
Abstract
Background
Tuberculosis (TB) affects one third of the global population, and TB of the central nervous system (CNS-TB) is the most severe form of tuberculosis which often associates with high mortality. The pro-inflammatory cytokine tumour necrosis factor (TNF) plays a critical role in the initial and long-term host immune protection against Mycobacterium tuberculosis (M. tuberculosis) which involves the activation of innate immune cells and structure maintenance of granulomas. However, the contribution of TNF, in particular neuron-derived TNF, in the control of cerebral M. tuberculosis infection and its protective immune responses in the CNS were not clear.
Methods
We generated neuron-specific TNF-deficient (NsTNF−/−) mice and compared outcomes of disease against TNFf/f control and global TNF−/− mice. Mycobacterial burden in brains, lungs and spleens were compared, and cerebral pathology and cellular contributions analysed by microscopy and flow cytometry after M. tuberculosis infection. Activation of innate immune cells was measured by flow cytometry and cell function assessed by cytokine and chemokine quantification using enzyme-linked immunosorbent assay (ELISA).
Results
Intracerebral M. tuberculosis infection of TNF−/− mice rendered animals highly susceptible, accompanied by uncontrolled bacilli replication and eventual mortality. In contrast, NsTNF−/− mice were resistant to infection and presented with a phenotype similar to that in TNFf/f control mice. Impaired immunity in TNF−/− mice was associated with altered cytokine and chemokine synthesis in the brain and characterised by a reduced number of activated innate immune cells. Brain pathology reflected enhanced inflammation dominated by neutrophil influx.
Conclusion
Our data show that neuron-derived TNF has a limited role in immune responses, but overall TNF production is necessary for protective immunity against CNS-TB.
Similar content being viewed by others


Background
Tuberculosis of the central nervous system (CNS-TB) is associated with significant mortality and high distressing levels of neurological morbidity, the majority of survivors suffering permanent neurologic complications [1–3]. It disproportionately affects children, especially in the developing world and immunocompromised individuals despite the availability of anti-tuberculosis treatment [3–5]. The lengthy duration of therapy for CNS-TB makes adherence difficult. In Sub-Saharan African and some Asian countries, tuberculosis is now one of the most common forms of bacterial meningitis due to the effects of HIV [6–8].
CNS-TB results from a rupture of subpial or subependymal foci which has been deposited earlier following lympho-haematogenous dissemination of Mycobacterium tuberculosis from the primary pulmonary infection, or rupture of an adjacent parenchymal focus [1, 9]. It is widely accepted that bacilli reach the CNS by a haematogenous route secondary to disease elsewhere in the body. Since the brain parenchyma and the meninges are physiologically and anatomically protected from the systemic circulation system by the blood–brain barrier (BBB), the mechanisms by which the bacilli initially invade this barrier are not clear. It is believed that M. tuberculosis can cross the BBB as a free organism or via infected peripheral myeloid cells [10]. However, the latter hypothesis seems controversial, as myeloid cellular traffic is severely restricted into the CNS prior to invasion by M. tuberculosis [11], however lymphoid cells in physiological state can bypass the BBB by entering the subarachnoid space via the meningeal veins or the choroid plexus [12, 13].
Tumour necrosis factor (TNF) has been shown to be critical in the pathogenesis of M. tuberculosis in the central nervous system [14–16]. TNF is synthesised by several cell types of haematopoietic origin, such as microglia/macrophages, neutrophils, dendritic cells and lymphocytes, and non-haematopoietic origin such as astrocytes and neurons [17–20]. The production of TNF in the CNS alters the BBB permeability and CSF leukocytosis in experimental bacterial meningitis [21, 22] and has been implicated in fostering the progression of CNS-TB in a rabbit model [16]. However, Tobin et al. [15] have shown using human and zebrafish models that either state, TNF deficiency or excessive TNF production, causes macrophage lysis, therefore placing M. tuberculosis in a permissive extracellular niche [15, 23, 24]. Within this context, it is important to understand the role of TNF in host immunity during M. tuberculosis infection in the CNS. Here, we generated neuron-specific TNF-deficient (NsTNF−/−) mice and investigated outcomes after intracerebral M. tuberculosis infection in comparative studies with TNFf/f control and global TNF−/− mice.
Methods
Mice
All mouse strains, TNFf/f (TNF-floxed wild-type [25]), Syn1-Cre (synapsin I promoter-driven Cre-transgenic, obtained from the Jackson Laboratory, http://jaxmice.jax.org/strain/003966.html), NsTNF−/− (neuron-derived TNF-deficient) and TNF−/− (TNF-deficient [25]) mice, were bred and maintained under specific pathogen-free conditions at the animal facility of the University of Cape Town (South Africa). Adult mice between 8 and 12 weeks of age were used, and infected mice were maintained under biosafety level 3 conditions. All animal procedures were approved by the Animal Research Ethics Committee (AEC reference number: 010/018), University of Cape Town, in accordance with the South African National Standard.
Genotyping PCR analysis
Genotyping of mouse strains was performed by PCR analysis of DNA extracted from tail biopsies. Genetic analysis of TNFf/f mice was previously described [25]. Syn1-Cre and NsTNF−/− mouse were genotyped for Cre expression using the primer sets IMR1084 (5′ GCG GTC TGG CAG TAA AAA CTA TC 3′) and IMR1085 (5′ GTG AAA CAG CAT TGC TGT CAC TT 3′), and the internal controls IMR7338 (5′ CTA GGC CAC AGA ATT GAA AGA TCT 3′) and IMR7339 (5′ GTA GGT GGA AAT TCT AGC ATC ATC C 3′). For the presence of the TNF loxP gene in NsTNF−/− mice, primers KO41 (5′ TGA GTC TGT CTT AAC TAA CC 3′) and KO42 (5′ CCC TTC ATT CTC AAG GCA CA 3′) were used [25].
Aerosol infection and immunohistochemistry of brain tissue
M. tuberculosis with green fluorescent expressing protein (M. tuberculosis H37Rv-GFP, provided by Joel Ernst, New York University School of Medicine, USA) was grown in Middlebrook 7H9 medium (Difco™ Laboratories) containing 25 μg/ml kanamycin and 0.5 % glycerol and enriched with 10 % OADC. Bacterial cultures were incubated at 37 °C and grown until log phase, aliquoted and stored at −80 °C. A frozen aliquot of M. tuberculosis was thawed, passed 30 times through a 29-G needle and diluted in sterile saline. Mice were infected by aerosol inhalation at a dose of 200–500 colony-forming units (CFUs)/lung under biosafety level 3 conditions using a Glas-Col Inhalation Exposure System Model A4224. For analysis, infected mice were deeply anaesthetised and transcardially perfused with 4 % paraformaldehyde. Brains were sectioned at 40 μm using a vibratome and processed for immunohistochemical staining. Neurons were labelled with polyclonal anti-MAP2 (1 μg/ml, Abcam) or anti-β-III-tubulin antibody (1 μg/ml, Abcam), and anti-rabbit Cy3-conjugated secondary antibody (1.5 μg/ml, Jackson ImmunoResearch Laboratories). The microglia/monocytes were labelled with monoclonal anti-CD11b antibody (Clone M1/70, 1 μg/ml, Abcam) and anti-rat Cy3-conjugated secondary antibodies (1.5 μg/ml, Jackson ImmunoResearch Laboratories). The sections were incubated with nuclear marker DAPI (Sigma) and then mounted in fluorescent mounting medium and images captured using a Zeiss 510LSM unit.
