
Abstract
Background
The aim of this study was to: (i) describe the abnormalities seen on brain imaging in a group of children with en coup de sabre (EDCS) with/without Parry-Romberg syndrome (PRS); and (ii) identify clinical predictors of brain imaging abnormalities.
Methods
This was a single centre (Great Ormond Street Hospital, London) retrospective case series of patients with ECDS/PRS seen from 2000 to 2018. We identified patients with cutaneous manifestations consistent with the clinical descriptions of ECDS/PRS. Presenting clinical, laboratory, and radiological brain findings are described. Results are expressed as medians and ranges or frequencies and percentages. Fisher’s exact test was used to identify clinical associations with magnetic resonance imaging (MRI) abnormalities.
Results
Fourteen patients were studied: 6 males and 8 females; median age 14 years (range 3–20). We observed neuroimaging abnormalities in 2/6 ECDS and 5/8 ECDS/PRS patients. White matter signal abnormality, dystrophic calcification, leptomeningeal enhancement, and sulcal crowding were the typical findings on brain imaging. A total of 50% of patients had no MRI abnormality despite some of these patients having neurological symptoms. The presence of seizures was significantly associated with ipsilateral enhanced white matter signalling on MRI (p < 0.05).
Conclusions
In summary, we observed several distinct radiographic patterns associated with ECDS/PRS. Seizure disorder was strongly associated with the presence of ipsilateral enhanced white matter signalling. Improved neuroimaging techniques that combine morphological with functional imaging may improve the detection rate of brain involvement in children with ECDS/PRS in the future.
Similar content being viewed by others

Background
En coup de sabre (ECDS, “from a blow of the sword”) is a type of linear scleroderma which begins as a line of cutaneous inflammation and develops into a line of fibrosis (as is the usual pattern for linear scleroderma anywhere on the body) [1]. Although ECDS tends to start on the scalp, this line may extend downwards to involve inferior areas such as the forehead, brow ridge, eyelid and its appendages, cheek, dental ridge, and chin [2]. Parry-Romberg syndrome (PRS) is a diffuse hemifacial atrophy [3]. It can be subtle at first but progresses over time to involve the skin, soft tissues, and underlying bone [3]. It almost always affects a larger area than ECDS, and affected skin has absent or minimal fibrosis which can be appreciated on examination and demonstrated on skin biopsy [3]. PRS also tends to become more prominent with age as the affected tissues fail to grow as well as the unaffected side [3]. ECDS and PRS may occur alone, but patients with ECDS may subsequently develop PRS as well [4]. Changes of cutaneous ECDS or PRS may be subtle on imaging studies, but 20% of these patients will have intracranial manifestations that may not correspond to the severity of soft-tissue involvement or neurological symptoms [5]. The recently published Single Hub Access Point for Paediatric Rheumatology (SHARE) consensus-based recommendations for the management of juvenile localised scleroderma recognised this lack of correlation between the severity of cutaneous disease and central nervous system (CNS) involvement and recommended that all patients with juvenile linear scleroderma such as ECDS and PRS involving the face and head, with or without signs of neurological involvement, should have magnetic resonance imaging (MRI) of the head at the time of diagnosis [6]. These recommendations did not, however, provide any guidance on the specific imaging findings associated with neurological symptoms in ECDS/PRS due to a lack of relevant data: most imaging studies to date have been reported in adults, with only single case reports or small cases series in childhood disease [7,8,9]. The aim of this study was therefore to: (i) describe the CNS imaging abnormalities seen in a group of children with ECDS with/without PRS; and (ii) identify clinical predictors for the development of these MRI abnormalities.
