
Abstract
Neuroinflammation is considered one of the cardinal features of Alzheimer’s disease (AD). Neuritic plaques composed of amyloid β and neurofibrillary tangle-laden neurons are surrounded by reactive astrocytes and microglia. Exposure of microglia, the resident myeloid cell of the CNS, to amyloid β causes these cells to acquire an inflammatory phenotype. While these reactive microglia are important to contain and phagocytose amyloid plaques, their activated phenotype impacts CNS homeostasis. In rodent models, increased neuroinflammation promoted by overexpression of proinflammatory cytokines can cause an increase in hyperphosphorylated tau and a decrease in hippocampal function. The peripheral immune system can also play a detrimental or beneficial role in CNS inflammation. Systemic inflammation can increase the risk of developing AD dementia, and chemokines released directly by microglia or indirectly by endothelial cells can attract monocytes and T lymphocytes to the CNS. These peripheral immune cells can aid in amyloid β clearance or modulate microglia responses, depending on the cell type. As such, several groups have targeted the peripheral immune system to modulate chronic neuroinflammation. In this review, we focus on the interplay of immunomodulating factors and cell types that are being investigated as possible therapeutic targets for the treatment or prevention of AD.
Similar content being viewed by others

Introduction
Neuroinflammation is considered one of the cardinal features of Alzheimer’s disease (AD). Early observations of neuritic plaques composed of amyloid β and neurofibrillary tangle-laden neurons surrounded by reactive astrocytes and microglia [1,2,3,4], as well as evidence that microglia exposed to amyloid β release proinflammatory factors such as interleukin 1 beta (IL-1β) and tumor necrosis factor alpha (TNFα) [5,6,7] that in turn could modify AD pathology [8,9,10,11,12], led to hypotheses positing chronic neuroinflammation as a driving feature of AD [13]. Subsequent investigations revealed a more complex picture, with increased recognition of factors and cell types involved in neuroinflammation, including the influence of peripheral immune mediators and their effects on AD pathology and associated cognitive function in animal models. These findings related to the role of neuroinflammation in AD have been reviewed in multiple articles [14,15,16,17,18]. In this review, we focus on the interplay between factors and cell types associated with neuroinflammation, including cytokines, microglia, and peripheral immune mediators, as well as some clinical studies, to describe how modulation of such processes might be exploited for prevention or treatment of AD.
Cytokines in AD and effects on pathology
Concentration of various proinflammatory cytokines including IL-1β, IL-6, IL-12, IL-18, and TNFα and anti-inflammatory cytokines such as interleukin-1 receptor antagonist (IL-1RA) and IL-10 have been found to increase in cerebrospinal fluid (CSF) of AD patients, pointing to an immune disturbance [19,20,21,22]. Whether production of these cytokines initiates AD or results from neurodegeneration and neuritic plaque deposition has not been conclusively demonstrated in humans. Murine models of AD have been useful in testing the effects of various cytokines on amyloid β and tau pathology. In general, most studies have found that overexpression of pro-inflammatory cytokines leads to a reduction of amyloid β plaque, possibly through microglial activity [10, 23,24,25]. Nonetheless, proinflammatory factors, such as IL-1β, IL-6, and TNFα, have also been shown to worsen tau hyperphosphorylation and thus could contribute to neurofibrillary tangle formation [8, 11, 26]. In addition, genetic studies have demonstrated that polymorphisms in various cytokines, such as IL-1β, IL-6, IL-12, IL-18, and TNFα, and cytokine receptors, such as the IL-1 receptor accessory protein, are associated with an increased risk of developing AD [27,28,29]. While the risk of developing AD due to these mutations is modest, they can hasten disease progression when concomitantly present with variants of apolipoprotein E (APOE) that are associated with late-onset AD [27]. Thus, cytokine production could be a potential therapeutic target to slow the progression of sporadic AD.
There is a possibility that AD progression is partially due to a dysregulation of sterile inflammation, such as the one that is induced in the brain with amyloid β accumulation. One of the components of the sterile inflammatory response is the formation of a multiprotein complex called the inflammasome, which is critical to convert precursors of IL-1β and IL-18 to the biologically active cytokines [30]. Fibrillar amyloid β has been shown to bind NOD-like receptor protein 3 (NLRP3), one of the pattern recognition receptors that regulates inflammasome formation in microglia and induces IL-1β production [31]. Deletion of NLRP3 ameliorated amyloid β pathology and improved cognitive impairment in APP/PS1 mice [32], perhaps by reducing cross-seeding of amyloid β [33]. Administration of NLRP3 inhibitors have decreased cerebral amyloid β and improved cognitive function in amyloidogenic models making inflammasome formation a promising therapeutic target [34,35,36]. Further discussion of the role of inflammasomes in AD and neurodegeneration can be found in recent reviews [37,38,39].
In addition to the proinflammatory changes that occur with aging and AD, there is evidence to suggest a dysregulation in anti-inflammatory pathways. Immunohistochemical studies have revealed that the neurotrophic factor TGF-β2 is localized in reactive astrocytes and microglia surrounding neuritic plaques [40]. In addition, increased concentrations of TGF-β2 have been detected in the AD patient CSF [41]. Expression of this cytokine in reactive glia may represent an attempt to curb the inflammatory response initiated by amyloid β accumulation. In AD patients, neuronal TGF-β2 signaling may be blunted due to decreased expression of TGFβRII that is present even in prodromal stages of AD [42]. Like proinflammatory cytokine modulation, overexpression of anti-inflammatory cytokines, such as IL-4, IL-13, and TGF-β1, has been shown to decrease amyloid beta plaque [43,44,45]. Stimulation with anti-inflammatory cytokines alternatively activates microglia, upregulating markers such as arginase 1 and YM1. One group demonstrated that hippocampal arginase 1 overexpression in rTg4510 tau transgenic mice led to a decrease in tau phosphorylation [46].
The role and modulation of microglia in AD
Microglia are the resident macrophages of the central nervous system. During homeostatic conditions, microglia survey the brain and participate in the maintenance and pruning of synapses [47, 48]. However, like macrophages in other tissues, microglia become activated when they encounter an offending stimulus or pathogen. In the context of AD, neuritic plaques are typically surrounded by extensive astrogliosis and microgliosis. Microglia surrounding plaques acquire an amoeboid morphology, which is associated with high phagocytic activity. Several lines of research demonstrated that amyloid β binds toll-like receptor 4 and its co-activator CD14, polarizing microglia towards an inflammatory phenotype which includes the release of proinflammatory cytokines like IL-1β, IL-6, and TNFα, as well as the chemokine CCL2 [6, 7, 49, 50]. As described above, these cytokines aid in the activation of other microglia to increase their phagocytic capabilities.
Amyloid β can exist as soluble oligomers or insoluble fibrils of the proteolytic products Aβ 1-40 or 1-42. While attention has largely focused on studying the effect of insoluble fibrils on neurons and glia, amyloid β oligomers are also thought to contribute to disease pathology. Aβ 1-42 oligomers, more so that Aβ 1-40 oligomers, have been shown to be neurotoxic and can elicit acute microgliosis and impairment in long-term potentiation when injected into the brain of wild-type mice [51, 52]. Furthermore, microglia are capable of phagocytizing all forms of amyloid β, including soluble oligomers [53]. Interestingly, the response of microglia to amyloid β is different depending on whether it exists as soluble oligomers or fibrillar plaques. In an in vitro study, oligomers stimulated increased levels of phagocytosis markers including activated Lyn and Syk kinase as well as p38MAPK compared to fibrils [54]. In addition, microglia treated with oligomers increased the production of cytokines IL-6 and CCL2 when compared to fibrils [54, 55]. Some investigators have considered amyloid β plaques to be a relatively benign aggregation of amyloid β that microglia contain to protect the brain from the more neurotoxic oligomers [56,57,58].
Microglia employ several strategies in order to contain and clear amyloid β pathology. Microglia surround amyloid β plaques in an effort to isolate them from the rest of the brain parenchyma [57]. In addition, microglia can release enzymes that aid in the degradation of amyloid β plaques. Complement activation is also important for amyloid β phagocytosis. There is increased C1 surrounding neuritic plaques in AD patients’ tissue samples [59]. In vitro studies have shown that human C1q is able to bind amyloid β and thus could initiate the complement cascade, allowing amyloid β plaques to be opsonized and facilitate phagocytosis by microglia [60]. Interferon γ-induced C3 upregulation in an amyloidogenic mouse model caused a decrease in amyloid β plaque size and number [61], and inhibition of C3 with sCrry or C3 knockout caused increased plaque deposition [62, 63]. However, upregulation of C1q and C3 also resulted in increased phagocytosis of synapses by microglia and appeared to be necessary for cognitive dysfunction in amyloidogenic models, indicating that this clearance mechanism might be a double-edged sword [64, 65]. C1q tags pathological tau-laden synapses, leading to microglial engulfment and synapse loss [66]. In addition, deletion of the C3a receptor in PS19 tau mice decreased neuroinflammation, attenuated tau pathology, and ameliorated disease-associated microglia and neurotoxic astrocyte signature [67].