Intracerebral infection and clinical scoring
M. tuberculosis strain H37Rv was grown at 37 °C in Middlebrook 7H9 broth containing 10 % OADC and 0.5 % Tween 80 until log phase, then aliquoted and stored at −80 °C. A frozen aliquot was thawed, passed 30 times through a 29-G needle and diluted in sterile saline. Intracerebral infection was performed using a stereotaxic approach of directly injecting M. tuberculosis H37Rv into the cerebral cortex. Prior to inoculation, a small burr hole was constructed anterior to the bregma and to the left of the midline in the skull exposing the dura mater. Five mice per strain were inoculated intracerebrally with 1 × 103–1 × 104 CFUs of M. tuberculosis H37Rv using a Hamilton syringe (Gastight no. 1701, Switzerland). The burr hole was sealed with bone wax and the skin sutured. Animals received a prophylactic pain killer for 3 days at 24-h intervals. The clinical scoring system was adapted from previous reports [26, 27]. Here, mice were weighed and scored daily for neurologic manifestations during the course of infection as follows: normal (no detectable signs) = 0; head tilt = 1; motility or decrease activity = 2; behaviour depression = 3; and moribund state = 4. Organs of infected mice were harvested and processed at 1, 2, 3 and 15 weeks post-infection. Haematoxylin and eosin (H&E)-stained brain sections at 3 weeks post-infection were reviewed and analysed by a pathologist who was blinded to the study.
Colony enumeration assay
Bacterial burdens in the brains, lungs and spleens of infected mice were determined at specific time points after infection with M. tuberculosis. Organs were weighed and homogenised in 0.04 % Tween 80 saline. Tenfold serial dilutions of organ homogenates were plated in duplicates on Middlebrook 7H10 (Becton, Dickinson and Company) agar plates containing 10 % OADC (Life Technologies, Gaithersburg, MD) and were incubated at 37 °C for 19–21 days. The concentration of M. tuberculosis was then determined by counting the CFUs.
Flow cytometry
To determine the expression level of TNF by neurons, TNFf/f, NsTNF−/− and TNF−/− mice were intracerebrally stimulated with 5 μg/ml of lipopolysaccharide (LPS) for 90 min. Mice were transcardially perfused with 4 % paraformaldehyde. For single-cell suspensions, brains were isolated and tissue passed through a 70-μm nylon cell strainer (Beckton and Dickinson) and washed 2× with phosphate-buffered saline (PBS) and the cell concentration was determined by counting in the presence of trypan blue. TNF expression in neurons was measured by intracellular staining through the addition of saponin buffer to permeabilise the cells, which were then labelled with polyclonal anti-β-III-tubulin antibody (20 μg/ml, Abcam) and an anti-rabbit PE-conjugated secondary antibody (1.5 μg/ml, Jackson ImmunoResearch Laboratories), and anti-TNF antibody (TNF:Alexa 647, Clone MP6-XT22, BD Pharmingen™).
To analyse surface marker expression in microglia, macrophages and dendritic cells, the following antibodies were used: CD11b:PerCP-Cy5-5 (Clone M1/70, BD Pharmingen™ [2 μg/ml]); CD11c:Alexa 700 (Clone HL3, BD Pharmingen™ [2.5 μg/ml]); CD45:APC (Clone 30-F11, BD Pharmingen™ [2 μg/ml]); CD80:FITC (16-10A1, BD Pharmingen™ [4 μg/ml]); CD86:V450 (Clone GL1, BD Horizon™ [4 μg/ml]) and MHCII/(I-A/I-E):PE (M5/114.15.2, BD Pharmingen™ [2 μg/ml]). Cells were washed with PBS/0.1 % bovine serum albumin (BSA)/0.01 % NaN3 and incubated with the appropriate antibodies for 20 min in the dark. Excess antibodies were removed by washing cells 2× with PBS/0.1 % BSA/0.01 % NaN3. Pelleted cells were fixed for 18–24 h in 2 % phosphate buffered formaldehyde and analysed on a FACSCalibur (Beckton Dickinson) flow cytometer using CellQuest software. Microglia were defined as CD11b+CD45low, macrophages as CD11b+CD45high and dendritic cells as CD11c+CD45high as previously described [28, 29].
Quantification of chemokines and cytokines
Supernatants from brain homogenates were prepared for cytokine and chemokine measurement by enzyme-linked immunosorbent assay (ELISA) after 3 weeks subsequent to intracerebral M. tuberculosis infection. The chemokines MCP-1, MIP-1α and RANTES, and the cytokines IL-1β, IL-12p70 and TNF (R&D Systems, Germany) were measured using commercially available ELISA reagents according to the manufacturer’s instructions. Chemokine and cytokine concentrations were measured by absorbance using a Versamax Microplate Reader (Molecular Devices, LLC, CA) with SoftMax software.
Statistical analysis
The data are presented as the mean ± standard error of the mean (SD). Statistical analysis was performed by one-way ANOVA and one-tailed t test for comparisons amongst the time points. For all tests, a p value of ≤0.05 was considered significant.
Results
TNF regulates M. tuberculosis dissemination into the brain but infection of neurons is TNF independent
TNF plays an important role in host immunity against M. tuberculosis dissemination and latent infection [30–32]. A key function of TNF in granuloma formation and maintenance has been described in models of TNF-gene deficiency or neutralisation [16, 33, 34], supported by clinical evidence observed in the rheumatoid arthritis patients who presented with TB reactivation after anti-TNF therapy [5]. Moreover, inhibition of TNF leads to M. tuberculosis dissemination which causes extrapulmonary TB including severe CNS-TB [35, 36]. To investigate whether TNF protects against M. tuberculosis dissemination to the CNS, we challenged wild-type (TNFf/f) and TNF−/− mice by aerosol inhalation with M. tuberculosis H37Rv-GFP. In contrast to TNFf/f mice, TNF−/− mice were unable to control pulmonary TB infection, which resulted in high bacilli burdens in the lungs and spleens (Fig. 1a), similar to previous reports [37, 38]. Further, we confirmed dissemination of M. tuberculosis bacilli from the lungs to the brains in the absence of TNF; however, bacilli in the brains of TNFf/f mice were not detectable by culture (Fig. 1a). The latter observation is supported by previous studies of Hernandez Pando et al. [39], who showed limited, if any, detectable bacilli in the brains of wild-type mice using the same M. tuberculosis H37Rv laboratory strain.

