instruments, San Diego, CA, USA).
Fear conditioning test
A fear conditioning test was conducted as described [23] on days 19 and 20 after surgery. All mice were placed in a chamber with a floor of stainless steel bars (LE111, Panlab, Barcelona, Spain) that had been wiped down with 70% ethanol. This chamber was designed to allow sound stimulation for the training of conditioned reflexes, as well as electrical stimulation on the feet using a constant-current generator. Mice were allowed to explore and habituate in the training environment for 3 min. Then they were stimulated with a monofrequency sound for 30 s (1 kHz, 80 dB), and during the last 2 s of this sound, a foot shock (1 mA) was delivered. After a 30-s pause, this pairing of sound and shock were delivered a second time. After 24 h, as a test of contextual memory, mice were allowed to enter the original chamber without any stimulation, and freezing behavior, defined as the absence of any movement other than that required for respiration for more than 3 s, was monitored using a SMART3.0 video tracking system (Panlab). Then, 2 h later, a cued fear conditioning test was conducted in which the animals were placed in the chamber and subjected to the monofrequency sound for 3 min without foot shock, and freezing behavior was monitored and recorded.
Western blot
Hippocampus tissues were homogenized and centrifuged at 13,000 g at 4 °C for 10 min. The supernatant was separated on SDS-PAGE gels, and then transferred to a nitrocellulose membrane (Hybond-ECL, Amersham Biosciences, Little Chalfont, UK). Membranes were blocked, then incubated overnight at 4 °C with antibodies against CXCL13 (1:100; Novus Biologicals, Littleton, CO, USA) or CXCR5 (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA); or antibodies against ERK (1:500), p-ERK (1:500), IL-1β (1:1000), or TNF-α (1:500) (Cell Signaling, Beverly, MA, USA). Next, blots were incubated with secondary antibodies (Sigma-Aldrich) conjugated to horseradish peroxidase. Protein bands were detected using enhanced chemiluminescence (Amersham Biosciences) and quantitated using Quantity One software (Bio-Rad Laboratories, Hercules, CA, USA). Band intensities were normalized to that of β-actin.
Reverse transcription-quantitative polymerase chain reaction
Total RNA was extracted from hippocampal tissue using Trizol (Invitrogen, Carlsbad, CA, USA). Total RNA (1 μg) was reverse-transcribed using a reverse transcription kit (no. DRR047A; Takara Bio, Otsu, Japan) according to the manufacturer’s protocol. Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) was performed using SYBR Green I dye detection (Takara Bio) in a Real-Time Detection system (Bio-Rad Laboratories). The following primers were used [13]: CXCL13 forward, 5′-GGC CAC GGT ATT CTG GAA GC-3′; CXCL13 reverse, 5′-ACC GAC AAC AGT TGA AAT CAC TC-3′; CXCR5 forward, 5′-TGG CCT TCTA CAG TAA CAG CA-3′; CXCR5 reverse, 5′-GCA TGA ATA CCG CCT TAA AGG AC-3′; GAPDH forward, 5′-GCT TGA AGG TGT TGC CCT CAG-3′; and GAPDH reverse, 5′-AGA AGC CAG CGT TCA CCA GAC-3′. PCR conditions included an initial step at 95 °C for 3 min, followed by 30 cycles at 95 °C for 1 min, 56 °C for 40 s, and 72 °C for 1 min. Melting curves were generated to verify amplification specificity. Quantification was performed using the 2-ΔΔCT method, and cycle threshold values were normalized to those of GAPDH.
Hematoxylin and eosin staining
Mice were perfused with 20-mL normal saline, followed by 20-mL 4% formaldehyde. Brain tissues were harvested and post-fixed with 4% paraformaldehyde at room temperature overnight and then embedded in paraffin. A series of 5-μm-thick coronal sections were deparaffinized, rehydrated, and stained by hematoxylin-eosin (HE). The stained sections were observed under an optical microscope (Olympus BX53, Tokyo, Japan) in order to assess histopathology in the hippocampal region.
Immunohistochemical staining
Brain tissue was immunostained as described [24]. It was isolated, fixed in 4% paraformaldehyde, placed into paraffin blocks, and cut coronally into 6-μm sections. Sections were deparaffinized, rehydrated, heated at 95 °C for 15 min for antigen retrieval, then incubated in 3% H2O2 for 25 min at room temperature to block endogenous peroxidases. After blocking by goat serum for 30 min, the sections were incubated overnight at 4 °C with primary antibodies against CXCL13 (1:100; ab272874, Abcam, Cambridge, UK) or CXCR5 (1:100; ab133706, Abcam) or p-ERK (1:250; 4695, CTS, Danvers, MA, USA). Sections were then incubated with secondary antibodies for 60 min at room temperature. A 3,3′-diaminobenzidine kit (Bioss, Beijing, China) was used to visualize antibody binding, and the sections were counterstained with hematoxylin. Photomicrographs were captured by a digital camera connected to a microscope (3D HISTECH, Budapest, Hungary). The immunoreactive cells in CA1, CA3, and dentate gyrus of hippocampus were counted using Image Pro-Plus 6.0 (Media Cybernetics, Bethesda, MD, USA) in three randomly selected fields on each section.
