
Abstract
Natural killer (NK) cells are unique from other immune cells in that they can rapidly kill multiple neighboring cells without the need for antigenic pre-sensitization once the cells display surface markers associated with oncogenic transformation. Given the dynamic role of NK cells in tumor surveillance, NK cell-based immunotherapy is rapidly becoming a "new force" in tumor immunotherapy. However, challenges remain in the use of NK cell immunotherapy in the treatment of solid tumors. Many metabolic features of the tumor microenvironment (TME) of solid tumors, including oxygen and nutrient (e.g., glucose, amino acids) deprivation, accumulation of specific metabolites (e.g., lactate, adenosine), and limited availability of signaling molecules that allow for metabolic reorganization, multifactorial shaping of the immune-suppressing TME impairs tumor-infiltrating NK cell function. This becomes a key barrier limiting the success of NK cell immunotherapy in solid tumors. Restoration of endogenous NK cells in the TME or overt transfer of functionally improved NK cells holds great promise in cancer therapy. In this paper, we summarize the metabolic biology of NK cells, discuss the effects of TME on NK cell metabolism and effector functions, and review emerging strategies for targeting metabolism-improved NK cell immunotherapy in the TME to circumvent these barriers to achieve superior efficacy of NK cell immunotherapy.
Similar content being viewed by others


Introduction
NK cells are cytotoxic lymphocytes of the innate immune system, accounting for approximately 15% of all circulating lymphocytes, and their activation is driven by a balance between activating and inhibitory signals, therefore, they do not need to be sensitized beforehand to lysate target cells and thus exert antitumor effects [1]. Activated NK cells induce metabolic changes and drive effector functions including the release of cytolytic particles containing perforin and granzymes or the killing of infected or transformed cells by ligating death-inducing receptors (TRAIL, FasL). They also contribute to the development of adaptive immune responses by producing various chemokines and pro-inflammatory cytokines [2, 3]. NK cells are therefore a valuable therapeutic tool in cancer immunotherapy and a variety of NK cell-based immunotherapies have been developed, some of which have been translationally applied in the clinic [4, 5]. Encouraging efficacy has been shown in hematologic malignancies (e.g., myeloid leukemia, chronic lymphocytic leukemia, and acute lymphoblastic leukemia), but efficacy in the treatment of solid tumors is usually poor [6]. One of the main limiting factors is immunosuppressive TME. The function, phenotype, activation, and persistence of tumor-infiltrating NK cells are impaired in nutrient-depleted, hypoxic, acidic TME, even leading to NK cell dysfunction or exhaustion. Consider that cellular metabolism provides the energy and biosynthetic requirements of cells to support their effector functions. We focused on the effects of inhibitory TME on energy expenditure and metabolic reprogramming of NK cells, where understanding and regulating NK cell metabolism is critical to optimize the efficacy of current and future NK cell immunotherapies. Here, we summarize the metabolic biology of NK cells, discuss the effects of TME on NK cell metabolism and effector functions, and review emerging strategies for targeting metabolic improvement of NK cell immunotherapy in the TME.
Immune metabolism of NK cells
Biological characteristics of NK cell metabolism
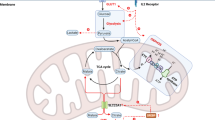
Cell destiny can be determined by metabolic flux. Different developmental stages of NK cells exhibit distinct metabolic fluxes. Developing mouse NK cells undergo several stages of maturation in the bone marrow and can be recognized based on CD11b and CD27 expression levels. They are mainly categorized into immature stage CD11blowCD27hi, CD11blowCD27hi NK cells, intermediate stage CD11b hiCD27 hi NK cells and final developmental maturation stage CD11bhiCD27low NK cells [7]. NK cells' metabolic activity varies dynamically during development, and as they continue to differentiate and mature, they become less dependent on glycolysis and cellular glucose uptake [8]. Whereas immature NK cells express the amino acid transporter CD98 and the transferrin receptor CD71, which gives them increased metabolic activity to sustain their proliferation [8]. It has been discovered that mTOR, a metabolic regulator, plays a crucial role in the integration of NK cell growth and metabolism. In mTOR-deficient mice, NK cell development is halted in the bone marrow at the CD11bhiCD27hi stage, and NK cells are almost nonexistent in peripheral organs [8]. Mature, differentiated CD11bhiCD27low NK cells arrest and enter a resting state. Resting mouse and human NK cells have similar low baseline metabolic rates, maintaining low levels of glycolysis and oxidative phosphorylation (OXPHOS), according to several metabolic profile investigations on the cells [9,10,11]. Resting mouse NK cells have a low basal metabolic rate and maintain low levels of glycolysis and OXPHOS, and the metabolic activity of NK cells does not increase significantly under short bursts of cytokine stimulation or receptor signaling. However, this low metabolic rate is important for maintaining an acute NK cell response, and inhibition of OXPHOS or glycolysis would result in almost complete elimination of interferon γ (IFN-γ) produced by receptor stimulation [12]. Elevated OXPHOS levels are seen in human NK cells activated with short-term IL-12/15 or IL-2 cytokines; these levels are crucial for effective ATP production, which is needed to activate human NK cell role [11]. Both human and murine NK cells respond to prolonged and sustained cytokine stimulation by strong metabolic changes that meet the energy requirements for NK cells to carry out their effector functions. These changes include increases in mitochondrial mass, glycolytic enzyme expression, key nutrient transport proteins (e.g., SLC2A1 and SLC1A5), and glycolysis rates and OXPHOS ratios [9, 11, 13]. The timescales of metabolic reprogramming in human and mouse lymphocytes, however, may differ, according to recent research, with human lymphocytes demonstrating longer metabolic reprogramming times—this is proven for T lymphocytes, but more evidence is required for NK cells [14]. The aforementioned results direct our emphasis toward the topic of how NK cells maintain increased levels of glycolysis and OXPHOS by reprogramming cellular metabolic pathways in response to cytokine stimulation. Glucose, the primary fuel for activated NK cells, is metabolized by aerobic glycolysis in the cytosol to pyruvate, which then generates lactate. Unlike other lymphocytes that metabolize pyruvate through the tricarboxylic acid (TCA) cycle, NK cells metabolize pyruvate across the mitochondrial membrane via the Citrate-malate shuttle (CMS) (Fig. 1), which drives the production of OXPHOS and adenosine triphosphate (ATP) [15], a unique metabolic conformation that is regulated by the sterol regulatory element binding protein (SREBP) transcription factor. It controls the expression of two key genes of the CMS: the ATP citrate lyase (ACLY) and the citrate-malate reverse transporter protein SLC25A1. Thus, SREBP activity is crucial for metabolic reprogramming and obtaining higher glycolysis and OXPHOS in NK cells [15]. Furthermore, unlike other types of lymphocytes, NK cells do not use glutamine as a fuel to drive OXPHOS [13].