Dissemination of M. tuberculosis to the brain in TNF−/− mice. a TNFf/f and TNF−/− mice were infected by aerosol inhalation at a dose of 200–500 CFUs/lung of M. tuberculosis, and mycobacterial burden in brains, lungs and spleens was determined at day 33 post-infection. b Foci of disseminated fluorescent bacilli (green) surrounded by CD11b+ microglia/macrophages (red) were present in the TNF−/− brain parenchyma. c, d The fluorescent image of intracellular M. tuberculosis GFP-expressing bacilli (green) found in the neurons (red) of TNF−/− mouse, labelled with c MAP2 and d β-III-tubulin. The orthogonal projection of confocal Z-stacks confirmed the cytosolic location of the bacilli in the x–y plane. All nuclei were labelled with DAPI (blue in b–d). Scale bars: b 20 μm; c, d 5 μm. The data represents two independent experiments
Pathogenesis of CNS-TB is thought to involve haematogenous spread and phagocytosis of bacilli by microglia which generates a robust immune response [1, 40]. TNF, as a pro-inflammatory cytokine mainly produced by microglia and astrocytes, has been proposed as the main cause of inflammation and neuropathology [2, 40], while others have suggested a neuroprotective role [41]. We have previously demonstrated that neurons can serve as host cells for M. tuberculosis [42]. Therefore, to determine if neuron infection is TNF dependent, we investigated whether dissemination of M. tuberculosis to the CNS of TNF−/− mice resulted in bacilli infection of neurons. Infected brain tissue sections presented predominantly with bacilli foci enriched with CD11b+ microglia/macrophages (Fig. 1b), most of which were intracellular. Significantly, we observed M. tuberculosis association with neurons. Three-dimensional orthogonal views of confocal images revealed that the green-expressing bacilli were embedded in the MAP2+ (Fig. 1c) or β-tubulin+ (Fig. 1d) neuronal cytoplasmic structures, similar to previously described observations in intracranially infected C57Bl/6 mice [42]. The data presented here together with previously published data [37] demonstrates an important role for TNF to regulate pulmonary immune responses against M. tuberculosis infection. Failure to control M. tuberculosis replication results in bacilli dissemination and subsequent infection of peripheral organs including the central nervous system where infection of neurons can occur independent of TNF.
Generation and characterisation of NsTNF−/− mice
Although our observations suggested TNF-independent neuronal invasion in the CNS, it was reported that TNF production, specifically by neurons, has an impact on disease outcome [43]. To investigate the role of neuron-derived TNF in CNS-TB immunity, we generated a mouse model of neuron-specific TNF deficiency (NsTNF−/−) through mating of Syn1-Cre mice [44] and TNF-floxed mice (TNFf/f) [25, 37]. We confirmed correct genotypes of the heterozygous Syn1-CreCre/wtTNFf/wt F1 offspring by PCR analysis. Typically, Syn1-CreCre/wtTNFf/wt mice displayed the 100-bp amplification product representative of the Cre gene (bottom panel, Fig. 2a) while both the TNF-floxed and normal TNF genes were present, represented by 400- and 350-bp amplification products, respectively (top panel, Fig. 2a). Syn1-CreCre/wtTNFf/wt F1 offspring was subsequently bred with TNFf/f mice to generate the homozygote neuronal cell type-specific TNF mice (Syn1-CreCre/wt TNFf/f), and offspring were analysed by tail biopsy analysis. The F2-generation Syn1-CreCre/wt TNFf/f mice displayed the 400-bp amplification fragment representative of the TNF-floxed gene while the normal wild-type TNF gene was absent (top panel, Fig. 2a), and the presence of a 100-bp amplification product specified the presence of the Cre gene. The data therefore confirmed the genotype of the new strain which was termed NsTNF−/− mice (bottom panel, Fig. 2a).

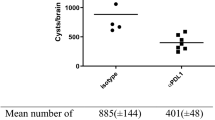
Analysis of NsTNF−/− mice. a Deletion of the neuron-specific TNF gene in NsTNF−/− mice was confirmed by PCR analysis. b TNF expression in neurons from TNFf/f, NsTNF−/− and TNF−/− mice was analysed by flow cytometry after LPS stimulation. c Mean fluorescence intensities of TNF expression in TNFf/f (green), NsTNF−/− (blue) and TNF−/− mice (red) are presented
We next assessed neuron-specific ablation of TNF. TNFf/f, TNF−/− and NsTNF−/− mice were inoculated with 3 μl LPS (5 μg/ml) in the left brain hemisphere by intracerebral inoculation. Single-cell suspensions, generated from harvested brains, were labelled and analysed for neurons expressing TNF. We confirmed expression of TNF in neurons of TNFf/f mice and the absence of TNF in both TNF−/− and NsTNF−/− mice through comparative TNF mean fluorescence intensity analysis which were both negligible in TNF−/− and NsTNF−/− mice (Fig. 2b), indicating almost complete neuron-specific ablation while the MFI for TNF was approximately 100-fold higher in TNFf/f mice (Fig. 2c). Thus, the successful generation of novel NsTNF−/− mice which presented with neuron-specific TNF ablation was confirmed by genetic and flow cytometric analysis.
TNF−/− mice are highly susceptible to cerebral M. tuberculosis infection while NsTNF−/− mice are resistant
Contention exists on the role of TNF in host protection against M. tuberculosis infection of the brain. Current literature argues that M. tuberculosis persistence is encouraged in the presence of TNF [45] whereas, in pulmonary infection, it is required to control bacterial replication [34, 38, 46]. Also, TNF was shown to be critical for protection against other central nervous system diseases [17, 47]. To resolve the apparent inconsistency in the role of TNF, particularly to address whether TNF contributes to immune protection against M. tuberculosis of the central nervous system, and to evaluate neurons as a potential cellular source of TNF in the cerebral immune response, TNFf/f, NsTNF−/− and TNF−/− mice were challenged with M. tuberculosis. Disease progression was clinically scored, and mortality and changes in body weight were recorded as indications of disease severity (Fig. 3). In contrast to TNFf/f mice which survived infection for 15 weeks, TNF−/− mice were highly susceptible and rapidly succumbed to infection within 3 weeks (Fig. 3b). Initial post-surgery body weight loss and recovery were observed between day 1 and day 5 after M. tuberculosis infection as previously reported [48, 49]. Susceptibility was accompanied by rapid loss of >20 % of the original body weight during the last week of illness as opposed to TNFf/f mice which displayed a steady increase in body weight during the same observation period (Fig. 3a). Similar to TNFf/f mice, NsTNF−/− mice were resistant and survived the duration of infection without any measured body weight loss. Brain, lung and spleen weights were measured as representative of organ inflammation during disease development. We observed enhanced brain inflammation in all three strains until 2 weeks post-infection, after which inflammation was sustained in TNFf/f and NsTNF−/− mice but significantly decreased (p < 0.05) in TNF−/− mice (Fig. 3a). Neurologic symptoms such as head tilt and anorexia were absent in both TNFf/f and NsTNF−/− mouse strains, contrasting the severe clinical neurologic symptoms prominent (p < 0.01) within 16–21 days post-infection in TNF−/− mice (Fig. 3d). It was therefore concluded that TNF was indispensable to generate protective immunity against M. tuberculosis infection of the central nervous system and unequivocally established the non-redundancy of TNF as a critical mediator of immune protection against CNS-TB. Nonetheless, while TNF was required for protection, neuron-specific TNF was functionally redundant.