Statistical analysis
Statistical analysis was carried out using GraphPad Prism Version 5.01 (Graph Pad Software, San Diego, CA, USA). Continuous variables were assessed using Shapiro-Wilk test for distribution. The analysis showed normal distribution for all variables. Thus, all continuous variables are shown as mean ± standard deviation (SD). Inter-group differences in Barnes maze training results were assessed for significance using two-way repeated-measures ANOVA, followed by the Bonferroni post hoc multiple comparison test. Inter-group differences on other variables were assessed using Student’s t test (for comparisons between two groups) or one-way ANOVA followed by Tukey’s multiple test (for comparisons among at least three groups). P < 0.05 was considered statistically significant.
Results
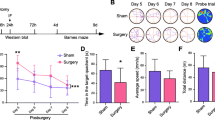
Surgery in mice impairs cognitive function and hippocampal morphology as a model of PNDs
Times in the Barnes maze decreased over the four training sessions in both control and surgery groups, and the mice that underwent surgery took longer to identify the target hole than controls (Fig. 2a). Surgery significantly prolonged latency time to identify the target hole at 1 or 8 days after training (Fig. 2b). Surgery shortened freezing times in the contextual fear conditioning test (Fig. 2c) without affecting freezing times in the cued fear conditioning test (Fig. 2d). HE staining also showed that surgery resulted in irregular neuronal arrangement, loss of morphology, and increases in neuronal swelling, nuclear pyknosis, and cellular vacuolization in the hippocampus (Fig. 2e).

Surgery in mice impairs cognitive function and hippocampal morphology as a model of PNDs. Adult male C57BL/6J mice were subjected to exploratory laparotomy (termed “Surgery”). Control mice did not undergo surgery. All mice were subjected to 4 days of training in the Barnes maze beginning 7 days after the surgery; memory phase testing was performed on days 11 and 18 post-surgery. a Time to find the target hole during the training sessions and b latency in identifying the target hole after training were measured. Fear conditioning tests were performed on days 19 and 20 after surgery. Freezing times were quantified c in context and d in response to a cue (n = 8 per group). e Micrographs of hippocampal sections stained with hematoxylin and eosin (n = 4 per group). Magnification, × 20 for regions CA1, CA3, and DG in hippocampus. *P < 0.05 vs. control
Surgery upregulates CXCL13 in mouse hippocampus
We checked CXCL13 mRNA levels in the hippocampus on days 1, , and 7 after surgery. Tissue from surgically treated mice had higher CXCL13 mRNA levels than control tissue at all timepoints (Fig. 3a). CXCL13 protein expression was higher in surgically treated animals than in controls on day 7 after surgery (Fig. 3b). Immunohistochemical staining of hippocampus showed more CXCL13-expressing neurons in the CA1, CA3, and dentate gyrus at 7 days after surgery than in controls (Fig. 3c).

Surgery up-regulates CXCL13 in mouse hippocampus. a Relative level of CXCL13 mRNA was measured by RT-qPCR in wild type mice at 1, 3, and 7 days after surgery. b Western blotting analysis of CXCL13 at 7 days after surgery. β-actin was used as an internal control (n = 8 per group). c Representative images of CXCL13 immunohistochemistry in the hippocampal CA1, CA3, and dentate gyrus regions. Positively stained cells are brown (red arrow). Magnification, × 5 for total hippocampus; × 20 for regions CA1, CA3, and DG in hippocampus. All experiments were performed with n = 4 per group. *P < 0.05 vs. control. CXCL13 C-X-C motif chemokine 13, DG dentate gyrus, RT-qPCR reverse transcription-quantitative polymerase chain reaction
CXCL13 knockdown alleviates PND-like cognitive deficits in mice
Mice that underwent surgery alone showed higher CXCL13 expression than controls (Fig. 4 a, b). To examine the role of CXCL13 in the development and maintenance of cognitive dysfunction, we injected CXCL13 shRNA into the lateral ventricle 3 days before surgery. CXCL13 knockdown significantly reduced CXCL13 expression in surgically treated mice, while scrambled RNA had no effect on CXCL13 expression (Fig. 4 a, b). Time to identify the target hole in the Barnes maze trial progressively decreased over the 4 days of training sessions in all groups (Fig. 4c). All surgically treated groups took significantly longer to find the target hole than the control group during training and testing, regardless of CXCL13 levels (Fig. 4 c, d ). However, mice transfected with CXCL13 shRNA prior to surgery identified the target hole faster than animals treated with surgery alone or with surgery and scrambled shRNA.