Methods
Patients
This was a retrospective case notes review collating anonymised clinical information obtained as part of routine clinical care. Diagnosis was made in patients with cutaneous symptoms consistent with the clinical descriptions of ECDS/PRS: lateral forehead scleroderma with/without hemifacial atrophy. We used electronic institutional clinical records to identify all patients with a diagnosis of ECDS or PRS seen at Great Ormond Street Hospital (GOSH), London, between January 2000 and December 2018. The demographic, clinical, and laboratory characteristics recorded were as follows: sex; age; age at diagnosis; ethnicity (established via the information provided by patients/parents upon registering with the hospital); neurological manifestations; other system involvement; presence of serum autoantibodies; cerebrospinal fluid (CSF) examination; and electroencephalogram (EEG). Therapies used were also recorded.
Neuroimaging acquisition and review
All patients had undergone basic magnetic resonance (MR) imaging including axial T2-weighted imaging, coronal fluid-attenuated inversion recovery (FLAIR) imaging, sagittal T1-weighted imaging and axial diffusion weighted imaging (DWI). All patients with MR imaging received gadolinium contrast on at least one follow up scan. Computed tomography (CT) or MR susceptibility weighted imaging (SWI) was available for the majority of patients to assess dense mineralisation.
MR imaging was performed at diagnosis in 13 patients, and 7 years after diagnosis in one patient without neurological manifestations. Follow up imaging was performed to assess for resolution or progression of imaging abnormalities at 6–12 month intervals and when there was acute neurological symptomatology.
Two neuroradiologists (KM and FK) blinded to the clinical status of the patients reviewed the imaging for the following specific abnormalities: white matter hyperintensities on T2-weighted imaging/FLAIR; calcification on CT/SWI; cerebral atrophy on T1-weighted imaging; and leptomeningeal enhancement post contrast administration. Any additional findings that were relevant were also recorded. Sequential imaging, where available, was also reviewed and assessed for white matter signal and calcification changes (stability, progression, improvement, and mixed responses).
Statistical analysis
Descriptive statistics were used: continuous variables were summarised as median and range; categorical variables were presented as frequencies and percentages. Parameters between groups were compared using the Fisher’s exact test. A two-sided p value < 0.05 was considered statistically significant. Statistical analysis was done with IBM SPSS statistics version 21.
Results
Demographics
A total of 14 patients were included in the study: 6 (43%) males and 8 (57%) females; median age 14 years (range 3–20 years). A total of 9/14 (64%) were white British, one was Pakistani, one was Indian, one was white Irish, and 2/14 (18%) were of unknown ethnic origin. Median age at the time of initial diagnosis was 7 years (range 5 months–14 years), with a median time from disease onset of 5 years (range 1–12 years). Six patients had ECDS alone; 8 patients had ECDS with PRS; and none had PRS alone.
Additional clinical features
A summary of clinical features additional to the cutaneous manifestations, related or unrelated to ECDS/PRS, is provided in Table 1.
Laboratory and other investigations
Antinuclear antibodies were positive in 7/12 (58% of patients tested; range 1:160 to 1:2560). Anti-smooth muscle antibodies were positive in 1/5 (20%); anti-rheumatoid factor antibodies were positive in 1/6 (17%); and anti-glutamic acid decarboxylase (GAD) antibodies were positive in 1/1. All other autoantibodies were negative in all patients tested (Supplementary Table S1).
CSF examination was performed in 3 patients: 2 were found to have normal levels of cells and protein; one patient was found to have positive CSF oligoclonal bands and low 5-methyltetrahydrofolate neurotransmitter (51 nmol/l, normal range 72–172).
EEG was performed in 5/6 patients with seizures. Four/5 (80% of EEGs) had documented abnormalities: all displayed intermittent slowing of the left posterior region; 3/4 (75%) displayed epileptiform activity, although this was only associated with a clinical event in one patient.
Imaging abnormalities and neurological manifestations
MRI abnormalities were noted in 2/6 ECDS and 5/8 ECDS/PRS patients (Table 2). A combined analysis of the imaging findings is presented (Tables 3 and 4; Fig. 1).