TNF−/− mice are highly susceptible to CNS-TB but NsTNF−/− mice are resistant. TNFf/f (black circle), NsTNF−/− (triangle), and TNF−/− (clear circle) mice were infected with M. tuberculosis H37Rv at a dose of 1 × 103–1 × 104 CFUs/brain by intracerebral inoculation. Body weight changes (a) and mortality (b) were measured and recorded for the experimental duration (n = 10–12 mice/group). c Brain weights of infected mice were recorded at 1, 2 and 3 weeks post-infection (n = 5–6 mice/group). d The clinical scores, indicative of the disease severity, are represented (n = 10–12 mice/group). The data represents three to five similar experiments. *p ≤ 0.05
Cerebral bacilli replication and dissemination are increased in TNF−/− mice but not in NsTNF−/− mice
The importance of TNF to control cerebral bacilli replication was assessed by comparing pathogen burden in the brains of M. tuberculosis-infected TNFf/f, NsTNF−/− and TNF−/− mice. We observed an increase in cerebral M. tuberculosis replication in TNFf/f mice during acute infection, burdens at week 3 post-infection being significantly higher compared to those at earlier time points (Fig. 4a). In contrast, differences in cerebral bacilli burden were already significantly higher (p < 0.01) in TNF−/− mice at 2 weeks post-infection which were sustained at 3 weeks post-infection. In contrast, NsTNF−/− mice controlled acute infection at burden levels equivalent to those in TNFf/f mice and were similarly significantly lower (p < 0.01) compared to those in TNF−/− mice (Fig. 4a). We confirmed differences in cerebral bacilli burden by image analysis of acid-fast bacilli staining (Fig. 5). In the brains of TNFf/f and NsTNF−/− mice, acid-fast bacilli were located either in the choroid plexus, in the ventricular space or in the perivascular region at 3 weeks post-infection. However, in TNF−/− mice, necrotic foci containing significantly higher numbers of bacilli were prominent in the brain parenchyma (Fig. 5). We then asked whether neuron-derived TNF is required to control chronic cerebral M. tuberculosis infection. We found no difference in cerebral bacilli burdens between TNFf/f and NsTNF−/− mice (Fig. 4b), indicating that TNF synthesis by neurons is not required to control infection. We next measured bacilli burdens in the lungs and spleens of infected mice to evaluate the role of TNF to control M. tuberculosis dissemination from the brain. Pulmonary and splenic infection was established early in the absence of TNF and bacilli burdens significantly higher than TNFf/f or NsTNF−/− mice (Fig. 4c, e), indicating that TNF is required to control early dissemination of infection. The comparative equivalence of bacilli burdens in both the lungs and spleens of TNFf/f and NsTNF−/− mice during acute (Fig. 4c, e) or chronic infection (Fig. 4d, f) suggested a redundancy of neuron-derived TNF to contain cerebral spread of infection.

TNF is required to control intracerebral M. tuberculosis replication but neuron-derived TNF is redundant. TNFf/f (black circle), NsTNF−/− (triangle) and TNF−/− (clear circle) mice (n = 5–6/group) were infected with M. tuberculosis at a dose of 1 × 103–1 × 104 CFUs/brain. Bacterial burdens in a, b brains; c, d spleens; and e, f lungs at 1, 2, 3 (left panel; a, c, e, respectively) and 15 weeks (right panel; b, d, e respectively) post-infection were determined. Data (mean ± SEM of the CFUs of 5–6 mice per time point) are representative of three similar experiments. *p ≤ 0.05; **p ≤ 0.01

Cerebral pathology is regulated by TNF but neuron-derived TNF is not required. a–c TNFf/f, d–f NsTNF−/− and g–i TNF−/− mice were infected with M. tuberculosis H37Rv at a dose of 1 × 103–1 × 104 CFUs/brain by intracerebral inoculation. Haematoxylin and eosin-stained coronal brain tissue sections at 3 weeks post-infection are presented (a, d, g). Ventriculitis of the choroid plexus is primarily lymphocytic in b TNFf/f and e NsTNF−/− mice while a predominant neutrophil infiltrate accompanied by necrosis is observed in h TNF−/− mice. Ziehl–Neelsen staining revealed acid-fast bacilli mainly in the choroid plexus (c, arrow) and ventricular space (f, arrow) in TNFf/f and e NsTNF−/− mice while infiltration of bacilli was frequently observed in brain parenchyma (i) in TNF−/− mice. The granuloma was found in TNF−/− mouse brain (circle in h, i). Scale bars 20 μm
TNF regulates cerebral inflammation but neuron-derived TNF is not required
We next examined the role of TNF to control cerebral inflammation by analysis of sectioned brain tissue for induced pathology in M. tuberculosis-infected mice at 3 weeks post-infection. In the brain sections taken from TNFf/f, NsTNF−/− and TNF−/− mice, we observed degrees of inflammation in different regions of the brain in all mouse strains (Fig. 5). Representative images of inflammation in the choroid plexus, ventricles and brain tissue are shown in Fig. 5b, e, h, respectively. Amongst the three mouse strains, TNF−/− mice exhibited the most severe neuropathology and uncontrolled inflammation where acute ventriculitis was characterised by a prominent neutrophil infiltrate which extended into the periventricular tissue (Fig. 5g). The choroid plexus in TNF−/− mice was infiltrated by a mixture of lymphocytes and neutrophils in contrast to a predominantly lymphocytic infiltrate in TNFf/f and NsTNF−/− mice. Also noted were associated foci of necrosis in the adjacent brain tissue with distinct areas of perivascular lymphocyte cuffing (Fig. 5h). Well-formed, discrete granulomas were present in TNFf/f and NsTNF−/− mice unlike in TNF−/− mice where granuloma formation was largely absent.
Therefore, TNF was required to regulate CNS inflammation during M. tuberculosis infection; moreover, it determined the quality of the inflammatory response.
TNF regulates recruitment and proliferation of innate immune cells
Early susceptibility of TNF−/− mice to cerebral M. tuberculosis infection suggested defective TNF-mediated function during initial innate immune responses. We postulated that TNF deficiency may impact on key innate immune cell function to promote the observed susceptible phenotype. We first investigated recruitment of macrophages and dendritic cells to the brain and evaluated proliferation of microglia, as central mediators of innate immune responses subsequent to M. tuberculosis challenge. Microglial proliferation and dendritic cell and macrophage recruitment occurred in all three mouse strains in response to M. tuberculosis infection (Fig. 6a–c). However, while cellular levels reached maximum at 2 weeks post-infection and were sustained at 3 weeks post-infection in TNFf/f and NsTNF−/− mice, TNF−/− mice displayed significantly higher levels at 2 weeks post-infection, the differential levels enhancing even further at 3 weeks post-infection.

TNF regulates innate immune cell proliferation and recruitment during CNS-TB. TNFf/f (black), NsTNF−/− (grey) and TNF−/− (clear) mice were infected with M. tuberculosis H37Rv at a dose of 1 × 103–1 × 104 CFUs/brain by intracerebral inoculation. The number of a microglia, b macrophages and c dendritic cells was analysed by flow cytometry at 0, 1, 2 and 3 weeks in infected brains. The chemokines d MIP-1α, e MCP-1 and f RANTES were measured by ELISA in the brains of infected mice at 3 weeks post-intracerebral infection. Data (mean ± SEM of the CFUs of 5–6 mice per time point) are representative of repeat experiments. *p ≤ 0.05; **p ≤ 0.01
We next investigated whether enhanced innate immune cell recruitment in TNF−/− mice was a consequence of heightened host cerebral chemokine induction in response to M. tuberculosis infection. We measured cerebral concentrations of MIP-1α, MCP-1 and RANTES in M. tuberculosis-infected mice representative of chemokines known to induce pro-inflammatory effects [50, 51]. We found escalating induction of each chemokine in the brains of infected mice over the duration of the experiment (data not shown). However, while TNFf/f and NsTNF−/− mice produced equivalent chemokine concentrations, TNF−/− mice synthesised significantly higher levels (p < 0.01) of MIP-1α, MCP-1 and RANTES at 3 weeks post-infection (Fig. 6d–f). The positive correlation between higher chemokine concentrations and enhanced cellular inflammation in TNF−/− mice suggests an unregulated TNF-dependent chemokine response driving uncontrolled cell recruitment in response to M. tuberculosis infection. Thus, while immune cell recruitment and proliferation were TNF dependent, neuron-derived TNF was redundant for this function.