CXCL13 knockdown alleviates PND-like cognitive deficits in mice. Mice were pretreated with CXCL13 shRNA or scrambled shRNA, subjected to surgery and then behavioral tests were performed 7 days after surgery. a Western blotting and densitometry of hippocampal CXCL13, as well as b RT-qPCR analysis of the corresponding mRNA. c Time to identify the target hole in training sessions and d latency time during memory phases were quantified in the Barnes maze. In the fear conditioning tests, freezing times were quantified e in context and f in response to a cue. All quantitative results (mean ± SD) shown were obtained with n = 8 animals per group. *P < 0.05 vs. control; #P < 0.05 vs. surgery. CXCL13 C-X-C motif chemokine 13, RT-qPCR reverse transcription-quantitative polymerase chain reaction, shRNA short hairpin RNA, sh-CRTL scrambled shRNA, sh-CXCL13 CXCL13 shRNA
All mice that underwent surgery exhibited shorter freezing times than controls in the contextual fear conditioning test (Fig. 4e), and CXCL13 knockdown partially reversed this surgery-induced impairment. In contrast, the groups did not differ significantly in freezing times in the hippocampal-independent cued fear conditioning test (Fig. 4f).
Surgery upregulates CXCR5 in mouse hippocampus
CXCR5 mRNA levels were higher in hippocampal tissue at 1, 3, and 7 days after surgery (Fig. 5a). Similarly, the level of corresponding protein was higher in surgically treated mice than controls at 7 days after surgery, as shown by Western blot and immunohistochemistry (Fig. 5 b, c ).

Surgery up-regulates CXCR5 in mouse hippocampus. a Relative level of CXCR5 mRNA was measured by RT-qPCR on days 1, 3, and 7 after surgery. b Western blotting analysis and densitometry of CXCR5 protein levels. β-actin was used as an internal control (n = 8 per group). c Representative images of CXCR5 immunohistochemistry in the hippocampal CA1, CA3, and dentate gyrus. Positively stained cells are brown (red arrow). Magnification, × 5 for total hippocampus; × 20 for hippocampal CA1, CA3, and DG (n = 4 per group). *P < 0.05 vs. control. CXCR5 C-X-C motif chemokine receptor 5, DG dentate gyrus, RT-qPCR reverse transcription-quantitative polymerase chain reaction
Deletion of CXCR5 alleviates PND-like cognitive deficits in mice
While all groups became increasingly faster with each training session in the Barnes maze trial, surgery impacted time more than CXCR5 levels (Fig. 6a). Surgically treated CXCR5−/− mice identified the target hole faster than surgically-treated WT mice (Fig. 6b).

Deletion of CXCR5 alleviates PND-like cognitive deficits in mice. WT or mice lacking the CXCR5 gene (CXCR5−/−) were subjected to surgery (denoted as “surgery”). Mice that did not undergo surgery were denoted “control.” All mice were tested in the Barnes maze and fear conditioning tests at 7 days after surgery. a Time to identify the target hole during training sessions and b latency time during memory phases of the Barnes maze. In the fear conditioning test, freezing times were quantified c in context and d in response to a cue. e Western blot and densitometry of hippocampal CXCR5, as well as f RT-qPCR analysis of the corresponding mRNA. All quantitative results (mean ± SD) shown were obtained with n = 8 animals per group. *P < 0.05 vs. WT + control; #P < 0.05 vs. WT + surgery. CXCR5 C-X-C motif chemokine receptor 5, RT-qPCR reverse transcription-quantitative polymerase chain reaction, WT wild-type
Although all mice that underwent surgery exhibited shorter freezing times than control mice in the contextual fear conditioning test, no differences were seen among the groups during the cued fear conditioning test (Fig. 6 c, d ). Surgically treated WT mice had higher CXCR5 mRNA and protein expression than control WT mice (Fig. 6 e, f ).
CXCR5 deficiency inhibits surgery-induced p-ERK activation and release of inflammatory cytokines in mouse hippocampus
WT and CXCR5−/− mice with surgery had significantly higher p-ERK expression in the hippocampus on day 7 after surgery than counterparts that did not undergo surgery (Fig. 7a). However, total ERK levels were unaffected. CXCR5 knockout reduced p-ERK levels in surgically treated mice but not control mice. Moreover, surgery induced an increase in the number of p-ERK-containing neurons in the hippocampus of WT mice on day 7, and less so in CXCR5−/− mice (Fig. 7b). There was no difference in the number of p-ERK-containing neurons in control animals of either genotype.

CXCR5 deficiency inhibits surgery-induced p-ERK activation and release of inflammatory cytokines in mouse hippocampus. a Western blotting analysis of total ERK and p-ERK (n = 8 per group). b Representative images of p-ERK immunohistochemistry in the hippocampus. Positive cells are stained brown. Magnification, × 5 for total hippocampus; × 20 for hippocampal CA1, CA3, and DG (n = 4 per group). c Western blot and densitometry of IL-1β and TNF-α in the hippocampus. β-actin was used as an internal control (n = 8 per group). *P < 0.05 vs. control; #P < 0.05 vs. surgery. CXCR5 C-X-C motif chemokine receptor 5, DG dentate gyrus, WT wild-type
TNF-α and IL-1β are important pro-inflammatory cytokines in regulating cognitive dysfunction in the CNS [25]. We checked TNF-α and IL-1β expression in the hippocampus on day 7 after surgery. WT mice with surgery had higher TNF-α and IL-1β expression than controls (Fig. 7c). Surgically treated CXCR5−/− mice had lower TNF-α and IL-1β expression than surgically treated WT mice.