Citrate-Malate Shuttle (drawed by Figdraw)
NK cells express a glucose transporter through which glucose is taken up into the cytoplasm (Glut1). Glucose is metabolized through glycolysis to pyruvate, which is enzyme-catalyzed to lactate and transported outside the cell via the monocarboxylic acid transporter protein (MCT). Some of the pyruvate enters the mitochondria, where most of it is further metabolized via the citrate-malate shuttle (CMS), and only a relatively small amount is metabolized via the tricarboxylic acid cycle (TCA). Pyruvate in the mitochondria is metabolized to Acetyl CoA, which produces NADH. Acetyl CoA binds to oxaloacetate to form citrate, which is then transported out of the mitochondria via the citrate transporter protein (SLC25A1). Citric acid in the cytoplasm is catalyzed by ATP citrate lyase to produce Acetyl CoA and oxaloacetate, which further generates NAD + , which serves as an essential cofactor for the glycolytic enzyme 3-phosphoglyceraldehyde dehydrogenase to maintain glycolysis. Oxaloacetate is converted to malate in the NAD + generating reaction and re-enters the mitochondria via SLC25A1, where it is converted back to oxaloacetate and NADH is produced in the mitochondria. Oxaloacetate can complete the cycle by reacting with another glucose-generating Acetyl CoA to form another citrate. NADH generated by CMS and TCA enters the electron transport chain (ETC), driving OXPHOS and efficient ATP production. In addition, NK cells activated under the stimulation of cytokines (IL15, IL-2, IL-12, IL-18) exhibit a significant increase in glycolysis rate, OXPHOS rate, mitochondrial mass, and metabolic flux to provide the energy support required for NK cell growth and effector molecule synthesis.
Metabolic characteristics of different subpopulations of NK cells
Human blood NK cells are divided into two main subpopulations: the CD56bright and CD56dim NK cells. CD56dim cells have higher cytotoxicity compared to CD56bright cells, whereas CD56bright NK cells are the main producers of cytokines, including IFN-γ and tumor necrosis factor-α (TNF-α) [1, 16]. The two have different metabolic profiles, with CD56bright cells being more metabolically active than CD56dim cells upon IL-2 or IL-12 stimulation. They preferentially upregulate nutrient receptors and show higher rates of glucose uptake. CD56bright cells require elevated levels of OXPHOS to support cytotoxicity and IFN-γ production in all NK cells [11]. In addition, human NK cells from blood and tissue-resident (e.g., spleen and liver) exhibit different metabolic profiles, and although tissue-resident NK cells also increase trophic receptor expression following stimulation, this increase is less than that of blood NK cells [17]. Human decidual NK cells are a unique type of tissue-resident NK cells. Integration of metabolomics and proteomics data revealed that 29 metabolites involved in the metabolism of glycerophospholipids and glutathione were significantly reduced in decidual NK cells compared to peripheral blood NK cells. In addition, decidual NK cells have an altered redox balance and tend to have more ROS, which may be associated with their reduced cytotoxicity [18].
NK cell metabolism in pathological states
Evidence suggests that NK cell metabolism is altered in chronic diseases such as cancer, obesity, and viral infections, which are key contributors to NK cell dysfunction. In obesity, NK cells take up lipids from the environment, which interferes with their cellular bioenergetics, leading to metabolic paralysis and a greatly reduced metabolic rate. This metabolic dysfunction is associated with peroxisome proliferator-activated receptor (PPAR)-driven lipid accumulation in NK cells, and PPARα/δ target genes are highly up-regulated in obesity, which inhibits mTOR-mediated glycolysis as well as downstream transcription of cytotoxic particles and IFN-γ production. In addition, obesity severely impedes the ability of NK cells to direct cleaved particles to tumor cells and degranulate them at the synapse, which is critical for NK cells to exert cytotoxicity and thus effectively kill tumors [19]. Glucose metabolism is vital for NK cell-mediated control of Mouse Cytomegalovirus (MCMV) infection, and when aerobic glycolysis is inhibited by cell-specific deletion of Lactate Dehydrogenase A (LDHA), the NK cell's ability to generate a robust effector and memory response to MCMV infection is dramatically diminished [20, 21].
How TME affects metabolic reprogramming of NK cells
TME refers to the surrounding microenvironment in which tumor cells exist, including surrounding blood vessels, immune cells, fibroblasts, bone marrow-derived inflammatory cells, various signaling molecules, and extracellular matrix (ECM) (Fig. 2) [22, 23]. The immune function of the TME can be shaped by competition for nutrients in the TME, inhibitory effects of accumulated metabolites, and signals that limit metabolic reorganization [24].

Tumor Microenvironment (drawed by Figdraw). The tumor microenvironment (TME) is the surrounding microenvironment in which tumor cells exist, including the surrounding vasculature, extracellular matrix (ECM), fibroblasts, myeloid-derived inflammatory cells, immune cells, and a variety of cytokines and signaling molecules. Cytokine signaling molecules released by immune cells infiltrated in the TME are involved in mediating immune-suppressive and/or immune-responsive events. DC Dendritic cell, NK CELL Natural killer cell, MDSC Myeloid-derived suppressor cell, CAF Cancer-associated fibroblast, TAM Tumor-associated macrophage, IL Interleukin, TNF-α Tumor necrosis factor-α, CXCL Chemokine, VEGF Vascular endothelial growth factor, TGF-β Transforming growth factor-β, Arg-1 Arginine-1, iNOS Inducible nitric oxide synthase, ROS Reactive oxygen species
Hypoxia and its metabolite buildup
Hypoxia
In rapidly growing and expanding tumors, the overall TME is characterized by hypoxia due to the increase in oxygen consumption and diffusion caused by proliferation and the incomplete internal vascular system of the tumor tissues, resulting in an insufficient supply of oxygen within the tumor tissues. In advanced cancer patients, hypoxic stress shapes NK cells into a tumor-resistant and immunosuppressive phenotype. A way for cells to adapt to hypoxia is to upregulate the protein expression of hypoxia-inducible factor 1α (HIF-1α). Under normoxia conditions, HIF-1α is thought to be dispensable for glycolysis and NK cell metabolic activation, as HIF1α-deficient mouse NK cells have normal levels of glycolysis and OXPHOS responses to IL2 + IL12 cytokine stimulation [13]. However, under hypoxic conditions, HIF-1α appears to be a central player in the activation of NK cell glycolysis. Stable expression of HIF-1α during hypoxia promotes the metabolic shift from OXPHOS to glycolysis in NK cells, overcoming hypoxia-mediated apoptosis and tumor cytotoxicity damage in NK cells maximizing NK cell effector function [25,26,27]. Notably, researchers have emphasized the importance of pre-cytokine such as IL-2 stimulation on the stable expression of HIF-1α in NK cells, which seems to provide new evidence to achieve high-functioning NK cells in hypoxic microenvironments for adoptive cell therapy [26, 27]. Furthermore, the HIF-1α metabolic pathway in resting NK cells differs from that in activated NK cells, where HIF-1α is a regulator of tryptophan metabolism and cellular nicotinamide adenine dinucleotide (NAD) levels, which prevents the generation of ROS during OXPHOS, thus blocking DNA damage and apoptosis of NK cells under homeostatic conditions [25]. Nevertheless, the question of whether HIF-1α promotes or impairs NK cell effector functions remains a controversial issue. Available evidence suggests that the absence of HIF-1α in NK cells is also beneficial, although this results in attenuated cytotoxicity while showing increased bioavailability of the angiogenic cytokine vascular endothelial growth factor (VEGF), which stimulates non-productive angiogenesis and thus inhibits tumor growth [28]. In addition, Ni et al. found that inhibition of HIF-1α released the antitumor activity of NK cells by establishing single-cell RNA sequencing in mice with conditional targeting deficiency of HIF-1α in NK cells, as evidenced by the inhibition of tumor growth, activation of markers, elevation of expression of the effector molecule IFN-γ, and enrichment of the NF-κB pathway in tumor-infiltrating NK cells [29]. HIF-1α regulation is complex, and the effects of hypoxia and HIF-1α may depend on microenvironmental conditions, which require further study.