TNF deficiency renders cerebral innate immune cells functionally defective
The increased presence of innate immune cells in the absence of TNF supports the potential for improved early control of cerebral M. tuberculosis, yet TNF−/− mice display a highly susceptible phenotype. We postulated that early susceptibility may be associated with defective function of innate immune cells during initial infection. We therefore investigated the capacity of microglia, macrophages and dendritic cells to present antigen by analysing expression of MHCII, CD80 and CD86 (Fig. 7). We observed comparable expression of CD80, CD86 and MHCII in TNFf/f and NsTNF−/− mice at each of the time points investigated, indicating that neuron-derived TNF does not regulate innate immune cell activation. In contrast, although TNF−/− mice presented with equivalent levels of immune-activated cells during the first 2 weeks of infection compared to TNFf/f and NsTNF−/− mice, they fail to sustain the required level of activation and significantly lower (p < 0.05) levels of activated innate immune cells were measured at 3 weeks post-infection.

Expression of CD80, CD86 and MHCII in microglia, macrophages and dendritic cells is TNF dependent. TNFf/f (black circle), NsTNF−/− (triangle) and TNF−/− (clear circle) mice were infected with M. tuberculosis H37Rv at a dose of 1 × 103–1 × 104 CFUs/brain by intracerebral inoculation. Single-cell suspensions of brains at 0, 1, 2 and 3 weeks post-infection were generated and microglia (a–c), macrophages (d–f) and dendritic cells (g–i) analysed for expression of CD80 (a, d, f), CD86 (b, e, h) and MHC II (c, f, i). Data (mean ± SEM of the CFUs of 5–6 mice per time point) are representative of repeat experiments. **p ≤ 0.01
To further investigate innate immune cell functionality, we measured levels of TNF, IL-1β and IL-12p70 in the brain homogenates of TNFf/f, NsTNF−/− and TNF−/− mice after 3 weeks of intracranial infection (Fig. 8). The induction of TNF in TNFf/f mice confirmed its requirement as part of initial protective immune responses, while equivalent expression in NsTNF−/− mice indicated neurons redundant as a TNF source (Fig. 8a). As expected, no TNF production was detected in TNF−/− mice (Fig. 8a). Similar levels of IL-1β (Fig. 8b) and IL-12p70 (Fig. 8c) production were observed in TNFf/f and NsTNF−/− infected brains. In contrast, the concentration of cerebral IL-1β and IL-12p70 in infected TNF−/− mice was significantly (p < 0.05) lower than in TNFf/f and NsTNF−/− mice. Therefore, the data indicates that TNF is required to regulate cytokine synthesised by innate immune cells but that neuron-derived TNF is not important.

TNF regulates synthesis of IL-1β and IL-12p70. TNFf/f (black), NsTNF−/− (grey) and TNF−/− (clear) mice were infected with M. tuberculosis H37Rv infection at a dose of 1 × 103–1 × 104 CFUs/brain by intracerebral inoculation. a TNF, b IL-1β and c IL-12p70 cerebral concentrations were measured by ELISA at 3 weeks post-infection. Data (mean ± SEM of the CFUs of 5–6 mice per time point) are representative of repeat experiments. *p ≤ 0.05
Discussion
TNF concentration forms a pivotal point of balance that determines outcome of disease during M. tuberculosis infection. Susceptibility is promoted either under conditions where excess TNF prevails or low TNF concentrations are present [15, 16, 23, 24]. Under such polar conditions, either macrophage/cell effector function is impaired which leads to intracellular mycobacterial replication and eventual cell lysis or excessive TNF levels promote macrophage necrosis, in so doing enrich conditions for bacilli growth. Diverse cell types can act as contributory sources of TNF (during pathogenic challenge) and may therefore be differential contributors to the TNF environment required for optimum mycobacterial control. It is known that microglia, as resident immune cells of the CNS, express TNF and that recruited dendritic cells and macrophages will similarly induce TNF during pathogen challenge [40, 52, 53]. Neurons, as the single most abundant cell type within the CNS, are capable of regulating immune responses during CNS insult [54, 55]. We have reported M. tuberculosis targeting of neurons and its capability to generate reactive immune responses [42]; however, its role in M. tuberculosis control remains largely untested. In the presence of pathogens, neurons induce specific cytokine signatures, TNF comprising a key component of many such signature responses. The synthesis of TNF by neurons was observed after systemic immune challenge [56]. Upon viral infection, both in vitro and clinical studies have shown TNF expression in neurons [57–60]. In other neurological diseases, upregulation of neuronal TNF expression has provided an indication to the severity of disease progression and inflammatory neuropathology [43, 61–63]. In view of these findings, we postulated that neuron-derived TNF may have a contributory role to regulate CNS immunity during M. tuberculosis infection. We therefore investigated the overall contribution of TNF and in particular the involvement of neuron-derived TNF in host-mediated immunity directed against M. tuberculosis during central nervous system infection. Several converging lines of study/evidence have indicated a neuroprotective role for TNF and its induction demonstrated to be a key determinant of disease outcome during cerebral infection [64]. In this study, we initially demonstrated that global TNF is critical to control M. tuberculosis dissemination to the CNS and regulates immune-mediated brain pathology during infection. Moreover, we demonstrate for the first time that TNF is an absolute requirement to mediate protective immunity against CNS-TB; however, neuron-derived TNF is considered redundant. Thus, host susceptibility to CNS-TB in the absence of TNF corroborates its significance in neuroprotective immunity. Although the findings in this study appear contrasting to those of Tsenova et al., where TNF was reported to promote mycobacterial onset of disease and pathogenesis [16], it is not unexpected and may rather reflect opposite ends of the TNF functionality spectrum as previously reported [23, 24]. TNF concentration in dynamic homeostasis is absolutely essential for effective control of M. tuberculosis infection, a shift in balance that favours either excessive or reduced levels being detrimental to the host that promotes conditions for mycobacterial persistence.
Mycobacterial infection of the CNS generates a robust immune response characterised by both pro-inflammatory and anti-inflammatory immune cellular and cytokine profiles [65]. While microglia were reported to be the primary source of TNF during CNS infection [65], we and others have shown that different cell types have hierarchical importance as sources of TNF in determining the outcome of tuberculosis disease [37]. TNF derived from T cells critically defines resolution of pulmonary infection, while TNF from macrophages has a transient role to control M. tuberculosis replication [37]. Our findings however ascribe neurons as a redundant cellular source of TNF to control M. tuberculosis infection or regulate immune pathology and suggest a far greater role for other resident or recruited cells to produce TNF. Interestingly, others have reported that TNF produced by neurons is redundant for mediating inflammation but that membrane TNF expressed on astrocytes has a functional role [66].