ERK-dependent production of pro-inflammatory cytokines in hippocampus mediates PND-like cognitive deficits in mice
Mice pretreated with PD98059 displayed shorter time to the target hole on all training days (Fig. 8a), and also had shorter latency than surgically-treated WT mice (Fig. 8b). PD98059 pretreatment also rescued PND-like effects in the contextual fear conditioning test (Fig. 8c), but made no difference in cued fear freezing (Fig. 8d). PD98059 pretreatment downregulated TNF-α and IL-1β expression in surgically treated mice (Fig. 8e).

ERK-dependent production of pro-inflammatory cytokines in hippocampus mediates PND-like cognitive deficits in mice. Mice subjected to surgery were pretreated with vehicle or PD98059, and behavioral tests were conducted 7 days after the surgery. a Time to find the target hole during training sessions and b latency time during memory phases were assessed in the Barnes maze. In the fear conditioning test, freezing times were quantified c in context and d in response to a cue. e Western blot and densitometry of hippocampal IL-1β and TNF-α protein levels. β-actin was used as an internal control. All quantitative results (mean ± SD) shown were obtained with n = 8 animals per group. *P < 0.05 vs. control; #P < 0.05 vs. surgery. TNF-α tumor necrosis factor alpha, IL-1β interleukin-1 beta
CXCL13 induces CXCR5/ERK-dependent release of pro-inflammatory cytokines in mouse hippocampus
Compared with control mice, all CXCL13 treatment groups took more time to identify the target hole across all days (Fig. 9 a, b ), and these impairments were partially reversed by deleting CXCR5. Additionally, CXCL13 treatment shortened freezing time in contextual fear conditioning, and it did so to a smaller extent in CXCR5−/− mice (Fig. 9c). No differences were found in the cued fear conditioning test (Fig. 9d). CXCL13 increased the levels of p-ERK, IL-1β, and TNF-α, and the effect was mitigated by CXCR5 deficiency (Fig. 9e).

CXCL13 induces CXCR5/ERK-dependent release of pro-inflammatory cytokines in mouse hippocampus. WT or CXCR5−/− mice received CXCL13 via intracerebroventricular injection. Seven days later, Barnes maze and fear conditioning tests were conducted. a Time to identify the target hole during training sessions and b latency time during memory phase were measured in the Barnes maze. Freezing times were quantified c in context and d in response to a cue. e Western blotting and densitometry of hippocampal p-ERK, total ERK, IL-1β, and TNF-α. Mice were injected with CXCL13 with or without PD98059 pretreatment, and behavioral tests were performed 7 days later. f Time to identify the target hole in training sessions and h latency time in memory phases of the Barnes maze. Freezing times were quantified i in context and j in response to a cue in fear conditioning tests. k Western blot and densitometry of IL-1β and TNF-α. All quantitative results (mean ± SD) shown were obtained with n = 8 animals per group. *P < 0.05 vs. vehicle + WT; #P < 0.05 vs. CXCL13 + WT; $P < 0.05 vs. vehicle; &P < 0.05 vs. CXCL13. CXCL13 C-X-C motif chemokine 13, CXCR5 C-X-C motif chemokine receptor 5, WT wild-type, TNF-α tumor necrosis factor alpha, IL-1β interleukin-1 beta
Pretreatment PD98059 partially attenuated CXCL13-induced cognitive dysfunction (Fig. 9 f–i ). Additionally, CXCL13 mice pretreated with PD98059 had lower expression of TNF-α and IL-1β than mice treated with CXCL13 alone (Fig. 9j).
Discussion
The present study explored whether chemokine CXCL13 and its receptor CXCR5 are involved in surgery-induced cognitive dysfunction in an animal model of PND. Our data clearly demonstrate that surgical intervention induced CXCL13 release and an increase in CXCR5 expression followed by cognitive impairment as observed in behavioral tests, and that inhibition of CXCL13/CXCR5 signaling attenuated surgery-induced cognitive impairment. We also investigated the role of ERK in surgery-induced neuroinflammation. Surgery induced ERK activation and ERK-dependent production of the inflammatory factors IL-1β and TNF-α in the hippocampus of mice, but this effect was repressed by CXCR5 knockout. Finally, i.c.v. injection of recombinant CXCL13 induced CXCR5/ERK-dependent upregulation of TNF-α and IL-1β as well as cognitive dysfunction. Altogether, our results suggest that CXCL13 induces PND, at least partially, by triggering hippocampal CXCR5-ERK signaling.