The ability of microglia to surround and phagocytize amyloid β plaques depends on the microglia-expressed gene Trem2. This gene encodes a protein called triggering receptor expressed on myeloid cells 2 that is expressed by innate immune cells. Among the functions of TREM2 are regulation of immune responses, expression of constitutive cytokines, and microglial response to neuronal injury [68, 69]. Mutations in TREM2 have been identified as a risk factor for the development of late-onset AD [70,71,72]. Mice deficient in TREM2 tend to have diffuse, irregularly-shaped plaques with multiple hotspots of Aβ 1-42 polymerization [73, 74]. Furthermore, these plaques were associated with increased neurodegeneration when compared to TREM2 sufficient mice [74]. In addition, TREM2 deficiency induced inflammation shown by an increase in IL-1β secretion and Iba1 staining in microglia [73,74,75]. Possibly because of the increase in neuroinflammation, TREM2 deficiency also exacerbated tau hyperphosphorylation in a tauopathy murine model [76]. These findings suggest that TREM2 activity regulates the inflammatory response by microglia and thus might be a target for modulation of their activation.
Microglia can also acquire an anti-inflammatory, neuroprotective phenotype in the context of AD. Evidence of the anti-inflammatory mechanisms in AD comes from studies that manipulated the neuroinflammatory environment in AD mouse models. Induction of neuroinflammation through acute intra-cortical lipopolysaccharide (LPS) injection or IL-1β overexpression in the hippocampus not only increased the number of proinflammatory nitric oxide synthase (iNOS) positive microglia, but also induced the expression of anti-inflammatory marker arginase 1 in some microglia [26, 45]. Interestingly, arginase 1-positive cells appeared to be better phagocytes of amyloid β when compared to iNOS-positive cells and IL-1β-dependent plaque clearance was inhibited using antibodies against the IL-4 receptor (IL-4Rα) [45]. Intrahippocampal injection of anti-inflammatory cytokines IL-4 and IL-13 also induced arginase 1 expression and reduced pathology in amyloidogenic murine models [44, 45]. Arginase 1 overexpression alone was able to cause a reduction in tau hyperphosphorylation, while arginase inhibition led to an increase in tau pathology [46]. What predisposes some microglia to move along the spectrum of proinflammatory and anti-inflammatory phenotypes in the context of AD is currently unknown. In spinal cord injury, inflammation induces expression of IL-4Rα and IL-4 mRNA [77]. Interestingly, IL-4 signaling correlates with not only an increase in arginase 1 signaling but also an increase in IL-1β and CCL2 expression that is not present in a microglia-specific IL-4Rα knockout mouse [77]. While initial studies sought to categorize these cells into either proinflammatory M1 cells or anti-inflammatory M2 cells, the reality is that microglial phenotypes show greater diversity during development and pathology [78,79,80,81], and the use of this simplistic nomenclature has been discouraged [82, 83].
Recently, careful attention has been given to describe microglia phenotypes. Advances such as single-cell RNA sequencing have revealed that immune cells in the brain comprise a more heterogeneous population than previously thought [84, 85]. In the healthy brain, homeostatic microglia are tightly regulated by TGF-β1 [86, 87]. Deletion of this gene leads to a downregulation of the homeostatic signature of microglia, which is characterized by the expression of P2ry12, Tmem119, Sall1, and Mertk, among other genes. Phenotypically, mice with absent TGF-β1 developed late-onset motor deficits and have synaptic plasticity and glutamate recycling abnormalities [86].
This TGF-β1-dependent state is also downregulated during neurodegenerative processes to give rise to a more phagocytic, inflammatory phenotype termed disease-associated microglia (DAM) or the microglia of neurodegenerative disease [88, 89]. In addition to a loss of homeostatic markers, the DAM phenotype has been characterized by an upregulation of phagocytic genes including Trem2, Apoe, Axl, Lpl, and Clec7a. DAM activation occurs in a stepwise manner with a TREM2-independent transitional stage followed by a TREM2-dependent state [88]. In AD, this program is specifically adopted by microglia surrounding amyloid β plaques in both humans and mouse models [89]. Interestingly, this program deviates from the M1 and M2 phenotype as DAM express both classic M2 markers, such as arginase 1 and YM1, and proinflammatory genes such as Il1b, PTGS2, Ccl2, Ccl5, Tspo, Msr1, and Cebpb [89]. In contrast, microglia stimulated with LPS or IFN γ, which are known inducers of the M1 phenotype, have low expression of Apoe, which is one of the most significantly upregulated genes in DAM cells [89].
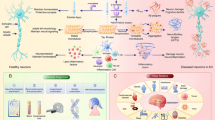
The phagocytic nature of the DAM program might be beneficial for the clearance or containment of proteinaceous deposits such as amyloid β. Similarly to the peripheral immune system, microglia have mechanisms to counterbalance this activation and return to homeostasis, including binding of CX3CR1 and CD200R to their respective ligands [90]. However, these immune checkpoints are downregulated during the course of DAM activation [88, 89]. Thus, with increased accumulation of amyloid β plaques, microglia may become dysregulated and unable to resume their neuroprotective functions, which include promoting neuronal health and secreting immunosuppressive factors, such as TGF-β1, which have been shown to prevent inflammatory cascades from persisting [91]. Furthermore, these phagocytic microglia may also partake in excessive synaptic pruning, contributing to memory loss [64]. Indeed, TREM2 deletion has been associated with an increase in the proportion of homeostatic microglia and a reduction in amyloid β plaques at early stages of amyloidogenesis [83, 89, 92]. Still, future studies evaluating the protective functions of DAM and the balance between pathological response and homeostasis offer a promising route for the development of comprehensive AD treatments that affect multiple disease hallmarks. In Fig. 1, we briefly summarize the role of microglia during homeostasis and AD neurodegeneration. Furthermore, microglial responses can be modulated by systemic inflammation and peripheral immune cells, which will be discussed later in this review.

Factors impacting microglial phenotype in the context of homeostasis and neurodegeneration. Adult microglia are characterized by the expression of P2ry12, Cx3cr1, and Tmem119, among others. In addition to their role in immune surveillance, microglia participate in central nervous system homeostasis and are known to release neurotrophic factors that promote neuronal health. During Alzheimer’s disease pathogenesis, microglia are activated by amyloid β through binding of TLR4 and, in a TREM2 dependent manner, downregulate expression of homeostatic genes and upregulate Apoe, Tyrobp, Trem2, and other genes, adopting the disease-associated microglia (DAM) program. DAM are effective at containing and phagocytosing amyloid β plaques. At the same time, DAM cease their neuroprotective functions and release neuroinflammatory factors such as IL-1β and TNFα that are directly damaging to neurons and lead to tau phosphorylation and subsequent cognitive decline. In addition to local factors, the peripheral immune system can modify these responses and induce the DAM program, indicating that this is a potential target through which the homeostatic and inflammatory balance can be modulated and perhaps used to promote beneficial outcomes in AD
In addition to changes during disease progression, microglia may also become dysfunctional and lose the capacity to return to their neuroprotective functions with aging [93]. Among the changes that occur in the aging brain are an increase in activation elements of microglia including toll-like receptor signaling and complement activation. In addition, there is a decrease of factors that curtail neuroinflammation including IL-10, fractalkine, and toll-interacting protein [20]. Careful morphological analysis of microglia in tissue from advanced AD patients revealed extensive cytorrhexis or fragmentation of microglia rather than amoeboid microglia [93]. Thus, late in disease, microglia might become unable to respond and control amyloid β pathology in addition to not being able to carry out their neuroprotective functions.
Peripheral immune cell contributions to AD progression and modulation of neuroinflammation
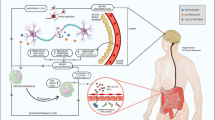
The central nervous system is considered an immune-privileged organ due to the blood-brain barrier (BBB) and the relatively low number of surveilling peripheral immune cells found within the brain parenchyma. Nonetheless, in the context of the chronic neuroinflammation that is present in AD, there is some evidence to suggest that peripheral immune cells infiltrate the CNS and accumulate near areas of pathology [94,95,96]. These peripheral immune cells include monocytes, which are highly phagocytic cells that migrate into areas of injury and proliferate and differentiate into macrophages to clear pathogens or cellular debris. Evidence suggests that inducing neuroinflammation in amyloidogenic mouse models by acute LPS injection or IL-1β overexpression leads to an increase of Ly6c+ CCR2+ monocyte migration into the central nervous system [97, 98]. Some studies have shown increased numbers of grafted monocytes associated with amyloid plaques in chimeric models and a dependence of amyloid clearance on the recruitment of CCR2+ cells [99,100,101]. Nonetheless, CCR2 knockout does not have an impact on IL-1β-induced clearance of amyloid β plaque by resident microglia and recent chimera studies that did not employ irradiation have shown very little evidence of migration [98]. Perhaps these monocytes could serve as a backup to resident microglial populations if the offending stimuli persist [102]. Interestingly, intracerebrally injected mesenchymal stem cells derived from the bone marrow or adipose tissue significantly reduced amyloid beta deposition and enhanced cognitive function, apparently by decreasing neuroinflammation [103, 104]. Thus, this pool of cells could be exploited to substitute for senescent microglia when they fail to slow AD progression [93, 105].