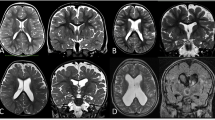
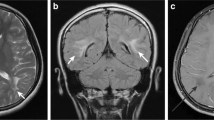
Magnetic resonance imaging (MRI) abnormalities in patients with Parry-Romberg syndrome and en coup de sabre. a-f T2-weighted and FLAIR MRI images showing enhanced white matter signalling. g-h T2-weighted MRI image showing porencephaly. i T2-weighted gradient echo MRI image showing multiple cavernomata. j T2-weighted gradient echo MRI image showing dystrophic calcification. k T1-weighted gadolinium enhanced MRI image showing leptomeningeal enhancement along the medial and lateral surfaces of the left frontal lobe. l T2-weighted MRI image showing sulcal crowding on the right
Six/14 patients demonstrated white matter signal abnormality which regressed over time in 3 cases, progressed in 2, and showed a mixed response in one where the signal improved in parts and progressed in other parts. One of these patients was found to have porencephalic dilatation of the left lateral ventricle at baseline along with ipsilateral Wallerian degeneration. These abnormalities were suggestive of an early life (potentially antenatal) insult, and developed into progressive hemispheric atrophy over time.
An unusual pattern of sulcal crowding subjacent to the morphea was shown in 3 of the 8 PRS patients. Whilst this is not typical of cortical malformation per se, the possibility of dysgyria cannot be excluded.
The presence of dystrophic calcification could be assessed in 6/7 patients with imaging abnormalities and was present in 5. Calcification was noted to be in parenchymal and/or leptomeningeal locations. Progressive calcification was demonstrable in 3 of these 5 cases.
Contrast enhancement was also assessed in 6/7 patients with imaging abnormalities and was found in 5 patients, all of whom also had dystrophic calcification. Contrast enhancement was noted to be in leptomeningeal locations except in one case where it was present surrounding the associated finding of multiple cavernomata.
All patients with neuroradiological findings had abnormalities ipsilateral to the scalp morphea, apart from the patient with cavernomata in whom the findings were bilateral.
Seven/14 (50%) displayed no imaging abnormality, despite some having neurological symptoms including headaches (n = 2) and seizures (n = 1); and psychiatric symptoms (n = 2).
Of the 7/14 (50%) with abnormal imaging, neurological symptoms included: seizures (n = 5); headaches (n = 2); cranial nerve palsy (n = 2); transient ischaemic attack (n = 1); myoclonus (n = 1); hyperaesthesia (n = 1); gait disturbance (n = 1); and psychiatric disorder (n = 2).
The only MRI abnormality in the 5 patients without neurological symptomatology was sulcal crowding in one patient.
Out of the 10 patients with neurological and/or psychiatric manifestations, 6 had abnormal imaging, all of which had follow up scans. There was no apparent correlation between changes to white matter signal and seizures over time.
Demographic, clinical and laboratory predictors of MRI brain abnormalities in ECDS/PRS
There were no associations identified between an abnormal MR brain scan and any demographic, clinical, or laboratory parameters (Table 3). The presence of neurological disease (p = 0.031), and specifically seizures (p = 0.026), were associated with the presence of ipsilateral enhanced white matter signalling on MR brain imaging (Table 4).
Treatment
The treatment received included: corticosteroids (30 mg/kg/day of IV methylprednisolone given as 3 pulsed doses, followed by 2 mg/kg/day oral prednisolone weaned over 3–6 months) in all patients; methotrexate (15 mg/m2/week subcutaneous) in all patients except one; and mycophenolate mofetil (600 mg/m2 twice a day oral) in 5 patients. The 5 patients with seizures received a variety of different antiepileptic medications.
Discussion
In this retrospective case series of 14 patients with ECDS/PRS we have characterised the MR brain abnormalities in ECDS/PRS with onset in childhood. A total of 50% of patients displayed MR brain abnormalities with the typical findings being white matter signal abnormality (43%), dystrophic calcification (36%), leptomeningeal enhancement (29%), and sulcal crowding (21%). We have shown that the development of seizures is significantly associated with ipsilateral enhanced white matter signalling on MRI brain (p < 0.05). Prospective studies are now needed to establish the relationship between the detection of MR brain abnormalities and the clinical course of these patients to determine whether escalation of anti-inflammatory treatment leads to prevention of neurological deterioration, such as the development of seizures.