Adenosine accumulation
Moreover, extracellular adenosine (eADO) buildup is brought on by hypoxia. Cancer cells release large amounts of ATP in hypoxic environments within tumors. These ATP molecules are then converted to adenosine (ADO) by the exonucleotidase enzymes CD39 and CD73, and ultimately the ADO is expelled externally [30, 31]. It has been suggested that the current purinergic signaling effect on the immune response is a balance between the pro-inflammatory, immunosurveillance effects of extracellular adenosine triphosphate (eATP) and the anti-inflammatory, immunosuppressive effects of eADO [32]. Exonucleotidase enzymes are more abundant in solid tumors than in non-malignant tissues, which promotes the hydrolysis of eATP to eADO [30]. G protein-coupled receptors A1, A2A, A2B, and A3 bind to eAOD to initiate downstream signaling pathways. A2A and A2b receptors bind to eAOD to inhibit immune cell activation by producing intracellular cyclic adenosine monophosphate (cAMP) and activating protein kinase A (PKA) during the subacute inflammatory phase that follows tissue damage [31]. The ability of IL-2/nkp46-activated NK cells to produce IFN-γ, tumor necrosis factor α (TNF-α), and macrophage colony-stimulating factor is dramatically inhibited by A2A-CAMP-PKA signaling, which in turn influences cytotoxicity [33]. Furthermore, it appears that ADO signaling has a multipathway effect on the ability of NK cells to kill. It has been discovered that ADO and its analogs hinder the cleavage and lysis of IL-2-activated NK cells by interfering with the killing pathways mediated by FAS ligand and perforin, which are accompanied by increased levels of cAMP [34]. Increased ATP hydrolysis was seen as a result of dysregulated CD39 and CD73 cell surface expression on non-malignant T and NK cell populations in an in vivo study of sezary syndrome. The effective killing of ADCC by NK cells and the restoration of non-malignant CD4 + and CD8 + T cell proliferation can both be achieved through inhibition of the CD39/CD73/ADO pathway [35]. Interestingly, NK cells show the highest amounts of A2A receptor expression, even though A2A receptors are found on the majority of immune cells [36]. Research has revealed that the A2A receptor functions as a checkpoint to restrict NK cell development by directly impeding NK cell maturation in the TME. The percentage of terminally developed NK cells in the TME can rise when the A2A receptor is conditionally deleted [37]. Thus, NK cell effector function may be restored and immune surveillance may be improved by targeting ADO-related signaling molecules in the purinergic signaling cascade.
Acidic TME shaped by glucose restriction and its metabolic end products
Glucose limitation
Most tumors are characterized by a high degree of glycolysis, even in the presence of oxygen, and cancer cells are thought to rely more on glycolysis than mitochondrial OXPHOS for energy production, i.e., aerobic glycolysis or the "Warburg effect". High glucose consumption in cancer cells leads to low levels of glucose available for TME and a lack of fuel for NK cell activation resulting in metabolic limitation, which affects NK cell activation, impairs IFN-γ production, and blunts the NK cell antitumor response [11, 38]. However, whether extrinsic cellular competition for nutrients or intrinsic cellular reprogramming leads to metabolic dysregulation of tumor-promoting immune cells remains a controversial issue. The study by Bradley et al. challenged the hypothesis that cancer cells induce immune cell nutrient deficiencies and dysfunction in the TME by competing for glucose with the TME. Their findings suggest that glucose is preferentially utilized by tumor-associated immune cells, in contrast to cancer cells that exhibit the greatest uptake of glutamine. Different cells in the TME have different nutrient uptake programs, which are cell-intrinsically programmed through mTORC1 signaling as well as glucose- and glutamine-related gene expression. This interfered with glutamine metabolism in cancer cells, which increases glucose uptake to generate energy using glutamine metabolism as a specific strategy to impede cancer cell growth while increasing glucose consumption and consequently altering the immune phenotype in TME [39].
LDH-derived lactic acid accumulation
Lactate dehydrogenase (LDH) is a key enzyme in glycolysis, and it catalyzes the reversible conversion of pyruvate to lactic acid and NADH to NAD, the last step in the glycolytic process [40] (Fig. 1). High serum LDH levels are frequently linked to a bad prognosis in a variety of cancer types, such as melanoma, bladder, nasopharyngeal, pancreatic, non-small-cell lung carcinoma, etc. [41,42,43,44,45]. Elevated serum LDH levels were linked to a lower chance of survival for solid tumors, specifically prostate cancer, renal cell carcinoma, and melanoma, according to a meta-analysis of 76 studies [46]. The potential of variations in serum LDH levels as biomarkers to forecast treatment outcomes and dynamically monitor therapy response has also been emphasized by several clinical investigations. In metastatic colorectal cancer, for instance, serum LDH can function as a measure of tumor angiogenesis activation, hence predicting the effectiveness of bevacizumab in suppressing angiogenesis [47]. LDH seems a biomarker for the anti-PD-1 immunotherapy result in esophageal squamous cell cancer [48]. Overexpression of LDH in tissues supports a range of malignant biological behaviors of tumor cells, including the epithelial-to-mesenchymal transition (EMT) [49], promotion of cell invasion and migration [50,51,52], angiogenesis [53], and cytoskeletal remodeling [54]. These findings are supported by numerous in vitro and in vivo experiments. It is also associated with resistance to anticancer therapies, and it has been found that impeding glycolytic metabolism in cancer cells through LDH inhibition can overcome resistance to chemotherapeutic agents [55,56,57,58,59,60]. Notably, serum LDH levels do not always correspond with the level of LDH in tumor tissues, and it is unclear if elevated serum LDH levels are a result of tumor cell leakage or the rapid growth of metastases destroying nonmalignant tissues [61]. Still, tissue LDH levels and serum LDH levels can cooperate and complement one another.