There is evidence to suggest migration of T lymphocytes into the brain of AD patients [106, 107]. As already noted, stimulation of microglia with amyloid β plaques in transgenic mouse models or injection of Aβ 1-42 oligomers lead to the release of proinflammatory cytokines such as IL-1β and TNFα. In vitro and in vivo studies have shown that these cytokines promote the production of chemokine CXCL8 by endothelial cells [108,109,110,111]. Interestingly, T cells of AD patients are enriched in the chemokine receptor CXCR2 which, upon binding to CXCL8, promotes T cell transmigration across endothelial barriers, including the blood-brain barrier [112]. This movement through the BBB was inhibited by anti-TNFα antibodies or inhibition of CXCR2 on T cells [112]. Other studies in rodent models have shown that once in the brain, T cells can modify microglia phenotype, increasing their motility and phagocytic activity through secretion of IFN γ [113]. Potentially, microglia could phagocytose and present amyloid β to T cells as a form of re-stimulation [114]. Whether this mechanism occurs in humans with AD and to what extent it is utilized remains an open question. Interestingly, RAG1 and RAG2 knockout mice, which lack B and T lymphocytes, demonstrated worse cognitive capabilities when compared to wild-type mice and increased amyloid β plaque deposition and microglia activation in amyloidogenic models [115, 116]. Nonetheless, these approaches have not yet teased out the specific contributions of different lymphocyte subtypes, which might explain differing results in some cases [117].
There are several types of T cells; among them are the classically defined CD8+ or cytotoxic T lymphocytes and CD4+ or helper T lymphocytes. The function of CD8 T cells is to infiltrate tissue and secrete enzymes such as perforin, granzymes, and granulysins that promote apoptosis of damaged or infected cells [118]. These cells can be found in AD patient brain tissue [96] and were shown in the hippocampus of tauopathy mouse models [119]. Indeed, inhibition of T cell infiltration was correlated with decreased microglial activation and cognitive impairment, without a significant impact on tau pathology [119].
In contrast to findings with cytotoxic T cells, T helper cells have been much more closely implicated in the response to AD pathology [116]. Th1 cells are responsible for promoting cellular responses to offending stimuli by activating macrophages and CD8+ T cells and their effector cytokine is IFNγ [120]. Contrastingly, Th2 cells are responsible for promoting humoral immune responses by inducing antibody production by B lymphocytes and some of their effector molecules are the anti-inflammatory cytokines IL-4 and IL-10, which inhibit Th1 responses [120]. In the context of AD, T helper cells have been shown to modulate microglial responses through contact or release of cytokines. Research into Th1 cells and IFNγ demonstrates that these cells can migrate into the brain parenchyma after immunization with Aβ 1-42 [95, 121, 122]. What they do once in the CNS is the subject of controversy. Some have argued that Th1 cells activate microglia to increase amyloid β clearance [113, 123], while others have claimed they increase pathology through IFNγ release [124]. This microglial activation also makes their processes more motile, which might help microglia encounter and surround amyloid β plaques more efficiently [124]. Interestingly, contrary to other proinflammatory cytokines, such as TNFα and IL-1β, overexpression of IFNγ using an AAV vector led to decreased tau pathology and increased neurogenesis [123]. However, the immune response of Th1 cells has been closely associated with the onset of meningoencephalitis in human subjects injected with an Aβ 1-42 vaccine, providing clear evidence that these cells increase proinflammatory responses [121, 125]. In contrast, induced Th2 cells may modulate glial responses with limited infiltration of the CNS, instead of acting from the choroid plexus or meningeal spaces [126,127,128,129]. Rodent studies have shown that when amyloid β specific Th2 cells are injected into the periphery, they modulate the cytokine profile of transgenic mice towards a more anti-inflammatory one by decreasing GM-CSF, TNFα, and IL-2 levels [128]. This peripheral modulation has been correlated with decreased microgliosis that is accompanied by improved cognitive performance [128]. Furthermore, Th2 cell activity might induce the promotion of Aβ 1-42 autoantibodies [130].
Attempts to harness the activity of the peripheral immune system to develop therapeutics for AD have been an ongoing subject of research. Active immunization with amyloid β was previously tried to promote clearance of AD pathology. While there is evidence that this therapy was able to improve cognition in some patients, this approach led to the development of meningoencephalitis in 6% of the subjects [131]. Since then, passive immunization approaches utilizing humanized antibodies against amyloid β, such as bapineuzumab and solanezumab, were developed and have since been tested in clinical trials [132, 133]. Many of these trials have concluded and most of the results are disappointing [134]. Ongoing trials are attempting to address whether treatment of individuals at risk at presymptomatic stages or utilizing higher doses can improve primary outcomes [134]. However, some have questioned whether targeting amyloid β production is sufficient to treat AD and if a revision of the amyloid hypothesis is needed [135, 136]. There has been a push to develop antibodies against tau oligomers, which are also known to be proinflammatory and may contribute to the spread of tau pathology [137]. Murine studies testing such approaches have succeeded in decreasing tau hyperphosphorylation after anti-tau oligomer administration [138, 139]. Currently, a study testing the humanized tau antibody ABBV-8E12 is enrolling patients in a phase II clinical trial [140].
In addition, others have proposed the use of the relapsing-remitting multiple sclerosis drug Copaxone or glatiramer acetate (GA) to treat AD [141,142,143]. GA is a mixture of peptides composed of the four amino acids found in myelin basic protein: glutamic acid, lysine, alanine, and tyrosine. While the mechanism of action of GA is still debated, evidence suggests that it is a stimulator of the Th2 response, which is beneficial in combating relapsing-remitting multiple sclerosis, possibly by suppressing the inflammatory, Th1 response [144,145,146]. Evaluation of serum and CNS of GA-treated mice showed increased levels of the anti-inflammatory cytokine IL-10 and brain-derived neurotrophic factor (BDNF), both of which are produced by activated Th2 cells [147,148,149]. In addition, not only has GA been effective in decreasing pathology in models of experimental autoimmune encephalomyelitis, but it has also shown benefits in Huntington’s disease models [149, 150]. GA use in amyloidogenic mouse models successfully reduced plaque load; however, the effects of GA on tau pathology remain untested [141,142,143].
The barriers that separate the periphery from the brain play an active role in regulating adaptive and innate immunity in the CNS. One of the most well-studied points of immune surveillance and trafficking into the CNS is the choroid plexus [151]. While leukocytes still have to receive chemoattractive signals to cross this structure, the fenestrated nature of the choroid plexus epithelium may be easier to traverse than the BBB. Recently, it has been shown that targeting the immune checkpoint protein programmed cell death 1 (PD-1) activates the choroid plexus epithelium and leads to an increase in monocyte trafficking through this structure, which hones to sites of amyloid deposition in the parenchyma [152]. This process is dependent on systemic T cell-derived IFNγ, most likely secreted by Th1 cells. Moreover, targeting immunosuppression mediated by regulatory T cells has a similar effect on immune trafficking through the choroid plexus in the 5xFAD model, which is correlated with improved cognition and pathology [142]. Nonetheless, it remains to be seen whether such approaches would be safe to consider long term and what effect they would have on neurofibrillary tangles. Uncontrolled T cell activity can not only increase hyperphosphorylation of tau but can also provoke more serious side effects such as encephalitis.
Systemic inflammation and AD immunomodulation
Systemic infection and inflammation in individuals with AD is associated with worse cognitive function and reduced hippocampal volume [153,154,155]. Specifically, high serum levels of inflammatory cytokines like IL-1β and TNFα are associated with disease progression. In addition, some have shown that systemic infection is associated with an increased risk of developing AD [33, 156]. Induction of systemic disorders such as osteoarthritis in APP/PS1 mice led to glial activation and exacerbation of amyloid pathology [157]. After peripheral infection of APP/PS1 mice with the respiratory pathogen Bordetella pertussis, increased brain infiltration of IFNγ and IL-17 producing T cells and natural killer cells was observed that correlated with increased glial activation and amyloid β plaque deposition [158]. Both of these rodent studies highlight the interconnections between the peripheral inflammatory state and the CNS. It is clear that changes in concentrations of peripheral cytokines and infiltration of peripheral cells can have lasting effects on the response of microglia to AD pathology.
Microglia are incredibly sensitive to perturbations in the brain environment by peripheral stimuli including inflammation and infections. Several studies have utilized intraperitoneal injections of bacterial LPS to evaluate the effects of infection in AD [159,160,161,162,163,164]. In a recent study, peripheral LPS induced microglial activation and phenotype changes, including an increase in markers of disease-associated microglia, such as APOE and CLEC7A [88, 165]. LPS administration also increased accumulation of amyloid precursor protein and its cleavage product amyloid β early in disease [159, 166]. Another study found that this accumulation of amyloid β was exacerbated by an altered amyloid β efflux through the BBB in a low-density lipoprotein receptor-related protein 1 (LRP1)-dependent manner [166]. Interestingly, while a single LPS injection increased pro-inflammatory cytokines in the blood and brain [167], a persistent inflammatory tone established by sequential LPS injections can lead microglia to become more tolerant of inflammatory stimuli and dampen their response [165]. This last study also found that mice that had tolerant microglia also had lower levels of cerebral amyloidosis.
Modulation of pro-inflammatory and anti-inflammatory signals in the periphery has been explored as a potential target to ameliorate AD pathology. Blockade of the classic proinflammatory cytokine TNFα decreased amyloid β [168, 169] and tau pathology [170]. Inhibition of TNFα production using the small molecule thalidomide or one of its derivatives improved cognition and CNS pathology in models of systemic disease [171, 172]. Likewise, modulation of TNFα by an orally administered isoindolin-1,3 dithione (IDT) caused a reduction in fibrillar amyloid and tau hyperphosphorylation in the 3xTg AD mouse [173]. Interestingly, IDT treatment was also associated with increased neutrophil infiltration while reducing TNFα expression in these cells. Lastly, TNFα receptor antibodies such as etanercept or infliximab have been tested in dementia rat models and showed beneficial effects on cognitive behavior and some evidence of decreased AD pathology [174]. A case report administering perispinal etanercept led to a rapid cognitive improvement [175]. Recently, a small randomized, placebo-control trial of subcutaneous etanercept for AD was completed. Researchers found that while there were no significant differences between placebo and treated patients, the study revealed interesting trends that might be worth following up on with a larger, more heterogeneous cohort [176]. In addition to TNFα, peripheral blockade of other proinflammatory cytokines such as IL-12 and IL-23 had similar effects on amyloid pathology and rodent behavior, indicating that there is more than one potential target for proinflammatory cytokine modulation that could be exploited [177].