CXCL13, a member of the chemokine family, is the major determinant for B cell recruitment to the intrathecal compartment during a range of human neurological disorders [6, 26]. Studies in animals and humans suggest that CXCL13 contributes to certain CNS disorders, and that blockade of this ligand might have therapeutic effects [25]. CXCL13 has been proposed as a biomarker of neuroinflammatory disease due to its high level in the CNS under a variety of conditions, including multiple sclerosis and neuromyelitis optica [7] as well as CNS lymphoma [27, 28]. Whether CXCL13 is involved in the pathogenesis of PNDs remains unclear. In the present study, CXCL13 mRNA in the hippocampus was upregulated 1 day after surgery and remained elevated for more than 7 days in surgically treated mice with cognitive impairment; Western blotting and immunostaining further showed upregulated CXCL13 protein in the hippocampus. In agreement with our results, several reports showed that CXCL13 was increased in dorsal root ganglion in a neuropathic pain model and hippocampus in CNS infection [29,30,31,32], and its high expression was detected mainly in microglia, macrophages, and endothelial cells [13, 33]. To examine the role of CXCL13 in the development and maintenance of cognitive dysfunction, we knocked it down specifically using shRNA. Knockdown effectively attenuated surgery-induced cognitive dysfunction. These data suggest the involvement of hippocampal CXCL13 in surgery-induced PNDs.
CXCR5 is responsible for the organization of B cell follicles and for directing T-helper cells to the lymphoid follicles [34,35,36,37]. In the CNS, CXCR5 is expressed in neuronal precursor cells, which migrate across brain endothelial cells upon exposure to CXCL13 [11]. CXCL13 and CXCR5 are involved in neurodegeneration and cognitive decline [9], which are related to a deficit in hippocampal neurogenesis [38, 39]. In previous work, we showed that CXCR5 may inhibit proliferation, differentiation, and survival of hippocampal neuronal stem cells in an animal model of sepsis, leading to learning and memory impairments [14]. Hippocampal CXCR5 upregulation causes sevoflurane anesthesia-induced cognitive dysfunction via PI3K/Akt signaling [40]. In the present study, we found that surgery elevated hippocampal CXCR5 mRNA and protein levels for more than 7 days in mice. CXCR5 deficiency attenuated surgery-induced cognitive dysfunction. Additionally, i.c.v. injection of recombinant CXCL13 triggered learning and memory deficits in surgically treated animals, which were attenuated in CXCR5−/− animals. These data suggest that CXCL13/CXCR5 signaling is necessary for the development and maintenance of PNDs.
CXCL13/CXCR5 signaling contributes to neurodegeneration in individuals with cognitive deficit disease [9], as well as in animal models with neuropathic pain [41]. In fact, animal studies have shown that CXCL13 and CXCR5 contribute to neuropathic pain via ERK signaling [13]. ERK is also activated in the hippocampus of animals anesthetized with sevoflurane or propofol, and these animals later show impaired learning and memory [42, 43]. Intra-prefrontal cortex administration of ERK inhibitor PD98059 in 6-month-old 3xTg mice reversed memory impairment, suggesting that ERK pathway alterations might at least partially explain memory deficits observed in an Alzheimer’s disease model [44]. Here, we found that surgery increased p-ERK levels in the hippocampus, and this upregulation was partially reversed by deleting CXCR5. Moreover, i.c.v. injection of CXCL13 induced CXCR5-dependent p-ERK upregulation in the hippocampus. Similarly, a recent study showed that CXCL13 activates ERK in the spinal cord through CXCR5 in a mouse model of diabetes-induced tactile allodynia [41]. These data suggest the important role of ERK in mediating CXCL13/CXCR5 signaling in the hippocampus.
Surgical trauma can cause neuroinflammation involving synaptic deficits, neuronal dysfunction and death, and impaired neurogenesis [45, 46]. ERK is known to mediate the expression of inflammatory mediators, including growth factors and pro-inflammatory cytokines (e.g., IL-1β) in an animal model of cognitive deficit [47]. IL-1β and TNF-α are important pro-inflammatory cytokines that mediate surgery-induced cognitive dysfunction [48]. We also found that surgery upregulated the pro-inflammatory cytokines IL-1β and TNF-α in the hippocampus, and this was partially reversed by deleting CXCR5 or knocking down ERK. Pretreatment with CXCL13 induced CXCR5/ERK-dependent release of pro-inflammatory cytokines in the hippocampus. Both CXCL13 and CXCR5 were upregulated in the brain in both patients with intractable epilepsy and a rat model of epilepsy with neuroinflammation [9]. Increased release of proinflammatory cytokines, including IL-1β and TNF-α, has been reported in the hippocampus of rodents after prolonged seizures [49], and contributes to cognitive impairment [50]. We did not observe spontaneous seizure in our experiments. Such a discrepancy warrants further investigation. Nevertheless, our findings data suggest that ERK-mediated release of pro-inflammatory cytokines in the hippocampus contributes to PNDs.
Previous studies showed that CXCR5−/− mice have more immature neurons in the dentate gyrus, an increase in baseline locomotor activity, and decreased anxiety-like behavior [39]. CXCR5−/− mice may also develop retinal degeneration with pathophysiological changes, such as activation of microglia and accumulation of inflammatory cells, loss of ZO-1 indicative of impaired blood-retinal barrier function after ischemia-reperfusion injury [51]. These changes could conceivably produce major impact on learning and memory, and thus bias the results in the current study.