One of the features of TME is the high enrichment of lactate, and LDHA has a considerable ability to affect immunity in TME by boosting lactate production. On the one hand, tumor-associated fibroblasts, the primary stromal cells in TME, use lactate produced by LDH as fuel. LDH facilitates the mutual nutrition exchange between stromal and tumor cells and encourages the growth of tumors [62]. Lactate, on the other hand, makes the TME acidic. Acidic TME inhibits the antitumor immune response, supports Treg cell differentiation and tumor-associated macrophage polarization toward M2, encourages the recruitment of myeloid suppressor cells, and prevents dendritic cell maturation, limits NK cell toxicity, which suppresses intrinsic and acquired immunity [61]. NK cells are affected by high TME lactate levels as follows: Lactate is a metabolic end product of glycolysis, and its high enrichment is one of the characteristics of the TME, which is increasingly being studied in closely related to the immunosuppressive microenvironment. Lactate causes acidification of TME, and lactate uptake by NK cells leads to intracellular acidification and impaired energy metabolism. 15 mM lactate has completely blocked IFN-γ production, and lactate concentrations higher than 20 mM led to apoptosis of T cells and NK cells [63]. Similar results were obtained in liver-resident NK cells treated with lactic acid [64]. Ge et al. verified in vivo and in vitro experiments in a pancreatic cancer model that the SIX1/LDHA axis promotes the accumulation of tumor lactate and thus inhibits the function of NK cells [65]. In addition, lactate-induced acidification inhibits the nuclear factor of activated T-cells (NFAT) for transcription, an important transcription factor involved in the transcriptional control of IFN-γ, which likewise leads to a decrease in IFN-γ production by NK cells [63]. Elevated lactate levels not only directly limit the cellular function of NK cells, but also indirectly inhibit NK cells by increasing the number of MDSCs.
Decreased available amino acids
Arginine
Tumor cells consume large amounts of amino acids, which synergize with tumor-associated cells to create a nutritionally depleted and immunosuppressive TME. Additionally, elevated catabolism of tryptophan and arginine (Arg) is a common hallmark of TME [66]. Previous data suggest that Arg plays a critical role in T cell activation and proliferation, and is also required for optimal proliferation of NK cells [67, 68]. Arg depletion reduces NK cell proliferation and IFN-γ production while decreasing NK ζ-chain expression inhibits activation signaling to control NK cell cytotoxicity [69]. A recent study indicated that mitochondria-derived damage-associated molecular pattern (mitoDAMP) inhibited NK cell-mediated cytotoxicity, IFN-γ production, T cell proliferation, and in vivo activation of antiviral T cells. Mass spectrometry analysis of mitoDAMP revealed enrichment of Arg and its enzymatic activity products, and further addition of Arg or arginase inhibitors could reverse the inhibitory effect of MitoDAMP preparations. Further supporting that Arg depletion is responsible for the altered immune polarity [70]. However, it has also been reported that NK cell granule exocytosis and cytotoxicity are not related to extracellular Arg [71]. Although the effects of Arg deprivation on NK cytotoxicity are controversial, they all point to the fact that lack of Arg impairs IFN-γ expression in NK cells through a post-transcriptional mechanism [69,70,71].
Tryptophan
Many cancer and tumor-associated cells in TME [e.g., tumor-associated macrophages (TAMs), tumor-associated dendritic cells (DCs), and fibroblasts] express increased indoleamine 2,3-dioxygenase (IDO) and tryptophan-2,3-dioxygenase (TDO), and large quantities of tryptophan are converted to kynurenine by the enzymes [72, 73]. On the one hand, this leads to tryptophan depletion and reduced availability of essential amino acids, inhibiting tumor-infiltrating lymphoid (TIL) cell responses. On the other hand, accumulation of kynurenine in TME enters TILs via the amino acid transporter protein SLC7A5 and impairs TIL proliferation and effector function [73]. In NK cells, exposure to IDO-derived kynurenine also induces apoptosis via reactive oxygen species-mediated pathways and also decreases the expression of the NK cell activation receptor NKG2D and natural cytotoxicity triggering receptor 1 (NCR1, also known as NKp46). But the limiting effect of kynurenine is selective and it does not affect the cytotoxicity effects mediated by NKp30 or CD16 [74, 75]. Inhibition of IDO may be a potential anticancer target. Silencing of IDO in ovarian cancer cells enhances cancer cell sensitivity to NK cells in vitro, and in vivo and manifests as inhibition of tumor growth, reduction of peritoneal dissemination, and promotion of NK cell accumulation in the tumor stroma [76].
Glutamine
Previously, we mentioned that cancer cells consume the highest amount of glutamine, while immune cells consume the highest amount of glucose [39]. The predatory uptake of glutamine by tumor cells in the TME results in limited utilization of glutamine by immune cells. In NK cells, SLC7A5-mediated glutamine uptake is required to regulate c-MYC-dependent NK cell activation. Glutamine-deficient NK cells exhibit reduced c-MYC protein expression, growth restriction, and impaired immune function. However, the inhibition of glutamine catabolism did not affect NK cells driving OXPHOS, suggesting that although glutamine is not an essential fuel for NK cells, glutamine is important for maintaining the signaling of metabolic regulators such as MYC [13, 77]. Targeting glutamine metabolism enhances NK cell-based therapies by impairing tumor fuel supply without decreasing NK cell function.