A comparison between our data and those from previous small case series on paediatric ECDS/PRS is present in Table 5 [9,10,11]. Our observations are consistent with previous studies in adults showing that white matter hyperintensities were the most commonly observed lesion in patients with ECDS/PRS and an abnormal MRI scan [8]. The exact pathophysiology of these focal white matter hyperintensities remains unclear, but it is likely they represent areas of inflammation, emphasizing their clinical relevance [12]. In line with previous studies, we have also observed a lack of MRI changes in some patients despite the presence of neurological symptoms [4]. We suggest this observation is clinically important since it emphasizes that a normal brain MRI does not exclude potentially reversible CNS involvement associated with ECDS/PRS. Better neuroimaging techniques that are capable of identifying the underlying pathological mechanisms (inflammatory and thrombotic) are needed to facilitate the diagnosis of CNS involvement in these diseases, particularly since treatment delay can be associated with high morbidity. Quantitative MRI techniques such as magnetic resonance spectroscopy (MRS), magnetisation transfer imaging (MTI), diffusion-weighted imaging (DWI), and perfusion-weighted imaging (PWI), as well as nuclear imaging techniques, such as single-photon emission computed tomography (SPECT), may have a role in detecting changes in ECDS/PRS, and could facilitate earlier diagnosis. The clinical relevance of these approaches therefore requires further validation in ECDS/PRS.
Histological data were not available for our study as brain biopsy is rarely performed in these cases due to the invasiveness of the procedure. Adult studies have suggested, however, that most of the CNS lesions in ECDS/PRS are likely inflammatory, either only affecting the parenchyma, or also the CNS vasculature causing vasculitis [12]. Interestingly, on the basis of their clinical presentation with severe epilepsy and the indication of an inflammatory process on brain biopsy, some patients with PRS have been diagnosed with a variant of Rasmussen encephalitis [13].
We detected no association between the presence of an abnormal MRI brain and specific demographic, clinical, or laboratory parameters. We have, however, identified an association between white matter hypersensitivities and seizures, albeit in a smaller exploratory analysis. Future larger studies are now needed to firmly establish the relationship between clinical endophenotypes and the risk of CNS inflammation and association with detectable MRI findings.
Our study is limited by all the usual caveats around small and largely descriptive retrospective case studies, an unavoidable situation since ECDS/PRS is so rare in the young. We used standardised protocols for conventional MRI sequences (such as T1-weighted [with and without contrast agents], T2-weighted, and FLAIR images), as utilised for routine care in our institution for at least 10 years. Further characterisation of the lesions with more recently introduced techniques and gadolinium enhancement may have increased the MRI detection yield. Detailed analysis of imaging findings of patients with PRS without ECDS would be interesting and needs to be addressed in future studies. It also remains challenging to determine whether the psychiatric manifestations we describe are related to the burden of chronic illness or are a true manifestation of CNS inflammation. Lastly, the relationship between skin disease activity and the development of MRI abnormalities needs to be assessed in future studies.
Conclusions
In summary, we observed several distinct radiographic patterns associated with ECDS/PRS but, importantly, some patients with neurological symptoms displayed no MRI brain abnormalities. Seizure disorder was strongly associated with the presence of enhanced white matter signalling ipsilateral to the cutaneous disease. Improved neuroimaging techniques that combine morphological with functional imaging may improve the detection rate of brain involvement in children with ECDS/PRS in the future.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Careta MF, Romiti R. Localized scleroderma: clinical spectrum and therapeutic update. An Bras Dermatol. 2015;90(1):62.
Tollefson MM, Witman PM. En coup de sabre morphea and parry-Romberg syndrome: a retrospective review of 54 patients. J Am Acad Dermatol. 2007;56(2):257–63.