Conclusions
These results from the current study indicated that CXCL13 and CXCR5 contribute to PNDs, possibly by activating ERK to trigger release of pro-inflammatory cytokines in the hippocampus. Thus, targeting the CXCL13/CXCR5/ERK pathway in the hippocampus may provide a novel therapeutic approach to treat these disorders.
Availability of data and materials
All data generated or analyzed during this study are available from the corresponding author upon reasonable request.
Abbreviations
- CNS:
-
Central nervous system
- CXCL13:
-
C-X-C motif chemokine 13
- CXCR5:
-
C-X-C motif chemokine receptor 5
- ERK:
-
Extracellular signal-regulated kinase
- i.c.v.:
-
Intracerebroventricular
- IL-1β:
-
Interleukin-1 beta
- PND:
-
Perioperative neurocognitive disorder
- RT-qPCR:
-
Reverse transcription-quantitative polymerase chain reaction
- shRNA:
-
Short hairpin RNA
- TNF-α:
-
Tumor necrosis factor alpha
- WT:
-
Wild-type
References
Evered L, Silbert B, Knopman DS, Scott DA, DeKosky ST, Rasmussen LS, Oh ES, Crosby G, Berger M, Eckenhoff RG, Nomenclature Consensus Working Grou. Recommendations for the nomenclature of cognitive change associated with anaesthesia and surgery-2018. Br J Anaesth. 2018;121(5):1005–12.
Fan D, Li J, Zheng B, Hua L, Zuo Z. Enriched environment attenuates surgery-induced impairment of learning, memory, and neurogenesis possibly by preserving BDNF expression. Mol Neurobiol. 2016;53(1):344–54.
Monk TG, Weldon BC, Garvan CW, Dede DE, van der Aa MT, Heilman KM, Gravenstein JS. Predictors of cognitive dysfunction after major noncardiac surgery. Anesthesiology. 2008;108(1):18–30.
Huang C, Irwin MG, Wong GTC, Chang RCC. Evidence of the impact of systemic inflammation on neuroinflammation from a non-bacterial endotoxin animal model. J Neuroinflammation. 2018;15(1):147.
Zheng B, Lai R, Li J, Zuo Z. Critical role of P2X7 receptors in the neuroinflammation and cognitive dysfunction after surgery. Brain Behav Immun. 2017;61:365–74.
Kowarik MC, Cepok S, Sellner J, Grummel V, Weber MS, Korn T, et al. CXCL13 is the major determinant for B cell recruitment to the CSF during neuroinflammation. J Neuroinflammation. 2012;9:93.
Alvarez E, Piccio L, Mikesell RJ, Klawiter EC, Parks BJ, Naismith RT, et al. CXCL13 is a biomarker of inflammation in multiple sclerosis, neuromyelitis optica, and other neurological conditions. Mult Scler. 2013;19(9):1204–8.
Bagaeva LV, Rao P, Powers JM, Segal BM. CXC chemokine ligand 13 plays a role in experimental autoimmune encephalomyelitis. J Immunol. 2006;176(12):7676–85.
Li R, Ma L, Huang H, Ou S, Yuan J, Xu T, et al. Altered expression of CXCL13 and CXCR5 in intractable temporal lobe epilepsy patients and pilocarpine-induced epileptic rats. Neurochem Res. 2017;42(2):526–40.
Krumbholz M, Theil D, Cepok S, Hemmer B, Kivisäkk P, Ransohoff RM, et al. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up-regulation is differentially linked to CNS immune cell recruitment. Brain. 2006;129(Pt 1):200–11.
Weiss N, Deboux C, Chaverot N, Miller F, Baron-Van Evercooren A, Couraud PO, et al. IL8 and CXCL13 are potent chemokines for the recruitment of human neural precursor cells across brain endothelial cells. J Neuroimmunol. 2010;223:131–4.
Moser B, Schaerli P, Loetscher P. CXCR5(+) T cells: follicular homing takes center stage in T-helper-cell responses. Trends Immunol. 2002;23(5):250–4.
Zhang Q, Cao DL, Zhang ZJ, Jiang BC, Gao YJ. Chemokine CXCL13 mediates orofacial neuropathic pain via CXCR5/ERK pathway in the trigeminal ganglion of mice. J Neuroinflammation. 2016;13(1):183.
Shen Y, Jing L, Zhang Y, Bao H, Vohra A, Si Y, et al. CXCR5 knockdown attenuates hippocampal neurogenesis deficits and cognitive impairment in a mouse model of sepsis-associated encephalopathy. Neuroscience. 2020;433:212–20.
El-Haibi CP, Singh R, Sharma PK, Singh S, Lillard JW Jr. CXCL13 mediates prostate cancer cell proliferation through JNK signalling and invasion through ERK activation. Cell Prolif. 2011;44(4):311–9.