Strategies for targeting immunometabolism to optimize NK cell immunotherapy
Targeted hypoxia and its metabolites
Targeted hypoxia
Although it is not yet clear whether the expression of HIF-1α in NK cells has a promoting or impairing effect on NK cell effector function, downregulation of HIF-1a signaling within the tumor itself can also enhance immune activity. A variety of HIF-1α inhibitors are being developed to improve cancer immunotherapy. For adoptive cellular therapy, preactivated NK cells using IL-2 can restore the killing potential of NK cells exposed to hypoxic TME. When NK cells are in an active proliferative state, the proliferative signaling of NK cells is sufficient to amplify hypoxia-induced activation of ERK1/2 and STAT3, thereby shifting to anti-apoptotic and pro-survival pathways to resist the deleterious hypoxic immunosuppressive environment [27]. In addition, dysfunctional vasculature in tumor cells contributes to hypoxia and tumor drug resistance, thus normalizing the vasculature to reduce hypoxia in the TME takes on a strong role. Treatment with a novel Fc-VEGF chimeric antibody drug (Fc-VFD) inhibits the secretion of pro-angiogenic factors VEGF-A and IL-6 by cancer cells in TME, suppressing excessive angiogenesis and overcoming hypoxia resistance in cancer cells. Single-cell RNA sequencing also revealed that it inhibited M2 macrophage polarization and increased immune cell infiltration, including cytotoxic T cells, NK cells, and M1 macrophages [78]. The use of the anti-vascular drug 5,6-Dimethylxanthenone-4-acetic Acid (DMXAA) in combination with the HIF-1α inhibitor Digoxin inhibited tumor growth and stimulated immunity in a melanoma model, demonstrating a significant increase in the percentage of CD8 cytotoxic lymphocytes and NK cells [79]. In addition, HIF-1α is associated with PD-L1, and HIF-1α binds to hypoxia-responsive elements in the PD-L1 promoter to regulate PD-L1 expression. Combination therapy with a PD-L1 antibody and a HIF-1α inhibitor may improve the immune response of patients and enhance immune efficacy [80]. Castration-resistant prostate cancer (CRPC) cells highly express PD-L1, and the cytotoxicity of NK cells against hypoxia-induced CRPC cells was enhanced by the inhibition of JAK1, STAT3 (upstream regulators of PD-L1) in combination with PD-L1 antibody [81].
Targeting adenosine
ADO has a key role in remodeling the immunosuppressive TME. The utilization of immunotherapy or inhibitors of signaling to promote immune cells against malignant tumors may be a promising therapeutic approach. Recently, a human anti-CD73 monoclonal antibody Oleclumab has completed phase I clinical trials and demonstrated a manageable safety profile when administered alone or in combination with Durvalumab and showed antitumor activity in immunotherapy-resistant tumor types [82, 83]. ARL67156, a dual inhibitor of CD39/73, enhanced the cytotoxic effects produced by NK tumor cell lines [84]. In addition, it was found that antagonism of A2A ADO receptors decreased the percentage of CD56bright NK cells and promoted the accumulation of highly cytotoxic CD56dim NK cells. This suggests that A2A receptor antagonism enhances adoptive NK cell immunotherapy [37]. There have been several reports using combinations of A2A receptor antagonists and NK cell-based therapies that can promote NK cell-mediated antitumor immunity [85,86,87,88].
Targeted glucose metabolism and its metabolic end products
Targeted glucose metabolism
In the TME where tumors consume glucose and have high rates of glycolysis, inhibition of glycolysis may be a promising therapeutic strategy to alleviate glucose deprivation and limit tumor growth in TME. Single or combination therapies with the glycolysis inhibitor 2-deoxyglucose (2-DG) have entered clinical trials [89, 90]. However, glucose is also a key fuel for the activation of NK cells, and one has to question whether targeting the glycolytic pathway would inhibit glycolysis in NK cells in the context of TME thereby impairing the ability of NK cells to kill tumor cells. The effect of 2-DG on NK cell cytotoxicity is currently unknown. One data showed decreased NK cell proliferation and cytotoxicity in CMV-infected mice treated with 2-DG [21]. In contrast, other data suggests that it is glucose starvation rather than 2-DG treatment that impairs NK cell cytotoxicity [91]. However, the preactivation of NK cells with cytokines for adoptive cell therapy may be able to overcome metabolic inhibition. It has been shown that even in the presence of 2-DG, NK cells preactivated with IL-12/15/18 exhibited higher cytotoxic activity than control NK cells. This suggests that cytokine-induced memory-like (CIML) NK cells can maintain higher anti-tumor activity under glycolysis-restricted conditions (e.g., TME) [92]. In addition to this, some inhibitors were used to assess the effect on glycolysis in NK cells. Administration of the aldose reductase inhibitor Fidarestat downregulated AKR1B10 expression in NK cells and promoted NK cell glycolysis to enhance its killing activity against hepatocellular carcinoma cells [93]. Adoptive transfer of NK cells treated with MB05032, an inhibitor of the gluconeogenic enzyme FBP1, restored NK cell glycolysis and effector functions and slowed down tumor growth in a murine lung cancer model [94].
Targeting lactate
Targeting lactate metabolism to regulate the pH of the TME is a promising approach due to the damage to NK cells caused by lactate and lactate-induced acidification of the TME. This includes both aspects of targeting lactate anabolism and targeting lactate transport. One key to the strategy of targeting lactate anabolism is LDH-A, a key enzyme that catalyzes the conversion of pyruvate to lactate during glycolysis. Several small molecule inhibitors targeting LDH-A have been applied in preclinical trials and shown to be effective in inhibiting tumor growth. These include substrate (pyruvate)-competitive inhibitors: oxalate [95]; cofactor (NADH)-competitive inhibitors: cotton phenol, quinoline 3-sulfonamides [96,97,98]; and dual competitive inhibitors (substrate and cofactor): N-hydroxy-indole (NHI) [99, 100]. Some of these molecules are in clinical trials, but no LDH-A inhibitor has yet been approved for clinical use, and the clinical utility of LDHA inhibitors may be limited by their non-selective toxicity or complex interactions with other cellular components. Second, targeting lactate transport. Cancer cells take advantage of the widespread expression of two monocarboxylic acid transporter (MCT) protein isoforms, namely, MCT1, which dominates lactate import, and MCT4, which dominates lactate export, to establish a lactate shuttle in cancer cells. The use of MCT inhibitors to block the release of lactate from cancer cells reduces TME acidification and causes lactate accumulation in cancer cells to reach toxic concentrations in tumor cells, reducing their growth and inducing apoptosis. An MCT1 inhibitor, AZD3965, is in clinical trials and modulates tumor immune cell infiltration, specifically increasing NK cell and DC cell abundance and maturation within the tumor [101, 102]. In addition, monotherapy using systemic bicarbonate buffering that neutralizes tumor acidity enhances IFN-γ expression in NK cells and increases the number of NK cells in tumor-growing lymphoid organs, and delays tumor growth in an NK cell-dependent manner [103]. However, the results of three phase I/IIa clinical trials of oral sodium bicarbonate tolerance (NCT01846429, NCT01198821, NCT01350583) suggest that it is not easy to implement mono buffered therapies in the clinic. The trials were unsustainable because patients were experiencing diminished taste and strong gastrointestinal reactions thus leading to poor compliance [104].