As discussed earlier, sterile inflammation has been implicated in AD progression. One of the cytokines playing a protective role in sterile inflammation is IL-33 [178]. IL-33 levels are low in the brains of AD patients, and serum levels of soluble ST2, the IL-33 receptor, are high in the brains of patients with mild cognitive impairment [179, 180]. Intraperitoneal injection of IL-33 ameliorated synaptic impairment and amyloid pathology in APP/PS1 mice [180]. Moreover, plaque-adjacent microglia in IL-33-injected mice showed increased expression of CD68, indicating increased phagolysosomal activity. In addition, there was an increase in levels of the enzyme neprilysin, which can aid in amyloid degradation, and decreased evidence of neuroinflammation, with lower levels of the molecules IL-1β, IL-6, and NLRP3 in the cortices of APP/PS1 mice [180]. IL-33 could be negatively regulating TLR4 activity by competing for MyD88, the binding partner of ST2 [181]. In another study, IL-33 deficient mice were found to develop severe neurodegeneration late in life that is characterized by abnormal accumulation of tau [182].
Activation of TLR9 with a peripheral injection of CpG oligodeoxynucleotides led to a reduction in both amyloid β and tau pathology in murine models of AD and thus could be beneficial [183,184,185]. TLR9 has been shown to regulate autoimmune responses in models of systemic lupus erythematosus [186]; therefore, it is possible that TLR9 in particular may regulate the inflammatory response in the context of AD, promoting an environment that leads to tissue repair. Taken together, there are many examples showing that alteration of peripheral inflammation by infectious agents or injections of cytokines or TLR ligands can have profound effects on brain AD pathology. More studies are needed to define mechanisms of CNS immunomodulation by peripheral factors. Nonetheless, the identification of such modulators is a promising and exciting route for therapeutic discovery. A recent review article by Jeffrey Cummings provides a comprehensive summary of immune-related and other therapeutics currently in the AD drug development pipeline [187].
Current evidence from traditional anti-inflammatory medications
Observational studies, such as case-control and incidence studies, have shown that regular use of non-selective NSAIDs is associated with a reduced relative risk of developing AD [188,189,190,191]. The majority of individuals followed in these observational studies suffered from an inflammatory condition, such as rheumatoid arthritis, which explains their long-term NSAID use. Thus, the cohorts observed in these studies only represent a proportion of the population at risk of developing AD. A review article by Imbimbo et al. [192] has delineated the following general trends of risk reduction found in observational studies: (1) risk was modified by the length of time NSAIDs were used with the lowest risk being associated with 2 years of use, while no effect was detected with 1 month of use; (2) the type of NSAID use, with non-aspirin NSAIDs having a higher risk reduction when compared to aspirin, an irreversible COX inhibitor; (3) NSAIDs that were found to be effective at reducing risk of AD also have been shown to decrease levels of Aβ-42 in murine models of AD such as ibuprofen, indomethacin, and diclofenac.
Unfortunately, randomized control trials (RCT) have found that the use of naproxen, diclofenac, or indomethacin, all of which have been shown to reduce AD risk in epidemiological studies, do not slow down the progression of AD when compared to placebo [192,193,194]. One of the challenges for these trials was the dropout rate of subjects due to gastrointestinal issues brought on by chronic NSAID use. In order to prevent side effects, some RCTs minimized the daily dose of NSAID administered to the treatment group, which might potentially explain the negative results [192]. Trials utilizing COX 2 selective NSAIDs like celecoxib and rofecoxib, which are less likely to cause gastrointestinal issues, but retain risks for deleterious cardiovascular events, have failed to demonstrate positive treatment benefits for AD or mild cognitive impairment [195, 196].
Another potential mechanism of action of NSAIDs is activation of the peroxisome proliferator-activated receptor gamma (PPARγ) [192, 197]. This receptor is a member of the nuclear factor family, and its activation regulates the transcriptional activation and repression of several genes. One of the cellular actions resulting from PPARγ activation is the decrease of inflammation by reducing the production of proinflammatory cytokines such as IL-1β, IL-6, and TNFα [197]. At high concentrations, some NSAIDs can serve as an agonist of this receptor. Indomethacin and ibuprofen can activate microglial PPARγ and reduce amyloid β induced release of proinflammatory cytokines [198]. Treatment of amyloidogenic murine models with pioglitazone, a direct agonist of PPARγ, reduces amyloid β 1-42 deposits in the hippocampus [199]. The TOMMORROW study was a multicenter trial created to evaluate pioglitazone as a preventative agent for the development of AD [200]. However, the study was recently halted due to inadequate treatment effect.
Conclusion
The contribution of specific pro-inflammatory and anti-inflammatory factors in AD is not straightforward, especially since the evaluation of cognition, amyloid β pathology, and neurofibrillary tangles yields conflicting results in mouse models. Furthermore, translating rodent studies that have modulated expression of specific cytokines in the CNS is challenging. In addition, studies that have shown promise, such as the beneficial effects of pioglitazone in mouse models of AD, do not always prove effective in humans. Nonetheless, the immune response is deeply tied to the development of pathology, and with advancing technologies, we are able to more fully dissect the complexity of this response and the effector cells that carry it out. Our knowledge of how microglia and peripheral immune cells interact has proved invaluable in understanding how this delicate balance goes awry in disease. Immunomodulation in AD offers multiple, promising pathways of investigation that might lead to therapeutics that can prevent or halt the development of amyloid and tau pathology and cognitive decline.
Abbreviations
- AD:
-
Alzheimer’s disease
- APOE:
-
Apolipoprotein E
- Aβ:
-
Amyloid β
- BBB:
-
Blood-brain barrier
- BDNF:
-
Brain-derived neurotrophic factor
- CNS:
-
Central nervous system
- COX 1:
-
Cyclooxygenase 1
- COX 2:
-
Cyclooxygenase 2
- CSF:
-
Cerebrospinal fluid
- DAM:
-
Disease-associated microglia
- GA:
-
Glatiramer acetate
- IFNγ:
-
Interferon γ
- IL-10:
-
Interleukin 10
- IL-12:
-
Interleukin 12
- IL-13:
-
Interleukin 13
- IL-17:
-
Interleukin 17
- IL-18:
-
Interleukin 18
- IL-1RA:
-
Interleukin-1 receptor antagonist
- IL-1β:
-
Interleukin 1β
- IL-33:
-
Interleukin 33
- IL-4:
-
Interleukin 4
- IL-4Rα:
-
Interleukin 4 receptor α
- IL-6:
-
Interleukin 6
- iNOS:
-
Nitric oxide synthase
- LPS:
-
Lipopolysaccharide
- NLRP3:
-
NACHT, LRR, and PYD domain-containing protein 3
- NSAIDs:
-
Non-steroidal anti-inflammatory drugs
- PPARγ:
-
Peroxisome proliferator-activated receptor γ
- RCT:
-
Randomized control trials
- TGF-β1:
-
Transforming growth factor β1
- TGF-β2:
-
Transforming growth factor β2
- TGFβRII:
-
Transforming growth factor β receptor 2
- Th1:
-
Type 1 T helper cells
- Th2:
-
Type 2 T helper cells
- TLR:
-
Toll-like receptor
- TNFα:
-
Tumor necrosis factor α
- TREM2:
-
Triggering receptor expressed on myeloid cells 2
References
Rogers J, Luber-Narod J, Styren SD, Civin WH. Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer’s disease. Neurobiol Aging. 1988;9:339–49.
McGeer PL, Akiyama H, Itagaki S, McGeer EG. Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neurosci Lett. 1989;107:341–6.
Sheng JG, Jones RA, Zhou XQ, McGinness JM, Van Eldik LJ, Mrak RE, et al. Interleukin-1 promotion of MAPK-p38 overexpression in experimental animals and in Alzheimer’s disease: potential significance for tau protein phosphorylation. Neurochem Int. 2001;39:341–8.
Hayes A, Thaker U, Iwatsubo T, Pickering-Brown SM, Mann DMA. Pathological relationships between microglial cell activity and tau and amyloid β protein in patients with Alzheimer’s disease. Neurosci Lett. 2002;331:171–4.
Benzing WC, Wujek JR, Ward EK, Shaffer D, Ashe KH, Younkin SG, et al. Evidence for glial-mediated inflammation in aged APP(SW) transgenic mice. Neurobiol Aging. 1999;20:581–9.
Mehlhorn G, Hollborn M, Schliebs R. Induction of cytokines in glial cells surrounding cortical beta-amyloid plaques in transgenic Tg2576 mice with Alzheimer pathology. Int J Dev Neurosci. 18:423–31.
Apelt J, Schliebs R. Beta-amyloid-induced glial expression of both pro- and anti-inflammatory cytokines in cerebral cortex of aged transgenic Tg2576 mice with Alzheimer plaque pathology. Brain Res. 2001;894:21–30.
Quintanilla RA, Orellana DI, González-Billault C, Maccioni RB. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp Cell Res. 2004;295:245–57.