Tolkachjov SN, Patel NG, Tollefson MM. Progressive hemifacial atrophy: a review. Orphanet J Rare Dis. 2015;10:39.
Stone J. Parry-Romberg syndrome: a global survey of 205 patients using the internet. Neurology. 2003;61(5):674.
Cory RC, Clayman DA, Faillace WJ, McKee SW, Gama CH. Clinical and radiologic findings in progressive facial hemiatrophy (parry-Romberg syndrome). Am J Neuroradiol. 1997;18(4):751–7.
Zulian F, Culpo R, Sperotto F, Anton J, Avcin T, Baildam EM, et al. Consensus-based recommendations for the management of juvenile localised scleroderma. Ann Rheum Dis. 2019;78(8):1019.
Moko SB, Mistry Y. Blandin de Chalain TM. Parry-Romberg syndrome: intracranial MRI appearances. J Cranio-Maxillofacial Surg. 2003;31(5):321–4.
Amaral TN, Peres FA, Lapa AT, Marques-Neto JF, Appenzeller S. Neurologic involvement in scleroderma: a systematic review. Semin Arthritis Rheum. 2013;43(3):335.
Chiu YE, Vora S, Kwon EKM, Maheshwari M. A significant proportion of children with morphea en coup de sabre and parry-Romberg syndrome have neuroimaging findings. Pediatr Dermatol. 2012;29(6):738–48.
Doolittle DA, Lehman VT, Schwartz KM, Wong-Kisiel LC, Lehman JS, Tollefson MM. CNS imaging findings associated with parry–Romberg syndrome and en coup de sabre: correlation to dermatologic and neurologic abnormalities. Neuroradiology. 2014;57(1):21.
Maloney E, Menashe SJ, Iyer RS, Ringold S, Chakraborty AK, Ishak GE. The central nervous system manifestations of localized craniofacial scleroderma: a study of 10 cases and literature review. Pediatr Radiol. 2018;48(11):1642.
Stone J, Franks AJ, Guthrie JA, Johnson MH. Scleroderma “en coup de sabre”: pathological evidence of intracerebral inflammation. J Neurol Neurosurg Psychiatry. 2001;70(3):382.
Paprocka J, Jamroz E, Adamek D, Marszal E, Mandera M. Difficulties in differentiation of parry-Romberg syndrome, unilateral facial sclerodermia, and Rasmussen syndrome. Childs Nerv Syst. 2006;22(4):409.
Acknowledgements
We thank all the doctors, nurses, and health care staff involved in the management of patients with ECDS and PRS at Great Ormond Street Hospital for Children NHS Foundation Trust.
Funding
Dr. Eleftheriou was supported by Versus Arthritis (grant 20164 and 21593). Dr. Eleftheriou also acknowledge support from Great Ormond Street Hospital Children’s Charity. All research at Great Ormond Street Hospital NHS Foundation Trust and UCL Great Ormond Street Institute of Child Health is made possible by the NIHR Great Ormond Street Hospital Biomedical Research Centre. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Author information
Authors and Affiliations
Contributions
DE, KM, LS, and MO conceived the presented idea. Data collection was performed by HK, EM, FK, KM, and DE. Data analysis was performed by HK, KM and DE. All authors wrote and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethical approval was given by the Institute of Child Health/ Great Ormond Street Research Ethics Committee for a retrospective case notes review; since this was a retrospective case note review using de-identified data, written consent from patients was not required.
Consent for publication
Since this was a retrospective case note review using de-identified data, written consent from patients was not required.
Competing interests
All authors declare no conflicts of interest relating to this work.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
Other autoantibodies tested.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Knights, H., Minas, E., Khan, F. et al. Magnetic resonance imaging findings in children with Parry-Romberg syndrome and en coup de sabre. Pediatr Rheumatol 19, 42 (2021). https://doi.org/10.1186/s12969-021-00512-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12969-021-00512-6