Targeted amino acid metabolism
Targeting arginine
Low Arg levels affect NK cell proliferation. The small molecule arginase inhibitor CB-1158 blocks Arg depletion by myelomonocytic arginase expression in the TME, activates NK cells, promotes their infiltration into the TME, and acts synergistically with checkpoint inhibition to promote tumor clearance [67]. Furthermore, in a study that included 18 colorectal cancer patients who underwent tumor resection, preoperative Arg supplementation was found to lead to an increase in tumor-infiltrating CD16 and CD56 NK cells by histopathological analysis of biopsies [105].
Targeting tryptophan
Based on its effects on NK cells as well as other immune cells, IDO inhibition has emerged as a potential target for anticancer therapies. Most inhibitors bind to IDO enzymes and prevent the conversion of tryptophan to kynurenine, resulting in toxic effects on NK cells and other immune substrates. The use of IDO/TDO-IN-2, a dual inhibitor targeting IDO3 and TDO1, reversed the inhibition of NK cell-mediated antibody-dependent cytotoxicity (ADCC) by oncolytic-positive cancer-associated fibroblasts and helped to attenuate Trastuzumab resistance in HER2 positive breast cancer [106]. In a study on sarcoma, an IDO inhibitor (GDC-0919) was in use. Although GDC-0919 alone or in combination with anti-PDL1 neither show antitumor activity nor affect tumor immune cell infiltration, GDC-0919 was found to induce downregulation of granzyme expression, as well as upregulation of inhibin subunit beta A (inhba) and E3 Ubiquitin ligase Dtx4 [107]. It has been reported that inhba plays a crucial role in inhibiting NK cell proliferation and granzyme B production, leading to impaired tumor susceptibility to NK cell-mediated killing [108]. These results indicate a potential role for the IDO pathway in controlling NK function [107]. By delivering immunogenic cell death (ICD) stimuli combined with interference with the immunometabolism effects of the IDO-1 pathway, a dual-delivery liposomal carrier with mitoxantrone and cholesterol fingolimod as prodrugs was developed, and the data showed that liposomal delivery was very effective for inducing a chemoimmunotherapeutic response with the involvement of NK, which has been successfully validated for chemoimmunotherapy in mouse models of breast and renal cancer [109]. In addition, multi-combination immunotherapy, i.e., nanoscale reduced graphene oxide-mediated photothermal therapy synergized with IDO inhibition and PD-L1 blockade, designed by Yan et al. was shown to directly kill tumor cells and induce synergistic anti-tumor immunity. Experiments in vivo further demonstrated that the immune response enhanced the production of tumor-infiltrating lymphocytes, including CD4 T cells, CD8 T cells, and NK cells, as well as INF-γ [110].
Targeted glutamine
As mentioned previously, we mentioned that targeting glutamine metabolism could enhance NK cell-based therapies by impairing tumor fuel supply without decreasing NK cell function. Based on this idea JHU083 was used in preclinical studies. JHU083 is a prodrug of 6-diazo-5-oxo-L-norleucine (DON), a glutamine antagonist that can interfere with tumor cell metabolism by inhibiting glutaminase and a variety of enzymes in tumors that require glutamine thereby increasing the availability of glucose and oxygen in the TME and reducing TME acidification [111]. These alterations are beneficial in supporting NK cell effector function. We have already known the importance of SLC7A5-dependent amino acid uptake in NK cells for the stable and sustained expression of c-MYC [13]. This has led researchers to focus on therapeutic strategies that target NK cells to upregulate amino acid transporter proteins to enhance metabolic adaptations in NK cells: ① Chimeric Antigen Receptor (CAR)-expressing NK cells armed with overexpressed transporter proteins; ② Overexpression of transporter protein positive regulators (e.g., c-MYC, YBX3) and knockdown/silencing of transporter protein negative regulators (e.g., MARCH1); ③ Enhance the expression of amino acid transporter proteins specifically using cytokines such as IL-15, IL-18 [72]. In addition, MYC in NK cells is degraded by glycogen synthase kinase 3 (GSK3), and several clinical trials (NCT01287520) targeting GSK3 inhibitors (NCT01214603) are ongoing [112, 113]. Notably, SREBP activity is not only critical for metabolic reprogramming and obtaining higher glycolysis and OXPHOS in NK cells, but is also required for c-MYC protein expression [15, 114]. Activation of SREBP transcription factors can be effectively inhibited by cholesterol and oxysterols, and fluoxetine, and betulinic acid alcohols have been shown to suppress SREBP activation and have shown promising antitumor effects in preclinical studies [115, 116].
Conclusion and outlook
Numerous NK cell-based immunotherapies, including ex vivo NK cell activation, NK cell overlay therapy, and CAR-NK cell therapy, are quickly developing in the context of molecularly targeted treatments. Among them, CAR-NK cell therapy is regarded as a milestone breakthrough in anti-tumor immunotherapy is CAR-NK cell treatment. Because of the special biological characteristics of NK cells, CAR-NK treatment has the distinct benefits of low toxicology and multi-cell origin. Even the use of allogeneic NK cells prevents allogeneic responses and lowers the risk of neurotoxicity and serious toxic side effects including acute cytokine release syndrome (CRS) and graft-versus-host disease (GVHD). Nevertheless, there are still several obstacles in the way of its clinical application in solid tumors, including those related to the creation of CAR-NK cells, their ability to migrate to the tumor site, and their survival and durability in immunosuppressed TME. In particular, the importance of "adaptive" regulation of CAR-NK cells in TME suggests that the eventual development of NK cell exhaustion remains a major obstacle to the success of NK cell immunotherapy treatments, which are closely related to TME. Understanding the interactions in the metabolic biology of NK cells and specific TMEs will help to identify the necessary NK cell modifications and the relevant choices of NK cell sources to optimize NK cell-based immunotherapies as a cancer immunotherapy boon.
In addition, immunometabolism is a fascinating area of research that may be key to successful cancer immunotherapy. However, targeting immune metabolism during immunotherapy is challenging. Because tumor cells and immune cells share similar metabolic pathways for energy acquisition, targeting tumor cell metabolism may impair immune cell metabolism in the context of TME and thus diminish their effector function. An example is the uncertainty of the glycolysis inhibitor 2-DG, which targets glucose metabolism, on NK cell metabolism. Future studies should deepen the differences between the metabolic mechanisms of cancer cells and immune cells in the context of TME and adjust the strategy of targeting metabolic regulation.