Li Y, Liu L, Barger SW, Griffin WST. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J Neurosci. 2003;23:1605–11.
Shaftel SS, Kyrkanides S, Olschowka JA, Miller JN, Johnson RE, O'Banion MK. Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest. 2007;117:1595-604
Ghosh S, Wu MD, Shaftel SS, Kyrkanides S, LaFerla FM, Olschowka JA, et al. Sustained interleukin-1 overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J Neurosci. 2013;33:5053–64.
Jee Y, Liu R, Bai X-F, Campagnolo DI, Shi F-D, Vollmer TL. Do Th2 cells mediate the effects of glatiramer acetate in experimental autoimmune encephalomyelitis? Int Immunol. 2006;18:537–44.
Griffin WST, Sheng JG, Royston MC, Gentleman SM, McKenzie JE, Graham DI, et al. Glial-neuronal interactions in Alzheimer’s disease: the potential role of a ‘cytokine cycle’ in disease progression. Brain Pathol. 2006;8:65–72.
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, et al. Inflammation and Alzheimer’s disease. Neurobiol Aging. 2000.
Wyss-Coray T, Rogers J. Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med. 2012;2:a006346.
Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14:388–405.
Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci. 2015;16:358–72.
Bronzuoli MR, Iacomino A, Steardo L, Scuderi C. Targeting neuroinflammation in Alzheimer’s disease. J Inflamm Res. 2016;9:199–208.
Ojala J, Alafuzoff I, Herukka S-K, van Groen T, Tanila H, Pirttilä T. Expression of interleukin-18 is increased in the brains of Alzheimer’s disease patients. Neurobiol Aging. 2009;30:198–209.
Cribbs DH, Berchtold NC, Perreau V, Coleman PD, Rogers J, Tenner AJ, et al. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: a microarray study. J Neuroinflammation. 2012;9:643.
Smith JA, Das A, Ray SK, Banik NL. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull. 2012;87:10–20.
Zheng C, Zhou X-W, Wang J-Z. The dual roles of cytokines in Alzheimer’s disease: update on interleukins, TNF-α, TGF-β and IFN-γ. Transl Neurodegener. 2016;5:7.
Shaftel SS, Griffin WST, O’Banion MK. The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective. J Neuroinflammation. 2008;5:7.
Yamamoto M, Kiyota T, Horiba M, Buescher JL, Walsh SM, Gendelman HE, et al. Interferon-γ and tumor necrosis factor-α regulate amyloid-β plaque deposition and β-secretase. Am J Pathol. 2007;170:680–92.
Chakrabarty P, Jansen-West K, Beccard A, Ceballos-Diaz C, Levites Y, Verbeeck C, et al. Massive gliosis induced by interleukin-6 suppresses Aβ deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. FASEB J. 2010;24:548–59.
Lee DC, Rizer J, Selenica M-LB, Reid P, Kraft C, Johnson A, et al. LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J Neuroinflammation. 2010;7:56.
Laws SM, Perneczky R, Wagenpfeil S, Müller U, Förstl H, Martins RN, et al. TNF polymorphisms in Alzheimer disease and functional implications on CSF beta-amyloid levels. Hum Mutat. 2005;26:29–35.
Ramanan VK, Risacher SL, Nho K, Kim S, Shen L, McDonald BC, et al. GWAS of longitudinal amyloid accumulation on 18F-florbetapir PET in Alzheimer’s disease implicates microglial activation gene IL1RAP. Brain. 2015;138:3076–88.
Mun M-J, Kim J-H, Choi J-Y, Jang W-C. Genetic polymorphisms of interleukin genes and the risk of Alzheimer’s disease: an update meta-analysis; 2016.
He Y, Hara H, Núñez G. Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem Sci. 2016;41(12):1012–21.
Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–65.
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2012;493:674–8.
Engelhart MJ, Geerlings MI, Meijer J, Kiliaan A, Ruitenberg A, van Swieten JC, et al. Inflammatory proteins in plasma and the risk of dementia. Arch Neurol. 2004;61:668.
Daniels MJD, Rivers-Auty J, Schilling T, Spencer NG, Watremez W, Fasolino V, et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat Commun. 2016;7:12504.
Dempsey C, Rubio Araiz A, Bryson KJ, Finucane O, Larkin C, Mills EL, et al. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain Behav Immun. 2017;61:306–16.
Yin J, Zhao F, Chojnacki JE, Fulp J, Klein WL, Zhang S, et al. NLRP3 inflammasome inhibitor ameliorates amyloid pathology in a mouse model of Alzheimer’s disease. Mol Neurobiol. 2018;55:1977–87.
Liu L, Chan C. The role of inflammasome in Alzheimer’s disease. Ageing Res Rev. 2014;15:6–15.
Heneka MT. Inflammasome activation and innate immunity in Alzheimer’s disease. Brain Pathol. 2017;27:220–2.
Heneka MT, McManus RM, Latz E. Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci. 2018;19:610–21.
Flanders KC, Lippa CF, Smith TW, Pollen D a, Sporn MB Altered expression of transforming growth factor β in Alzheimer's disease. Neurology. 1995 Aug;45(8):1561–1569.
Chao CC, Ala TA, Hu S, Crossley KB, Sherman RE, Peterson PK, et al. Serum cytokine levels in patients with Alzheimer’s disease. Clin Diagn Lab Immunol. 1994;1:433–6.
Tesseur I, Zou K, Esposito L, Bard F, Berber E, Van Can J, et al. Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer’s pathology. J Clin Invest. 2006;116:3060–9.
Wyss-Coray T, Lin C, Yan F, Yu G-Q, Rohde M, McConlogue L, et al. TGF-β1 promotes microglial amyloid-β clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:612–8.
Kawahara K, Suenobu M, Yoshida A, Koga K, Hyodo A, Ohtsuka H, et al. Intracerebral microinjection of interleukin-4/interleukin-13 reduces β-amyloid accumulation in the ipsilateral side and improves cognitive deficits in young amyloid precursor protein 23 mice. Neuroscience. 2012;207:243–60.
Cherry JD, Olschowka JA, O’Banion MK. Arginase 1+ microglia reduce Aβ plaque deposition during IL-1β-dependent neuroinflammation. J Neuroinflammation. 2015;12:203.
Hunt JBJ, Nash KR, Placides D, Moran P, Selenica M-LB, Abuqalbeen F, et al. Sustained arginase 1 expression modulates pathological tau deposits in a mouse model of tauopathy. J Neurosci. 2015;35:14842–60.
Lannes N, Eppler E, Etemad S, Yotovski P, Filgueira L. Microglia at center stage: a comprehensive review about the versatile and unique residential macrophages of the central nervous system. Oncotarget. 2017;8:114393–413.
Li Q, Barres BA. Microglia and macrophages in brain homeostasis and disease. Nat Rev Immunol. 2017;18:225–42.
Ishizuka K, Kimura T, Igata-Yi R, Katsuragi S, Takamatsu J, Miyakawa T. Identification of monocyte chemoattractant protein-1 in senile plaques and reactive microglia of Alzheimer’s disease. Psychiatry Clin Neurosci. 1997;51:135–8.
Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, et al. Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998;152:307–17.
Benilova I, Karran E, De Strooper B. The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci. 2012;15:349–57.
Goure WF, Krafft GA, Jerecic J, Hefti F. Targeting the proper amyloid-beta neuronal toxins: a path forward for Alzheimer’s disease immunotherapeutics. Alzheimers Res Ther. 2014;6:42.
Parvathy S, Rajadas J, Ryan H, Vaziri S, Anderson L, Murphy GM. Aβ peptide conformation determines uptake and interleukin-1α expression by primary microglial cells. Neurobiol Aging. 2009;30:1792–804.
Sondag CM, Dhawan G, Combs CK. Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J Neuroinflammation. 2009;6:1.
El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, et al. CD36 mediates the innate host response to beta-amyloid. J Exp Med. 2003;197:1657–66.
Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, et al. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci. 2008;28:4283–92.
Condello C, Yuan P, Schain A, Grutzendler J. Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat Commun. 2015;6:6176.
Zhao J, Nussinov R, Ma B. Mechanisms of recognition of amyloid-β (Aβ) monomer, oligomer, and fibril by homologous antibodies. J Biol Chem. 2017;292:18325–43.
McGeer PL, McGeer EG, Yasojima K. Complement, neuroinflammation and neuronal degeneration in Alzheimer disease. Neurosci Basis Dement. 2001:15–20.
Webster SD, Tenner AJ, Poulos TL, Cribbs DH. The mouse C1q A-chain sequence alters beta-amyloid-induced complement activation. Neurobiol Aging. 1999;20:297–304.
Chakrabarty P, Ceballos-Diaz C, Beccard A, Janus C, Dickson D, Golde TE, et al. IFN-γ promotes complement expression and attenuates amyloid plaque deposition in amyloid precursor protein transgenic mice. J Immunol. 2010;184:5333–43.
Wyss-Coray T, Yan F, Lin AH-T, Lambris JD, Alexander JJ, Quigg RJ, et al. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc Natl Acad Sci. 2002;99:10837–42.
Maier M, Peng Y, Jiang L, Seabrook TJ, Carroll MC, Lemere CA. Complement C3 deficiency leads to accelerated amyloid beta plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J Neurosci. 2008;28:6333–41.
Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352:712–6.
Shi Q, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, et al. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci Transl Med. 2017;9:eaaf6295.