Nonetheless, the overall requirement of TNF for control of both mycobacterial replication and immune-mediated pathology was evident and the rapid rate of host susceptibility suggested immune deficiency during early infection. Brain pathology, particularly at end-stage disease, was characterised by increased inflammation of dendritic cells and macrophages, and microglial proliferation which was underpinned by an enhanced pro-inflammatory chemokine environment in the absence of TNF. These observations align with earlier reports of a regulatory role for TNF in inflammation during M. tuberculosis infection [67]. In earlier studies, we reported on a deregulated overall cytokine and chemokine response in the absence of TNF during pulmonary tuberculosis [37]. We show here a neutrophil-dominant pathology in TNF−/− mice also associates with a similar unregulated increase in chemokine synthesis during CNS-TB. Moreover, a lethal neutrophil-driven inflammatory response associated with, amongst others, MIP-1α in the absence of microRNA 223 in pulmonary tuberculosis was reported [68]. Taken together, the data suggest that perturbation of controlled host immune regulation which promotes a dominant neutrophilic response leads to increase susceptibility with fatal consequences. Selective inhibition of neutrophil recruitment under conditions of immune suppression during tuberculosis should therefore be considered.
Microglia, macrophages and dendritic cells as the main facilitators of early innate immune protection are activated in a TNF-dependent manner [69, 70]. We therefore postulated that early host susceptibility of TNF−/− mice was the result of defective activation of innate antigen-presenting immune cells during early infection. Activation of macrophages, dendritic cells and microglia within the first 2 weeks of infection appeared to be independent of TNF with equivalent levels of activated cells evident in TNFf/f, NsTNF−/− and TNF−/− mice; however, the ability to subsequently sustain the required measures of activated cells was TNF dependent. These cells which are critical effectors of early innate immunity were rendered functionally defective in the absence of TNF. Initial cytokine signature responses by innate immune cells include synthesis of IL-12 and IL-1 [71, 72], both of which are critical to control tuberculosis infection [14, 73–75]. Mice deficient for either cytokine develop rapid onset of disease and succumb to infection, while patients with defective IL-12 signalling display susceptibility to tuberculosis infection. Maturation of antigen-presenting cells is characterised by the expression of, amongst others, MHCII and CD80 [76] during M. tuberculosis infection and is mediated largely by the presence of IL-1 and TNF [77, 78]. Moreover, IL-12 synthesis by mature activated dendritic cells is dependent on TNF-mediated signalling [78] and is a key cytokine required to restrict bacilli replication. Here, we observed that maturation of microglia, dendritic cells and macrophages initially occurs independent of TNF; however, sustained cell activation was TNF dependent. The deficiency in IL-12 and IL-1β synthesis in the CNS when TNF is absent argues strongly for functional failure of innate immune cells and inability to effectively participate in control of M. tuberculosis. Moreover, the reduction of MHCII- and CD80-expressing cells indicated reduced capacity to initiate M. tuberculosis-specific Th1 protective immune responses required for control of intracellular bacilli, and hence predisposed the host to a susceptible phenotype.
Conclusion
In conclusion, the findings in this study corroborate earlier reports of the global importance of TNF in immune-mediated responses against tuberculosis and for the first time demonstrate its importance to control M. tuberculosis replication within the CNS. We further showed that neuron-derived TNF is not required for this process.
Abbreviations
- BBB:
-
Blood–brain barrier
- BCG:
-
Bacillus Calmette–Guérin
- CCL:
-
Chemokine C–C motif ligand
- CD:
-
Cluster of differentiation
- CFU:
-
Colony-forming unit
- CNS:
-
Central nervous system
- CNS-TB:
-
Tuberculosis of the central nervous system
- CSF:
-
Cerebrospinal fluid
- GFP:
-
Green fluorescent protein
- H&E:
-
Haematoxylin and eosin
- IL:
-
Interleukin
- LPS:
-
Lipopolysaccharide
- MAP:
-
Microtubule-associated protein
- MCP:
-
Monocyte chemotactic protein
- MHC:
-
Major histocompatibility complex
- MIP:
-
Macrophage inflammatory protein
- M. tuberculosis :
-
Mycobacterium tuberculosis
- NsTNF−/− :
-
Neuron-specific TNF-deficient
- TB:
-
Tuberculosis
- TNF:
-
Tumour necrosis factor
- TNF−/− :
-
Tumour necrosis factor-deficient
- TNFf/f :
-
Tumour necrosis factor-floxed
References
Be NA, Kim KS, Bishai WR, Jain SK. Pathogenesis of central nervous system tuberculosis. Curr Mol Med. 2009;9:94–9.
Garg RK. Tuberculosis of the central nervous system. Postgrad Med J. 1999;75:133–40.
Rock RB, Olin M, Baker CA, Molitor TW, Peterson PK. Central nervous system tuberculosis: pathogenesis and clinical aspects. Clin Microbiol Rev. 2008;21:243–61. table of contents.
Donald PR. The chemotherapy of tuberculous meningitis in children and adults. Tuberculosis (Edinb). 2010;90:375–92.
Gardam MA, Keystone EC, Menzies R, Manners S, Skamene E, Long R, et al. Anti-tumour necrosis factor agents and tuberculosis risk: mechanisms of action and clinical management. Lancet Infect Dis. 2003;3:148–55.
Vinnard C, Macgregor RR. Tuberculous meningitis in HIV-infected individuals. Curr HIV/AIDS Rep. 2009;6:139–45.
Xiao J, Gao G, Li Y, Zhang W, Tian Y, Huang Y, et al. Spectrums of opportunistic infections and malignancies in HIV-infected patients in tertiary care hospital, China. PLoS One. 2013;8:e75915.
Misra UK, Kalita J, Bhoi SK. Spectrum and outcome predictors of central nervous system infections in a neurological critical care unit in India: a retrospective review. Trans R Soc Trop Med Hyg. 2014;108:141–6.
Leonard JM, Des Prez RM. Tuberculous meningitis. Infect Dis Clin North Am. 1990;4:769–87.
Drevets DA, Leenen PJ, Greenfield RA. Invasion of the central nervous system by intracellular bacteria. Clin Microbiol Rev. 2004;17:323–47.
Ransohoff RM, Kivisakk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol. 2003;3:569–81.
Kivisakk P, Mahad DJ, Callahan MK, Trebst C, Tucky B, Wei T, et al. Human cerebrospinal fluid central memory CD4+ T cells: evidence for trafficking through choroid plexus and meninges via P-selectin. Proc Natl Acad Sci U S A. 2003;100:8389–94.
Krumbholz M, Theil D, Steinmeyer F, Cepok S, Hemmer B, Hofbauer M, et al. CCL19 is constitutively expressed in the CNS, up-regulated in neuroinflammation, active and also inactive multiple sclerosis lesions. J Neuroimmunol. 2007;190:72–9.
Mastroianni CM, Paoletti F, Lichtner M, D’Agostino C, Vullo V, Delia S. Cerebrospinal fluid cytokines in patients with tuberculous meningitis. Clin Immunol Immunopathol. 1997;84:171–6.
Tobin DM, Roca FJ, Oh SF, McFarland R, Vickery TW, Ray JP, et al. Host genotype-specific therapies can optimize the inflammatory response to mycobacterial infections. Cell. 2012;148:434–46.
Tsenova L, Bergtold A, Freedman VH, Young RA, Kaplan G. Tumor necrosis factor alpha is a determinant of pathogenesis and disease progression in mycobacterial infection in the central nervous system. Proc Natl Acad Sci U S A. 1999;96:5657–62.