Availability of data and materials
Not applicable.
Abbreviations
- NK:
-
Natural killer
- TME:
-
Tumor microenvironment
- OXPHOS:
-
Oxidative phosphorylation
- IFN-γ:
-
Interferon γ
- TCA:
-
Tricarboxylic acid
- CMS:
-
Citrate-malate shuttle
- SREBP:
-
Sterol regulatory element binding protein
- ATP:
-
Adenosine triphosphate
- ACLY:
-
ATP citrate lyase
- PPAR:
-
Peroxisome proliferator-activated receptor
- MCMV:
-
Mouse cytomegalovirus
- LDHA:
-
Lactate dehydrogenase A
- ECM:
-
Extracellular matrix
- HIF-1α:
-
Hypoxia-inducible factor 1α
- NAD:
-
Nicotinamide adenine dinucleotide
- VEGF:
-
Vascular endothelial growth factor
- PKA:
-
Protein kinase A
- NFAT:
-
Uclear factor of activated T-cells
- Arg:
-
Arginine
- mitoDAMP:
-
Mitochondria-derived damage-associated molecular pattern
- IDO:
-
Indoleamine 2,3-dioxygenase
- TDO:
-
Tryptophan-2,3-dioxygenase
- TIL:
-
Tumor-infiltrating lymphoid
- NCR1:
-
Natural cytotoxicity triggering receptor 1
- CRPC:
-
Castration-resistant prostate cancer
- 2-DG:
-
2-Deoxyglucose
- CIML:
-
Cytokine-induced memory-like
- NHI:
-
N-hydroxy-indole
- MCT:
-
Monocarboxylic acid transporter
- ADCC:
-
Antibody-dependent cytotoxicity
- ICD:
-
Immunogenic cell death
- CAR:
-
Chimeric antigen receptor
- GSK3:
-
Glycogen synthase kinase 3
- DMXAA:
-
5,6-Dimethylxanthenone-4-acetic acid
- Inhba:
-
Inhibin subunit beta a
- TNF-α:
-
Tumor necrosis factor-α
- ADO:
-
Adenosine
- eATP:
-
Extracellular adenosine triphosphate
- eADO:
-
Extracellular adenosine
- cAMP:
-
Cyclic adenosine monophosphate
- LDH:
-
Lactate dehydrogenase
- EMT:
-
Epithelial-to-mesenchymal transition
- NFAT:
-
Nuclear factor of activated T-cells
- CRS:
-
Cytokine release syndrome
- GVHD:
-
Graft-versus-host disease
References
Cooper MA, Fehniger TA, Caligiuri MA. The biology of human natural killer-cell subsets. Trends Immunol. 2001;22:11.
Martinet L, Smyth MJ. Balancing natural killer cell activation through paired receptors. Nat Rev Immunol. 2015;15:4.
Vivier E, Tomasello E, Baratin M, Walzer T, Ugolini S. Functions of natural killer cells. Nat Immunol. 2008;9:5.
Liu S, Galat V, Galat Y, Lee YKA, Wainwright D, Wu J. NK cell-based cancer immunotherapy: from basic biology to clinical development. J Hematol Oncol. 2021;14:1.
Laskowski TJ, Biederstädt A, Rezvani K. Natural killer cells in antitumour adoptive cell immunotherapy. Nat Rev Cancer. 2022;22:10.
Tong L, Jiménez-Cortegana C, Tay AHM, Wickström S, Galluzzi L, Lundqvist A. NK cells and solid tumors: therapeutic potential and persisting obstacles. Mol Cancer. 2022;21:1.
Chiossone L, Chaix J, Fuseri N, Roth C, Vivier E, Walzer T. Maturation of mouse NK cells is a 4-stage developmental program. Blood. 2009;113:22.
Marçais A, Cherfils-Vicini J, Viant C, Degouve S, Viel S, Fenis A, et al. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat Immunol. 2014;15:8.
Keppel MP, Saucier N, Mah AY, Vogel TP, Cooper MA. Activation-specific metabolic requirements for NK Cell IFN-γ production. J Immunol. 2015;194:4.
Schafer JR, Salzillo TC, Chakravarti N, Kararoudi MN, Trikha P, Foltz JA, et al. Education-dependent activation of glycolysis promotes the cytolytic potency of licensed human natural killer cells. J Allergy Clin Immunol. 2019;143:1.
Keating SE, Zaiatz-Bittencourt V, Loftus RM, Keane C, Brennan K, Finlay DK, et al. Metabolic reprogramming supports IFN-γ Production by CD56bright NK cells. J Immunol. 2016;196:6.
O’Brien KL, Finlay DK. Immunometabolism and natural killer cell responses. Nat Rev Immunol. 2019;19:5.
Loftus RM, Assmann N, Kedia-Mehta N, O’Brien KL, Garcia A, Gillespie C, et al. Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat Commun. 2018;9:1.
Renner K, Geiselhöringer AL, Fante M, Bruss C, Färber S, Schönhammer G, et al. Metabolic plasticity of human T cells: preserved cytokine production under glucose deprivation or mitochondrial restriction, but 2-deoxy-glucose affects effector functions. Eur J Immunol. 2015;45:9.
Assmann N, O’Brien KL, Donnelly RP, Dyck L, Zaiatz-Bittencourt V, Loftus RM, et al. Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat Immunol. 2017;18:11.
Romagnani C, Juelke K, Falco M, Morandi B, D’Agostino A, Costa R, et al. CD56brightCD16- killer Ig-like receptor- NK cells display longer telomeres and acquire features of CD56dim NK cells upon activation. J Immunol. 2007;178:8.
Salzberger W, Martrus G, Bachmann K, Goebels H, Heß L, Koch M, et al. Tissue-resident NK cells differ in their expression profile of the nutrient transporters Glut1, CD98 and CD71. PLoS ONE. 2018;13:7.
Wang P, Liang T, Zhan H, Zhu M, Wu M, Qian L, et al. Unique metabolism and protein expression signature in human decidual NK cells. Front Immunol. 2023. https://doi.org/10.3389/fimmu.2023.1136652.
Michelet X, Dyck L, Hogan A, Loftus RM, Duquette D, Wei K, et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat Immunol. 2018;19:12.
Sheppard S, Santosa EK, Lau CM, Violante S, Giovanelli P, Kim H, et al. Lactate dehydrogenase A-dependent aerobic glycolysis promotes natural killer cell anti-viral and anti-tumor function. Cell Rep. 2021;35:9.