Bu HL, Xia YZ, Liu PM, Guo HM, Yuan C, Fan XC, et al. The roles of chemokine CXCL13 in the development of bone cancer pain and the regulation of morphine analgesia in rats. Neuroscience. 2019;406:62–72.
Yang Y, Zhang X, Cui H, Zhang C, Zhu C, Li L. Apelin-13 protects the brain against ischemia/reperfusion injury through activating PI3K/Akt and ERK1/2 signaling pathways. Neurosci Lett. 2014;568:44–9.
Denner LA, Rodriguez-Rivera J, Haidacher SJ, Jahrling JB, Carmical JR, Hernandez CM, et al. Cognitive enhancement with rosiglitazone links the hippocampal PPARγ and ERK MAPK signaling pathways. J Neurosci. 2012;32(47):16725–35a.
Jahrling JB, Hernandez CM, Denner L, Dineley KT. PPARγ recruitment to active ERK during memory consolidation is required for Alzheimer's disease-related cognitive enhancement. J Neurosci. 2014;34(11):4054–63.
Pan K, Li X, Chen Y, Zhu D, Li Y, Tao G, et al. Deferoxamine pre-treatment protects against postoperative cognitive dysfunction of aged rats by depressing microglial activation via ameliorating iron accumulation in hippocampus. Neuropharmacology. 2016;111:180–94.
Ni J, Wang X, Chen S, Liu H, Wang Y, Xu X, et al. MicroRNA let-7c-5p protects against cerebral ischemia injury via mechanisms involving the inhibition of microglia activation. Brain Behav Immun. 2015;49:75–85.
Liang P, Shan W, Zuo Z. Perioperative use of cefazolin ameliorates postoperative cognitive dysfunction but induces gut inflammation in mice. J Neuroinflammation. 2018;15(1):235.
Ji MH, Tang H, Luo D, Qiu LL, Jia M, Yuan HM, et al. Environmental conditions differentially affect neurobehavioral outcomes in a mouse model of sepsis-associated encephalopathy. Oncotarget. 2017;8(47):82376–89.
Hu S, Hu M, Liu J, Zhang B, Zhang Z, Zhou FH, et al. Phosphorylation of Tau and α-synuclein induced neurodegeneration in MPTP mouse model of Parkinson's disease. Neuropsychiatr Dis Treat. 2020;16:651–63.
Huber AK, Irani DN. Targeting CXCL13 during neuroinflammation. Adv Neuroimmune Biol. 2015;6(1):1–8.
Campbell DJ, Kim CH, Butcher EC. Chemokines in the systemic organization of immunity. Immunol Rev. 2003;195:58–71.
Smith JR, Braziel RM, Paoletti S, Lipp M, Uguccioni M, Rosenbaum JT. Expression of B-cell-attracting chemokine 1 (CXCL13) by malignant lymphocytes and vascular endothelium in primary central nervous system lymphoma. Blood. 2003;101(3):815–21.
Fischer L, Korfel A, Pfeiffer S, Kiewe P, Volk HD, Cakiroglu H, et al. CXCL13 and CXCL12 in central nervous system lymphoma patients. Clin Cancer Res. 2009;15(19):5968–73.
Wu XB, Cao DL, Zhang X, Jiang BC, Zhao LX, Qian B, et al. CXCL13/CXCR5 enhances sodium channel current density via p38 MAP kinase in primary sensory neurons following inflammatory pain. Sci Rep. 2016;6:34836.
Jiang BC, Cao DL, Zhang X, Zhang ZJ, He LN, Li CH, et al. CXCL13 drives spinal astrocyte activation and neuropathic pain via CXCR5. J Clin Invest. 2016;126(2):745–61.
Strong JA, Xie W, Coyle DE, Zhang JM. Microarray analysis of rat sensory ganglia after local inflammation implicates novel cytokines in pain. PLoS One. 2012;7:e40779.
Narayan K, Dail D, Li L, Cadavid D, Amrute S, Fitzgerald-Bocarsly P, et al. The nervous system as ectopic germinal center: CXCL13 and IgG in lyme neuroborreliosis. Ann Neurol. 2005;57(6):813–23.
Laurent C, Dorothee G, Hunot S, Martin E, Monnet Y, Duchamp M, et al. Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain. 2017;140(1):184–200.
Muller G, Hopken UE, Lipp M. The impact of CCR7 and CXCR5 on lymphoid organ development and systemic immunity. Immunol Rev. 2003;195:117–35.
Burkle A, Niedermeier M, Schmitt-Graff A, Wierda WG, Keating MJ, Burger JA. Overexpression of the CXCR5 chemokine receptor, and its ligand, CXCL13 in B-cell chronic lymphocytic leukemia. Blood. 2007;110:3316–25.
Legler DF, Loetscher M, Roos RS, Clark-Lewis I, Baggiolini M, Moser B. B cell-attracting chemokine 1, a human CXC chemokine expressed in lymphoid tissues, selectively attracts B lymphocytes via BLR1/CXCR5. J Exp Med. 1998;187(4):655–60.