Hayasaka D, Shirai K, Aoki K, Nagata N, Simantini DS, Kitaura K, et al. TNF-alpha acts as an immunoregulator in the mouse brain by reducing the incidence of severe disease following Japanese encephalitis virus infection. PLoS One. 2013;8:e71643.
Figiel I, Dzwonek K. TNFalpha and TNF receptor 1 expression in the mixed neuronal-glial cultures of hippocampal dentate gyrus exposed to glutamate or trimethyltin. Brain Res. 2007;1131:17–28.
Henderson RA, Watkins SC, Flynn JL. Activation of human dendritic cells following infection with Mycobacterium tuberculosis. J Immunol. 1997;159:635–43.
Sawant KV, McMurray DN. Guinea pig neutrophils infected with Mycobacterium tuberculosis produce cytokines which activate alveolar macrophages in noncontact cultures. Infect Immun. 2007;75:1870–7.
Ramilo O, Saez-Llorens X, Mertsola J, Jafari H, Olsen KD, Hansen EJ, et al. Tumor necrosis factor alpha/cachectin and interleukin 1 beta initiate meningeal inflammation. J Exp Med. 1990;172:497–507.
Saukkonen K, Sande S, Cioffe C, Wolpe S, Sherry B, Cerami A, et al. The role of cytokines in the generation of inflammation and tissue damage in experimental gram-positive meningitis. J Exp Med. 1990;171:439–48.
Clay H, Volkman HE, Ramakrishnan L. Tumor necrosis factor signaling mediates resistance to mycobacteria by inhibiting bacterial growth and macrophage death. Immunity. 2008;29:283–94.
Roca FJ, Ramakrishnan L. TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell. 2013;153:521–34.
Grivennikov SI, Tumanov AV, Liepinsh DJ, Kruglov AA, Marakusha BI, Shakhov AN, et al. Distinct and nonredundant in vivo functions of TNF produced by T cells and macrophages/neutrophils: protective and deleterious effects. Immunity. 2005;22:93–104.
Tsenova L, Harbacheuski R, Sung N, Ellison E, Fallows D, Kaplan G. BCG vaccination confers poor protection against M. tuberculosis HN878-induced central nervous system disease. Vaccine. 2007;25:5126–32.
Tsenova L, Ellison E, Harbacheuski R, Moreira AL, Kurepina N, Reed MB, et al. Virulence of selected Mycobacterium tuberculosis clinical isolates in the rabbit model of meningitis is dependent on phenolic glycolipid produced by the bacilli. J Infect Dis. 2005;192:98–106.
Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J Immunol. 1995;154:4309–21.
Sedgwick JD, Schwender S, Imrich H, Dorries R, Butcher GW, ter Meulen V. Isolation and direct characterization of resident microglial cells from the normal and inflamed central nervous system. Proc Natl Acad Sci U S A. 1991;88:7438–42.
Jacobs M, Samarina A, Grivennikov S, Botha T, Allie N, Fremond C, et al. Reactivation of tuberculosis by tumor necrosis factor neutralization. Eur Cytokine Netw. 2007;18:5–13.
Yasui K. Immunity against Mycobacterium tuberculosis and the risk of biologic anti-TNF-alpha reagents. Pediatr Rheumatol Online J. 2014;12:45.
Quesniaux VF, Jacobs M, Allie N, Grivennikov S, Nedospasov SA, Garcia I, et al. TNF in host resistance to tuberculosis infection. Curr Dir Autoimmun. 2010;11:157–79.
Botha T, Ryffel B. Reactivation of latent tuberculosis infection in TNF-deficient mice. J Immunol. 2003;171:3110–8.
Bean AG, Roach DR, Briscoe H, France MP, Korner H, Sedgwick JD, et al. Structural deficiencies in granuloma formation in TNF gene-targeted mice underlie the heightened susceptibility to aerosol Mycobacterium tuberculosis infection, which is not compensated for by lymphotoxin. J Immunol. 1999;162:3504–11.
Lynch K, Farrell M. Cerebral tuberculoma in a patient receiving anti-TNF alpha (adalimumab) treatment. Clin Rheumatol. 2010;29:1201–4.
Seong SS, Choi CB, Woo JH, Bae KW, Joung CL, Uhm WS, et al. Incidence of tuberculosis in Korean patients with rheumatoid arthritis (RA): effects of RA itself and of tumor necrosis factor blockers. J Rheumatol. 2007;34:706–11.
Allie N, Grivennikov SI, Keeton R, Hsu NJ, Bourigault ML, Court N, et al. Prominent role for T cell-derived tumour necrosis factor for sustained control of Mycobacterium tuberculosis infection. Sci Rep. 2013;3:1809.
Dambuza I, Allie N, Fick L, Johnston N, Fremond C, Mitchell J, et al. Efficacy of membrane TNF mediated host resistance is dependent on mycobacterial virulence. Tuberculosis (Edinb). 2008;88:221–34.
Hernandez Pando R, Aguilar D, Cohen I, Guerrero M, Ribon W, Acosta P, et al. Specific bacterial genotypes of Mycobacterium tuberculosis cause extensive dissemination and brain infection in an experimental model. Tuberculosis (Edinb). 2010;90:268–77.
Rock RB, Hu S, Gekker G, Sheng WS, May B, Kapur V, et al. Mycobacterium tuberculosis-induced cytokine and chemokine expression by human microglia and astrocytes: effects of dexamethasone. J Infect Dis. 2005;192:2054–8.
Figiel I. Pro-inflammatory cytokine TNF-alpha as a neuroprotective agent in the brain. Acta Neurobiol Exp (Wars). 2008;68:526–34.
Randall PJ, Hsu NJ, Lang D, Cooper S, Sebesho B, Allie N, et al. Neurons are host cells for Mycobacterium tuberculosis. Infect Immun. 2014;82:1880–90.
Janelsins MC, Mastrangelo MA, Park KM, Sudol KL, Narrow WC, Oddo S, et al. Chronic neuron-specific tumor necrosis factor-alpha expression enhances the local inflammatory environment ultimately leading to neuronal death in 3xTg-AD mice. Am J Pathol. 2008;173:1768–82.
Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, et al. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 2001;15:859–76.
Tsenova L, Sokol K, Freedman VH, Kaplan G. A combination of thalidomide plus antibiotics protects rabbits from mycobacterial meningitis-associated death. J Infect Dis. 1998;177:1563–72.
Allie N, Alexopoulou L, Quesniaux VJ, Fick L, Kranidioti K, Kollias G, et al. Protective role of membrane tumour necrosis factor in the host’s resistance to mycobacterial infection. Immunology. 2008;125:522–34.
Chertoff M, Di Paolo N, Schoeneberg A, Depino A, Ferrari C, Wurst W, et al. Neuroprotective and neurodegenerative effects of the chronic expression of tumor necrosis factor alpha in the nigrostriatal dopaminergic circuit of adult mice. Exp Neurol. 2011;227:237–51.
Kirby ED, Jensen K, Goosens KA, Kaufer D. Stereotaxic surgery for excitotoxic lesion of specific brain areas in the adult rat. J Vis Exp. 2012;(65):4079. doi:10.3791/4079.
Weiergraber M, Henry M, Hescheler J, Smyth N, Schneider T. Electrocorticographic and deep intracerebral EEG recording in mice using a telemetry system. Brain Res Brain Res Protoc. 2005;14:154–64.