Dejanovic B, Huntley MA, De Mazière A, Meilandt WJ, Wu T, Srinivasan K, et al. Changes in the synaptic proteome in tauopathy and rescue of tau-induced synapse loss by C1q antibodies. Neuron. 2018;100:1322–1336.e7.
Litvinchuk A, Wan Y-W, Swartzlander DB, Chen F, Cole A, Propson NE, et al. Complement C3aR inactivation attenuates tau pathology and reverses an immune network deregulated in tauopathy models and Alzheimer’s disease. Neuron. 2018;100:1337–1353.e5.
Leavy O. TREM2 in Alzheimer disease. Nat Rev Immunol. 2015;15:201.
Mazaheri F, Snaidero N, Kleinberger G, Madore C, Daria A, Werner G, et al. TREM2 deficiency impairs chemotaxis and microglial responses to neuronal injury. EMBO Rep. 2017;18:1186–98.
Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368:107–16.
Cuyvers E, Bettens K, Philtjens S, Van Langenhove T, Gijselinck I, van der Zee J, et al. Investigating the role of rare heterozygous TREM2 variants in Alzheimer's disease and frontotemporal dementia. Neurobiol Aging. 2014;35:726.e11–9.
Cheng-Hathaway PJ, Reed-Geaghan EG, Jay TR, Casali BT, Bemiller SM, Puntambekar SS, et al. The Trem2 R47H variant confers loss-of-function-like phenotypes in Alzheimer’s disease. Mol Neurodegener. 2018;13:29.
Xiang X, Werner G, Bohrmann B, Liesz A, Mazaheri F, Capell A, et al. TREM2 deficiency reduces the efficacy of immunotherapeutic amyloid clearance. EMBO Mol Med. 2016;8:992–1004.
Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, et al. TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron. 2016;90:724–39.
Hickman SE, El Khoury J. TREM2 and the neuroimmunology of Alzheimer’s disease. Biochem Pharmacol. 2014;88:495–8.
Jiang T, Tan L, Zhu X-C, Zhou J-S, Cao L, Tan M-S, et al. Silencing of TREM2 exacerbates tau pathology, neurodegenerative changes, and spatial learning deficits in P301S tau transgenic mice. Neurobiol Aging. 2015;36:3176–86.
Fenn AM, Hall JCE, Gensel JC, Popovich PG, Godbout JP. IL-4 signaling drives a unique arginase+/IL-1 + microglia phenotype and recruits macrophages to the inflammatory CNS: consequences of age-related deficits in IL-4R after traumatic spinal cord injury. J Neurosci. 2014;34:8904–17.
Weekman EM, Sudduth TL, Abner EL, Popa GJ, Mendenhall MD, Brothers HM, et al. Transition from an M1 to a mixed neuroinflammatory phenotype increases amyloid deposition in APP/PS1 transgenic mice. J Neuroinflammation. 2014;11:127.
Wilcock DM. Neuroinflammatory phenotypes and their roles in Alzheimer’s disease. Neurodegener Dis. 2014;13:183–5.
Matcovitch-Natan O, Winter DR, Giladi A, Vargas Aguilar S, Spinrad A, Sarrazin S, et al. Microglia development follows a stepwise program to regulate brain homeostasis. Science. 2016;353:aad8670.
Friedman BA, Srinivasan K, Ayalon G, Meilandt WJ, Lin H, Huntley MA, et al. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep. 2018;22:832–47.
Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci. 2016;19:987–91.
Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13.
Li Q, Cheng Z, Zhou L, Darmanis S, Neff NF, Okamoto J, et al. Developmental heterogeneity of microglia and brain myeloid cells revealed by deep single-cell RNA sequencing. Neuron. 2019;101:207–223.e10.
Masuda T, Sankowski R, Staszewski O, Böttcher C, Amann L, Scheiwe C, et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. 2019;566:388.
Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, et al. Identification of a unique TGF-β-dependent molecular and functional signature in microglia. Nat Neurosci. 2014;17:131–43.
Bohlen CJ, Bennett FC, Tucker AF, Collins HY, Mulinyawe SB, Barres BA. Diverse requirements for microglial survival, specification, and function revealed by defined-medium cultures. Neuron. 2017;94:759–773.e8.
Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169:1276–1290.e17.
Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, El Fatimy R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47:566–581.e9.
Biber K, Neumann H, Inoue K, Boddeke HWGM. Neuronal ‘on’ and ‘off’ signals control microglia. Trends Neurosci. 2007;30:596–602.
Li MO, Wan YY, Sanjabi S, Robertson A-KL, Flavell RA. Transforming growth factor-β regulation of immune responses. Annu Rev Immunol. 2006;24:99–146.
Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med. 2015;212:287–95.
Streit WJ, Braak H, Xue Q-S, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer’s disease. Acta Neuropathol. 2009;118:475–85.
Fiala M, Liu QN, Sayre J, Pop V, Brahmandam V, Graves MC, et al. Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer’s disease brain and damage the blood-brain barrier. Eur J Clin Investig. 2002;32:360–71.
Town T, Tan J, Flavell RA, Mullan M. T-cells in Alzheimer’s disease. NeuroMolecular Med. 2005;7:255–64.
Merlini M, Kirabali T, Kulic L, Nitsch RM, Ferretti MT. Extravascular CD3+ T cells in brains of Alzheimer disease patients correlate with tau but not with amyloid pathology: an immunohistochemical study. Neurodegener Dis. 2018;18:49–56.
Malm TM, Koistinaho M, Pärepalo M, Vatanen T, Ooka A, Karlsson S, et al. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to β-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis. 2005;18:134–42.
Rivera-Escalera F, Matousek SB, Ghosh S, Olschowka JA, O’Banion MK. Interleukin-1β mediated amyloid plaque clearance is independent of CCR2 signaling in the APP/PS1 mouse model of Alzheimer’s disease. Neurobiol Dis. 2014;69:124–33.
Simard AR, Soulet D, Gowing G, Julien J-P, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron. 2006;49:489–502.
El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, et al. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–8.
Naert G, Rivest S. Hematopoietic CC-chemokine receptor 2 (CCR2) competent cells are protective for the cognitive impairments and amyloid pathology in a transgenic mouse model of Alzheimer’s disease. Mol Med. 2012;18:297–313.
Thériault P, ElAli A, Rivest S. The dynamics of monocytes and microglia in Alzheimer’s disease. Alzheimers Res Ther. 2015;7:41.
Lee JK, Jin HK, Endo S, Schuchman EH, Carter JE, Bae J. Intracerebral transplantation of bone marrow-derived mesenchymal stem cells reduces amyloid-beta deposition and rescues memory deficits in Alzheimer’s disease mice by modulation of immune responses. Stem Cells. 2010;28(2):329–43.
Ma T, Gong K, Ao Q, Yan Y, Song B, Huang H, et al. Intracerebral transplantation of adipose-derived mesenchymal stem cells alternatively activates microglia and ameliorates neuropathological deficits in Alzheimer’s disease mice. Cell Transplant. 2013;22:113–26.
Hawkes CA, McLaurin J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc Natl Acad Sci U S A. 2009;106:1261–6.
Itagaki S, McGeer PL, Akiyama H. Presence of T-cytotoxic suppressor and leucocyte common antigen positive cells in Alzheimer’s disease brain tissue. Neurosci Lett. 1988;91:259–64.
Togo T, Akiyama H, Iseki E, Kondo H, Ikeda K, Kato M, et al. Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J Neuroimmunol. 2002;124:83–92.
Lou J, Ythier A, Burger D, Zheng L, Juillard P, Lucas R, et al. Modulation of soluble and membrane-bound TNF-induced phenotypic and functional changes of human brain microvascular endothelial cells by recombinant TNF binding protein I. J Neuroimmunol. 1997;77:107–15.
Zhang W, Smith C, Howlett C, Stanimirovic D. Inflammatory activation of human brain endothelial cells by hypoxic astrocytes in vitro is mediated by IL-1β. J Cereb Blood Flow Metab. 2000;20:967–78.
Eisenhauer PB, Jacewicz MS, Conn KJ, Koul O, Wells JM, Fine RE, et al. Escherichia coli Shiga toxin 1 and TNF-α induce cytokine release by human cerebral microvascular endothelial cells. Microb Pathog. 2004;36:189–96.
Galanakis E, Di Cello F, Paul-Satyaseela M, Kim KS. Escherichia coli K1 induces IL-8 expression in human brain microvascular endothelial cells. Eur Cytokine Netw. 2006;17:260–5.
Liu YJ, Guo DW, Tian L, Shang DS, Zhao WD, Li B, et al. Peripheral T cells derived from Alzheimer’s disease patients overexpress CXCR2 contributing to its transendothelial migration, which is microglial TNF-α-dependent. Neurobiol Aging. 2010;31:175–88.
Fisher Y, Nemirovsky A, Baron R, Monsonego A. T cells specifically targeted to amyloid plaques enhance plaque clearance in a mouse model of Alzheimer’s disease. PLoS One. 2010;5:e10830.
Schetters STT, Gomez-Nicola D, Garcia-Vallejo JJ, Van Kooyk Y. Neuroinflammation: microglia and T cells get ready to tango. Front Immunol. 2017;8:1905.
Kipnis J, Cohen H, Cardon M, Ziv Y, Schwartz M. T cell deficiency leads to cognitive dysfunction: implications for therapeutic vaccination for schizophrenia and other psychiatric conditions. Proc Natl Acad Sci U S A. 2004;101:8180–5.