Kim CH, Rott LS, Clark-Lewis I, Campbell DJ, Wu L, Butcher EC. Subspecialization of CXCR5+ T cells: B helper activity is focused in a germinal center-localized subset of CXCR5+ T cells. J Exp Med. 2001;193(12):1373–81.
Chapman KZ, Ge R, Monni E, Tatarishvili J, Ahlenius H, Arvidsson A, et al. Inflammation without neuronal death triggers striatal neurogenesis comparable to stroke. Neurobiol Dis. 2015;83:1–15.
Stuart MJ, Corrigan F, Baune BT. Knockout of CXCR5 increases the population of immature neural cells and decreases proliferation in the hippocampal dentate gyrus. J Neuroinflammation. 2014;11:31.
Su R, Sun P, Zhang D, Xiao W, Feng C, Zhong L. Neuroprotective effect of miR-410-3p against sevoflurane anesthesia-induced cognitive dysfunction in rats through PI3K/Akt signaling pathway via targeting C-X-C motif chemokine receptor 5. Genes Genomics. 2019;41(10):1223–31.
Liu S, Liu X, Xiong H, Wang W, Liu Y, Yin L, et al. CXCL13/CXCR5 signaling contributes to diabetes-induced tactile allodynia via activating pERK, pSTAT3, pAKT pathways and pro-inflammatory cytokines production in the spinal cord of male mice. Brain Behav Immun. 2019;80:711–24.
Zhu QL, Luo Y, Xue QS, Zhang FJ, Yu BW. Different doses of sevoflurane facilitate and impair learning and memory function through activation of the ERK pathway and synthesis of ARC protein in the rat hippocampus. Brain Res. 1678;2018:174–9.
Li GF, Li ZB, Zhuang SJ, Li GC. Inhibition of microRNA-34a protects against propofol anesthesia-induced neurotoxicity and cognitive dysfunction via the MAPK/ERK signaling pathway. Neurosci Lett. 2018;675:152–9.
Feld M, Krawczyk MC, Sol Fustiñana M, Blake MG, Baratti CM, Romano A, et al. Decrease of ERK/MAPK overactivation in prefrontal cortex reverses early memory deficit in a mouse model of Alzheimer's disease. J Alzheimers Dis. 2014;40(1):69–82.
Alam A, Hana Z, Jin Z, Suen KC, Ma D. Surgery, neuroinflammation and cognitive impairment. EBioMedicine. 2018;37:547–56.
Cibelli M, Fidalgo AR, Terrando N, Ma D, Monaco C, Feldmann M, et al. Role of interleukin-1beta in postoperative cognitive dysfunction. Ann Neurol. 2010;68(3):360–8.
Saeed M, Ghadiri A, Hadizadeh F, Attaranzadeh A, Alavi MS, Etemad L. Cinnamaldehyde improves methamphetamine-induced spatial learning and memory deficits and restores ERK signaling in the rat prefrontal cortex. Iran J Basic Med Sci. 2018;21(12):1316–21.
Li Z, Liu F, Ma H, White PF, Yumul R, Jiang Y, et al. Age exacerbates surgery-induced cognitive impairment and neuroinflammation in Sprague-Dawley rats: the role of IL-4. Brain Res. 1665;2017:65–73.
Garcia-Curran MM, Hall AM, Patterson KP, Shao M, Eltom N, Chen K, et al. Dexamethasone attenuates hyperexcitability provoked by experimental febrile status epilepticus. eNeuro. 2019;6(6) ENEURO.0430-19.2019:1-17.
Dube CM, Zhou JL, Hamamura M, Zhao Q, Ring A, Abrahams J, et al. Cognitive dysfunction after experimental febrile seizures. Exp Neurol. 2009;215(1):167–77.
Cao X, Li W, Liu Y, Huang H, Ye CH. The anti-inflammatory effects of CXCR5 in the mice retina following ischemia-reperfusion injury. Biomed Res Int. 2019;2019:3487607.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (81873954 and 81971872), Nanjing Medical Science and Technical Development Foundation (QRX17019 and YKK18105) and Six Talent Peaks Project in Jiangsu (WSW-106).
Author information
Authors and Affiliations
Contributions
SYN (Yanna Si) conceived and designed the study. SYN (Yanan Shen) performed the majority of the experiments. DJY and CMM assisted with tissue sample collection. ZY and XY analyzed the data. SYN (Yanna Si) and SYN (Yanan Shen) drafted and edited the manuscript. BHG and CLH provided critical discussion and participated in the revision. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No human data or tissues were used in this study. All animal experimental protocols and handling procedures were approved by the Institutional Animal Care and Use Committee of Nanjing Medical University.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Shen, Y., Zhang, Y., Chen, L. et al. Chemokine CXCL13 acts via CXCR5-ERK signaling in hippocampus to induce perioperative neurocognitive disorders in surgically treated mice. J Neuroinflammation 17, 335 (2020). https://doi.org/10.1186/s12974-020-02013-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-020-02013-x