Jiang Y, Beller DI, Frendl G, Graves DT. Monocyte chemoattractant protein-1 regulates adhesion molecule expression and cytokine production in human monocytes. J Immunol. 1992;148:2423–8.
Crane IJ, McKillop-Smith S, Wallace CA, Lamont GR, Forrester JV. Expression of the chemokines MIP-1alpha, MCP-1, and RANTES in experimental autoimmune uveitis. Invest Ophthalmol Vis Sci. 2001;42:1547–52.
Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–94.
Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR, et al. Role of microglia in central nervous system infections. Clin Microbiol Rev. 2004;17:942–64. table of contents.
Chavarria A, Cardenas G. Neuronal influence behind the central nervous system regulation of the immune cells. Front Integr Neurosci. 2013;7:64.
Tian L, Rauvala H, Gahmberg CG. Neuronal regulation of immune responses in the central nervous system. Trends Immunol. 2009;30:91–9.
Breder CD, Hazuka C, Ghayur T, Klug C, Huginin M, Yasuda K, et al. Regional induction of tumor necrosis factor alpha expression in the mouse brain after systemic lipopolysaccharide administration. Proc Natl Acad Sci U S A. 1994;91:11393–7.
Cowan EP, Alexander RK, Daniel S, Kashanchi F, Brady JN. Induction of tumor necrosis factor alpha in human neuronal cells by extracellular human T-cell lymphotropic virus type 1 Tax. J Virol. 1997;71:6982–9.
Rostasy K, Monti L, Lipton SA, Hedreen JC, Gonzalez RG, Navia BA. HIV leucoencephalopathy and TNFalpha expression in neurones. J Neurol Neurosurg Psychiatry. 2005;76:960–4.
Nazmi A, Dutta K, Basu A. RIG-I mediates innate immune response in mouse neurons following Japanese encephalitis virus infection. PLoS One. 2011;6:e21761.
Kumar M, Verma S, Nerurkar VR. Pro-inflammatory cytokines derived from West Nile virus (WNV)-infected SK-N-SH cells mediate neuroinflammatory markers and neuronal death. J Neuroinflammation. 2010;7:73.
Sairanen T, Carpen O, Karjalainen-Lindsberg ML, Paetau A, Turpeinen U, Kaste M, et al. Evolution of cerebral tumor necrosis factor-alpha production during human ischemic stroke. Stroke. 2001;32:1750–8.
Kiaei M, Kipiani K, Calingasan NY, Wille E, Chen J, Heissig B, et al. Matrix metalloproteinase-9 regulates TNF-alpha and FasL expression in neuronal, glial cells and its absence extends life in a transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol. 2007;205:74–81.
Knoblach SM, Fan L, Faden AI. Early neuronal expression of tumor necrosis factor-alpha after experimental brain injury contributes to neurological impairment. J Neuroimmunol. 1999;95:115–25.
Faber M, Bette M, Preuss MA, Pulmanausahakul R, Rehnelt J, Schnell MJ, et al. Overexpression of tumor necrosis factor alpha by a recombinant rabies virus attenuates replication in neurons and prevents lethal infection in mice. J Virol. 2005;79:15405–16.
Lee J, Ling C, Kosmalski MM, Hulseberg P, Schreiber HA, Sandor M, et al. Intracerebral Mycobacterium bovis bacilli Calmette-Guerin infection-induced immune responses in the CNS. J Neuroimmunol. 2009;213:112–22.
Akassoglou K, Probert L, Kontogeorgos G, Kollias G. Astrocyte-specific but not neuron-specific transmembrane TNF triggers inflammation and degeneration in the central nervous system of transgenic mice. J Immunol. 1997;158:438–45.
Zganiacz A, Santosuosso M, Wang J, Yang T, Chen L, Anzulovic M, et al. TNF-alpha is a critical negative regulator of type 1 immune activation during intracellular bacterial infection. J Clin Invest. 2004;113:401–13.
Dorhoi A, Iannaccone M, Farinacci M, Fae KC, Schreiber J, Moura-Alves P, et al. MicroRNA-223 controls susceptibility to tuberculosis by regulating lung neutrophil recruitment. J Clin Invest. 2013;123:4836–48.
Kuno R, Wang J, Kawanokuchi J, Takeuchi H, Mizuno T, Suzumura A. Autocrine activation of microglia by tumor necrosis factor-alpha. J Neuroimmunol. 2005;162:89–96.
Sriram K, Matheson JM, Benkovic SA, Miller DB, Luster MI, O’Callaghan JP. Deficiency of TNF receptors suppresses microglial activation and alters the susceptibility of brain regions to MPTP-induced neurotoxicity: role of TNF-alpha. FASEB J. 2006;20:670–82.
Xin G, Su Y, Gao YL, Zhang H, Wang GF, Li KS. Lipopolysaccharide enhances asymmetrical production of cytokines and nitric oxide by left and right cerebral cortical microglial cells in BALB/C mice. Cell Biochem Funct. 2011;29:50–4.
Saliba E, Henrot A. Inflammatory mediators and neonatal brain damage. Biol Neonate. 2001;79:224–7.
Misra UK, Kalita J, Srivastava R, Nair PP, Mishra MK, Basu A. A study of cytokines in tuberculous meningitis: clinical and MRI correlation. Neurosci Lett. 2010;483:6–10.
Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature. 2014;511:99–103.
Tso HW, Lau YL, Tam CM, Wong HS, Chiang AK. Associations between IL12B polymorphisms and tuberculosis in the Hong Kong Chinese population. J Infect Dis. 2004;190:913–9.
Mihret A, Mamo G, Tafesse M, Hailu A, Parida S. Dendritic cells activate and mature after infection with Mycobacterium tuberculosis. BMC Res Notes. 2011;4:247.
Hanekom WA, Mendillo M, Manca C, Haslett PA, Siddiqui MR, Barry 3rd C, et al. Mycobacterium tuberculosis inhibits maturation of human monocyte-derived dendritic cells in vitro. J Infect Dis. 2003;188:257–66.
Keeton R, Allie N, Dambuza I, Abel B, Hsu NJ, Sebesho B, et al. Soluble TNFRp75 regulates host protective immunity against Mycobacterium tuberculosis. J Clin Invest. 2014;124:1537–51.
Acknowledgements
This study was supported by the National Research Foundation (South Africa), Deutscher Akademischer Austausch Dienst (Germany), the South African Medical Research Council, University of Cape Town and National Health Laboratory Service (South Africa). We thank Marilyn Tyler and Lizette Fick for their sterling contribution to the histopathology. We are grateful to A. Professor Dirk Lang and Susan Cooper for their contribution to image analysis. Special thanks are due to Faried Abbass for technical support. We thank the staff of the Division of Immunology and the Animal Unit at the University of Cape Town for their contribution to animal care and technical support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NMF, NJH, RK, BS and NA were involved in the experiments on the intracerebral infection and the subsequent analysis. NJH and PR performed the experiments on aerosol infection and neural immunohistochemistry. DG contributed to the analysis of histopathology. VQ, BR and LK contributed to the experimental design. MJ oversaw the project. NMF, NJH and MJ prepared the figures and wrote the manuscript. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Francisco, N.M., Hsu, NJ., Keeton, R. et al. TNF-dependent regulation and activation of innate immune cells are essential for host protection against cerebral tuberculosis. J Neuroinflammation 12, 125 (2015). https://doi.org/10.1186/s12974-015-0345-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-015-0345-1