Marsh SE, Abud EM, Lakatos A, Karimzadeh A, Yeung ST, Davtyan H, et al. The adaptive immune system restrains Alzheimer’s disease pathogenesis by modulating microglial function. Proc Natl Acad Sci. 2016;113:E1316–25.
Späni C, Suter T, Derungs R, Ferretti MT, Welt T, Wirth F, et al. Reduced β-amyloid pathology in an APP transgenic mouse model of Alzheimer’s disease lacking functional B and T cells. Acta Neuropathol Commun. 2015;3:71.
Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity. 2011;35:161–8.
Laurent C, Dorothée G, Hunot S, Martin E, Monnet Y, Duchamp M, et al. Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain. 2017;140:184–200.
Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–57.
Orgogozo J-M, Gilman S, Dartigues J-F, Laurent B, Puel M, Kirby LC, et al. Subacute meningoencephalitis in a subset of patients with AD after Aβ42 immunization. Neurology. 2003;61:46–54.
McQuillan K, Lynch MA, Mills KHG. Activation of mixed glia by Aβ-specific Th1 and Th17 cells and its regulation by Th2 cells. Brain Behav Immun. 2010;24:598–607.
Mastrangelo MA, Sudol KL, Narrow WC, Bowers WJ. Interferon-{gamma} differentially affects Alzheimer’s disease pathologies and induces neurogenesis in triple transgenic-AD mice. Am J Pathol. 2009;175:2076–88.
Browne TC, McQuillan K, McManus RM, O’Reilly J-A, Mills KHG, Lynch MA. IFN-γ production by amyloid β-specific Th1 cells promotes microglial activation and increases plaque burden in a mouse model of Alzheimer’s disease. J Immunol. 2013;190:2241–51.
Monsonego A, Imitola J, Petrovic S, Zota V, Nemirovsky A, Baron R, et al. A beta-induced meningoencephalitis is IFN-gamma-dependent and is associated with T cell-dependent clearance of A beta in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2006;103:5048–53.
Town T, Vendrame M, Patel A, Poetter D, DelleDonne A, Mori T, et al. Reduced Th1 and enhanced Th2 immunity after immunization with Alzheimer’s β-amyloid 1-42. J Neuroimmunol. 2002;132:49–59.
Nikolic WV, Bai Y, Obregon D, Hou H, Mori T, Zeng J, et al. Transcutaneous beta-amyloid immunization reduces cerebral beta-amyloid deposits without T cell infiltration and microhemorrhage. Proc Natl Acad Sci U S A. 2007;104:2507–12.
Cao C, Arendash GW, Dickson A, Mamcarz MB, Lin X, Ethell DW. Abeta-specific Th2 cells provide cognitive and pathological benefits to Alzheimer’s mice without infiltrating the CNS. Neurobiol Dis. 2009;34:63–70.
Baruch K, Schwartz M. CNS-specific T cells shape brain function via the choroid plexus. Brain Behav Immun. 2013;34:11–6.
Henkel AW, Dittrich PS, Groemer TW, Lemke EA, Klingauf J, Klafki HW, et al. Immune complexes of auto-antibodies against Aβ1-42 peptides patrol cerebrospinal fluid of non-Alzheimer’s patients. Mol Psychiatry. 2007;12:601–10.
Nicoll JAR, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide: a case report. Nat Med. 2003;9:448–52.
Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;73:2061–70.
Siemers ER, Friedrich S, Dean RA, Gonzales CR, Farlow MR, Paul SM, et al. Safety and changes in plasma and cerebrospinal fluid amyloid β after a single administration of an amyloid β monoclonal antibody in subjects sith Alzheimer disease. Clin Neuropharmacol. 33:–67.
van Dyck CH. Anti-amyloid-β monoclonal antibodies for Alzheimer’s disease: pitfalls and promise. Biol Psychiatry. 2018;83:311–9.
Giacobini E, Gold G. Alzheimer disease therapy—moving from amyloid-β to tau. Nat Rev Neurol. 2013;9:677–86.
Castello MA, Jeppson JD, Soriano S. Moving beyond anti-amyloid therapy for the prevention and treatment of Alzheimer’s disease. BMC Neurol. 2014;14:169.
Sigurdsson EM. Tau-focused kmmunotherapy for Alzheimer’s disease and related tauopathies. Curr Alzheimer Res. 2009;6(5):446–50.
Boimel M, Grigoriadis N, Lourbopoulos A, Haber E, Abramsky O, Rosenmann H. Efficacy and safety of immunization with phosphorylated tau against neurofibrillary tangles in mice. Exp Neurol. 2010;224:472–85.
Yanamandra K, Kfoury N, Jiang H, Mahan TE, Ma S, Maloney SE, et al. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron. 2013;80:402–14.
West T, Hu Y, Verghese PB, Bateman RJ, Braunstein JB, Fogelman I, et al. Preclinical and clinical development of ABBV-8E12, a humanized anti-tau antibody, for treatment of Alzheimer’s disease and other tauopathies. J Prev Alzheimer’s Dis. 2017;4:236–41.
Butovsky O, Koronyo-Hamaoui M, Kunis G, Ophir E, Landa G, Cohen H, et al. Glatiramer acetate fights against Alzheimer’s disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proc Natl Acad Sci U S A. 2006;103:11784–9.
Baruch K, Rosenzweig N, Kertser A, Deczkowska A, Sharif AM, Spinrad A, et al. Breaking immune tolerance by targeting Foxp3+ regulatory T cells mitigates Alzheimer’s disease pathology. Nat Commun. 2015;6:7967.
Koronyo Y, Salumbides BC, Sheyn J, Pelissier L, Li S, Ljubimov V, et al. Therapeutic effects of glatiramer acetate and grafted CD115+ monocytes in a mouse model of Alzheimer’s disease. Brain. 2015;138:2399–422.
Chen M, Gran B, Costello K, Johnson K, Martin R, Dhib-Jalbut S. Glatiramer acetate induces a Th2-biased response and crossreactivity with myelin basic protein in patients with MS. Mult Scler J. 2001;7:209–19.
Aharoni R, Meshorer A, Sela M, Arnon R. Oral treatment of mice with copolymer 1 (glatiramer acetate) results in the accumulation of specific Th2 cells in the central nervous system. J Neuroimmunol. 2002;126:58–68.
Vieira PL, Heystek HC, Wormmeester J, Wierenga EA, Kapsenberg ML. Glatiramer acetate (copolymer-1, copaxone) promotes Th2 cell development and increased IL-10 production through modulation of dendritic cells. J Immunol. 2003;170:4483–8.
Ziemssen T, Schrempf W. Glatiramer acetate: mechanisms of action in multiple sclerosis. Int Rev Neurobiol. 2007;79:537–70.
Chen M, Valenzuela RM, Dhib-Jalbut S. Glatiramer acetate-reactive T cells produce brain-derived neurotrophic factor. J Neurol Sci. 2003;215:37–44.
Reick C, Ellrichmann G, Tsai T, Lee D-H, Wiese S, Gold R, et al. Expression of brain-derived neurotrophic factor in astrocytes - beneficial effects of glatiramer acetate in the R6/2 and YAC128 mouse models of Huntington’s disease. Exp Neurol. 2016;285:12–23.
Corey-Bloom J, Jia H, Aikin AM, Thomas EA. Disease modifying potential of glatiramer acetate in Huntington’s disease. J Huntingtons Dis. 2014;3:311–6.
Wilson EH, Weninger W, Hunter CA. Trafficking of immune cells in the central nervous system. J Clin Invest. 2010;120:1368–79.
Baruch K, Deczkowska A, Rosenzweig N, Tsitsou-Kampeli A, Sharif AM, Matcovitch-Natan O, et al. PD-1 immune checkpoint blockade reduces pathology and improves memory in mouse models of Alzheimer’s disease. Nat Med. 2016;22:135–7.
Holmes C, El-Okl M, Williams AL, Cunningham C, Wilcockson D, Perry VH. Systemic infection, interleukin 1beta, and cognitive decline in Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2003;74:788–9.
Yaffe K, Kanaya A, Lindquist K, Simonsick EM, Harris T, Shorr RI, et al. The metabolic syndrome, inflammation, and risk of cognitive decline. JAMA. 2004;292:2237.
Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S, et al. Systemic inflammation and disease progression in Alzheimer disease. Neurology. 2009;73:768–74.
Eikelenboom P, Hoozemans JJ, Veerhuis R, van Exel E, Rozemuller AJ, van Gool WA. Whether, when and how chronic inflammation increases the risk of developing late-onset Alzheimer’s disease. Alzheimers Res Ther. 2012;4:15.
Kyrkanides S, Tallents RH, Miller J-NH, Olschowka ME, Johnson R, Yang M, et al. Osteoarthritis accelerates and exacerbates Alzheimer’s disease pathology in mice. J Neuroinflammation. 2011;8:112.
McManus RM, Higgins SC, Mills KHG, Lynch MA. Respiratory infection promotes T cell infiltration and amyloid-β deposition in APP/PS1 mice. Neurobiol Aging. 2014;35:109–21.
Sheng JG, Bora SH, Xu G, Borchelt DR, Price DL, Koliatsos VE. Lipopolysaccharide-induced-neuroinflammation increases intracellular accumulation of amyloid precursor protein and amyloid β peptide in APPswe transgenic mice. Neurobiol Dis. 2003;14:133–45.
Quinn J, Montine T, Morrow J, Woodward WR, Kulhanek D, Eckenstein F. Inflammation and cerebral amyloidosis are disconnected in an animal model of Alzheimer’s disease. J Neuroimmunol. 2003;137:32–41.
Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong J-S, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–62.
Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB, Oh KW, et al. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation. 2008;5:37.
Jaeger LB, Dohgu S, Sultana R, Lynch JL, Owen JB, Erickson MA, et al. Lipopolysaccharide alters the blood-brain barrier transport of amyloid β protein: a mechanism for inflammation in the progression of Alzheimer’s disease. Brain Behav Immun. 2009;23:507–17.
Carret-Rebillat A-S, Pace C, Gourmaud S, Ravasi L, Montagne-Stora S, Longueville S, et al. Neuroinflammation and Aβ accumulation linked to systemic inflammation are decreased by genetic PKR down-regulation. Sci Rep. 2015;5:8489.
Wendeln A-C, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. 2018;556:332–8.
Erickson MA, Hartvigson PE, Morofuji Y, Owen JB, Butterfield DA, Banks WA. Lipopolysaccharide impairs amyloid β efflux from brain: altered vascular sequestration, cerebrospinal fluid reabsorption, peripheral clearance and transporter function at the blood-brain barrier. J Neuroinflammation. 2012;9:150.
Erickson MA, Banks WA. Cytokine and chemokine responses in serum and brain after single and repeated injections of lipopolysaccharide: multiplex quantification with path analysis. Brain Behav Immun. 2011;25:1637–48.
McAlpine FE, Lee J-K, Harms AS, Ruhn KA, Blurton-Jones M, Hong J, et al. Inhibition of soluble TNF signaling in a mouse model of Alzheimer’s disease prevents pre-plaque amyloid-associated neuropathology. Neurobiol Dis. 2009;34:163–77.
MacPherson KP, Sompol P, Kannarkat GT, Chang J, Sniffen L, Wildner ME, et al. Peripheral administration of the soluble TNF inhibitor XPro1595 modifies brain immune cell profiles, decreases beta-amyloid plaque load, and rescues impaired long-term potentiation in 5xFAD mice. Neurobiol Dis. 2017;102:81–95.
Chang R, Yee K-L, Sumbria RK. Tumor necrosis factor α inhibition for Alzheimer’s disease. J Cent Nerv Syst Dis. 2017;9:1179573517709278.
Gabbita SP, Srivastava MK, Eslami P, Johnson MF, Kobritz NK, Tweedie D, et al. Early intervention with a small molecule inhibitor for tumor nefosis factor-α prevents cognitive deficits in a triple transgenic mouse model of Alzheimer’s disease. J Neuroinflammation. 2012;9:578.
Tweedie D, Ferguson RA, Fishman K, Frankola KA, Van Praag H, Holloway HW, et al. Tumor necrosis factor-α synthesis inhibitor 3,6′-dithiothalidomide attenuates markers of inflammation. Alzheimer pathology and behavioral deficits in animal models of neuroinflammation and Alzheimer’s disease J Neuroinflammation. 2012;9:575.
Gabbita SP, Johnson MF, Kobritz N, Eslami P, Poteshkina A, Varadarajan S, et al. Oral TNFα modulation alters neutrophil infiltration, improves cognition and diminishes tau and amyloid pathology in the 3xTgAD mouse model. PLoS One. 2015;10:e0137305.
Kübra Elçioğlu H, Kabasakal L, Tufan F, Elçioğlu ÖH, Solakoglu S, Kotil T, et al. Effects of systemic Thalidomide and intracerebroventricular Etanercept and Infliximab administration in a Streptozotocin induced dementia model in rats. Acta Histochem. 2015;117:176–81.
Tobinick EL, Gross H. Rapid cognitive improvement in Alzheimer’s disease following perispinal etanercept administration. J Neuroinflammation. 2008;5:2.
Butchart J, Brook L, Hopkins V, Teeling J, Püntener U, Culliford D, et al. Etanercept in Alzheimer disease: a randomized, placebo-controlled, double-blind, phase 2 trial. Neurology. 2015;84:2161–8.
vom Berg J, Prokop S, Miller KR, Obst J, Kälin RE, Lopategui-Cabezas I, et al. Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s disease–like pathology and cognitive decline. Nat Med. 2012;18:1812–9.
Molofsky AB, Savage AK, Locksley RM. Interleukin-33 in tissue homeostasis, injury, and inflammation. Immunity. 2015;42:1005–19.
Chapuis J, Hot D, Hansmannel F, Kerdraon O, Ferreira S, Hubans C, et al. Transcriptomic and genetic studies identify IL-33 as a candidate gene for Alzheimer’s disease. Mol Psychiatry. 2009;14:1004–16.
Fu AKY, Hung K-W, Yuen MYF, Zhou X, Mak DSY, Chan ICW, et al. IL-33 ameliorates Alzheimer’s disease-like pathology and cognitive decline. Proc Natl Acad Sci U S A. 2016;113:E2705–13.
Kakkar R, Lee RT. The IL-33/ST2 pathway: therapeutic target and novel biomarker. Nat Rev Drug Discov. 2008;7:827–40.
Carlock C, Wu J, Shim J, Moreno-Gonzalez I, Pitcher MR, Hicks J, et al. Interleukin33 deficiency causes tau abnormality and neurodegeneration with Alzheimer-like symptoms in aged mice. Transl Psychiatry. 2017;7:e1164.
Scholtzova H, Kascsak RJ, Bates KA, Boutajangout A, Kerr DJ, Meeker HC, et al. Induction of toll-like receptor 9 signaling as a method for ameliorating Alzheimer’s disease-related pathology. J Neurosci. 2009;29:1846–54.
Scholtzova H, Chianchiano P, Pan J, Sun Y, Goñi F, Mehta PD, et al. Amyloid β and tau Alzheimer’s disease related pathology is reduced by Toll-like receptor 9 stimulation. Acta Neuropathol Commun. 2014;2:101.
Scholtzova H, Do E, Dhakal S, Sun Y, Liu S, Mehta PD, et al. Innate immunity stimulation via Toll-like receptor 9 ameliorates vascular amyloid pathology in Tg-SwDI mice with associated cognitive benefits. J Neurosci. 2017;37:936–59.
Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–28.
Cummings J, Lee G, Ritter A, Zhong K. Alzheimer’s disease drug development pipeline: 2018. Alzheimer’s Dement (N Y). 2018;4:195–214.
Breitner JCS, Welsh KA, Helms MJ, Gaskell PC, Gau BA, Roses AD, et al. Delayed onset of Alzheimer’s disease with nonsteroidal anti-inflammatory and histamine H2 blocking drugs. Neurobiol Aging. 1995;16:523–30.
In’t Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, et al. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N Engl J Med. 2001;345:1515–21.
Etminan M, Gill S, Samii A. Effect of non-steroidal anti-inflammatory drugs on risk of Alzheimer’s disease: systematic review and meta-analysis of observational studies. BMJ. 2003;327:128.
Townsend KP, Praticò D. Novel therapeutic opportunities for Alzheimer’s disease: focus on nonsteroidal anti-inflammatory drugs. FASEB J. 2005;19:1592–601.
Imbimbo BP, Solfrizzi V, Panza F. Are NSAIDs useful to treat Alzheimer’s disease or mild cognitive impairment? Front Aging Neurosci. 2010;2.
ADAPT Research Group AR, Martin BK, Szekely C, Brandt J, Piantadosi S, JCS B, et al. Cognitive function over time in the Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT): results of a randomized, controlled trial of naproxen and celecoxib. Arch Neurol. 2008;65:896–905.
Alzheimer’s Disease Anti-inflammatory Prevention Trial Research Group TADAPTR. Results of a follow-up study to the randomized Alzheimer’s Disease Anti-inflammatory Prevention Trial (ADAPT). Alzheimers Dement. 2013;9:714–23.
Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, et al. Effects of Rofecoxib or Naproxen vs Placebo on Alzheimer disease progression. JAMA. 2003;289:2819.
Soininen H, West C, Robbins J, Niculescu L. Long-term efficacy and safety of celecoxib in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2007;23:8–21.
Landreth GE, Heneka MT. Anti-inflammatory actions of peroxisome proliferator-activated receptor gamma agonists in Alzheimer’s disease. Neurobiol Aging. 2001;22:937–44.
Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, et al. A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature. 2001;414:212–6.
Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C, et al. Acute treatment with the PPARγ agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ1–42 levels in APPV717I transgenic mice. Brain. 2005;128:1442–53.
Budur K, Welsh-Bohmer K, Burns D, Chiang C, O’Neil J, Runyan G, et al. A pharmacogenetics-supported clinical trial to delay onset of mild cognitive impairment due to Alzheimer's disease using low-dose pioglitazone: an update on the TOMMORROW study. Alzheimers Dement. 2014;10:P809–10.
Acknowledgements
Not applicable
Funding
DADS is a student in the University of Rochester Medical Scientist Training Program (T32 GM007536) and is currently supported by The University of Rochester Toxicology Training Grant (T32 ES7026) MKO and JAO receive research support from the National Institutes of Health (R01 AG30149) and the National Aeronautics and Space Administration (NNX16AE07G).
Availability of data and materials
Not applicable
Author information
Authors and Affiliations
Contributions
DADS researched the literature and drafted the manuscript. JAO and MKO critically reviewed and edited the work. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Dionisio-Santos, D.A., Olschowka, J.A. & O’Banion, M.K. Exploiting microglial and peripheral immune cell crosstalk to treat Alzheimer’s disease. J Neuroinflammation 16, 74 (2019). https://doi.org/10.1186/s12974-019-1453-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-019-1453-0