Mah AY, Rashidi A, Keppel MP, Saucier N, Moore EK, Alinger JB, et al. Glycolytic requirement for NK cell cytotoxicity and cytomegalovirus control. JCI insight. 2017;2:23.
Pitt JM, Marabelle A, Eggermont A, Soria JC, Kroemer G, Zitvogel L. Targeting the tumor microenvironment: removing obstruction to anticancer immune responses and immunotherapy. Ann Oncol. 2016;27:8.
Arneth B. Tumor microenvironment. Medicina (Kaunas). 2019;56:1.
O’Sullivan D, Sanin DE, Pearce EJ, Pearce EL. Metabolic interventions in the immune response to cancer. Nat Rev Immunol. 2019;19:5.
Pelletier A, Nelius E, Fan Z, Khatchatourova E, Alvarado-Diaz A, He J, et al. Resting natural killer cell homeostasis relies on tryptophan/NAD(+) metabolism and HIF-1α. EMBO Rep. 2023;24:6.
Cluff E, Magdaleno CC, Fernandez E, House T, Swaminathan S, Varadaraj A, et al. Hypoxia-inducible factor-1 alpha expression is induced by IL-2 via the PI3K/mTOR pathway in hypoxic NK cells and supports effector functions in NKL cells and ex vivo expanded NK cells. Cancer Immunol Immunother CII. 2022;71:8.
Lim SA, Moon Y, Shin MH, Kim TJ, Chae S, Yee C, et al. Hypoxia-driven HIF-1α activation reprograms pre-activated NK cells towards highly potent effector phenotypes via ERK/STAT3 pathways. Cancers. 2021;13:8.
Krzywinska E, Kantari-Mimoun C, Kerdiles Y, Sobecki M, Isagawa T, Gotthardt D, et al. Loss of HIF-1α in natural killer cells inhibits tumour growth by stimulating non-productive angiogenesis. Nat Commun. 2017;8:1.
Ni J, Wang X, Stojanovic A, Zhang Q, Wincher M, Bühler L, et al. Single-cell RNA sequencing of tumor-infiltrating NK cells reveals that inhibition of transcription factor HIF-1α unleashes NK cell activity. Immunity. 2020;52:6.
Allard B, Allard D, Buisseret L, Stagg J. The adenosine pathway in immuno-oncology. Nat Rev Clin Oncol. 2020;17:10.
Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol. 2016;16:3.
Sek K, Mølck C, Stewart GD, Kats L, Darcy PK, Beavis PA. Targeting adenosine receptor signaling in cancer immunotherapy. Int J Mol Sci. 2018;19:12.
Raskovalova T, Lokshin A, Huang X, Jackson EK, Gorelik E. Adenosine-mediated inhibition of cytotoxic activity and cytokine production by IL-2/NKp46-activated NK cells: involvement of protein kinase A isozyme I (PKA I). Immunol Res. 2006;36:1–3.
Raskovalova T, Huang X, Sitkovsky M, Zacharia LC, Jackson EK, Gorelik E. Gs protein-coupled adenosine receptor signaling and lytic function of activated NK cells. J Immunol. 2005;175:7.
Sonigo G, Bozonnat A, Dumont M, Thonnart N, Ram-Wolff C, de Masson A, et al. Involvement of the CD39/CD73/adenosine pathway in T-cell proliferation and NK cell-mediated antibody-dependent cell cytotoxicity in Sézary syndrome. Blood. 2022;139:17.
Zahavi D, Hodge JW. Targeting immunosuppressive adenosine signaling: a review of potential immunotherapy combination strategies. Int J Mol Sci. 2023;24:10.
Young A, Ngiow SF, Gao Y, Patch AM, Barkauskas DS, Messaoudene M, et al. A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Can Res. 2018;78:4.
Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162:6.
Reinfeld BI, Madden MZ, Wolf MM, Chytil A, Bader JE, Patterson AR, et al. Cell-programmed nutrient partitioning in the tumour microenvironment. Nature. 2021;593:7858.
Comandatore A, Franczak M, Smolenski RT, Morelli L, Peters GJ, Giovannetti E. Lactate Dehydrogenase and its clinical significance in pancreatic and thoracic cancers. Semin Cancer Biol. 2022;86:93.
Gong T, Liu J, Jiang J, Zhai YF, Wu CM, Ma C, et al. The role of lactate deshydrogenase levels on non-small cell lung cancer prognosis: a meta-analysis. Cell Mol Biol. 2019;65:1.
Gu S, Yang C. Serum lactate dehydrogenase level predicts the prognosis in bladder cancer patients. BMC Urol. 2023;23:1.
Yu SL, Xu LT, Qi Q, Geng YW, Chen H, Meng ZQ, et al. Serum lactate dehydrogenase predicts prognosis and correlates with systemic inflammatory response in patients with advanced pancreatic cancer after gemcitabine-based chemotherapy. Sci Rep. 2017. https://doi.org/10.1038/srep45194.
Zhang C, Zhan Z, Fang Y, Ruan Y, Lin M, Dai Z, et al. Prognostic nutritional index and serum lactate dehydrogenase predict the prognosis of nasopharyngeal carcinoma patients who received intensity-modulated radiation therapy. J Cancer Res Clin Oncol. 2023;149:20.
Zhang Y, Liu B, Kotenko S, Li W. Prognostic value of neutrophil-lymphocyte ratio and lactate dehydrogenase in melanoma patients treated with immune checkpoint inhibitors: a systematic review and meta-analysis. Medicine. 2022;101:32.
Petrelli F, Cabiddu M, Coinu A, Borgonovo K, Ghilardi M, Lonati V, et al. Prognostic role of lactate dehydrogenase in solid tumors: a systematic review and meta-analysis of 76 studies. Acta Oncol. 2015;54:7.
Marmorino F, Salvatore L, Barbara C, Allegrini G, Antonuzzo L, Masi G, et al. Serum LDH predicts benefit from bevacizumab beyond progression in metastatic colorectal cancer. Br J Cancer. 2017;116:3.
Wang X, Zhang B, Chen X, Mo H, Wu D, Lan B, et al. Lactate dehydrogenase and baseline markers associated with clinical outcomes of advanced esophageal squamous cell carcinoma patients treated with camrelizumab (SHR-1210), a novel anti-PD-1 antibody. Thoracic cancer. 2019;10:6.
Zhao J, Huang X, Xu Z, Dai J, He H, Zhu Y, et al. LDHA promotes tumor metastasis by facilitating epithelial-mesenchymal transition in renal cell carcinoma. Mol Med Rep. 2017;16:6.
Baumann F, Leukel P, Doerfelt A, Beier CP, Dettmer K, Oefner PJ, et al. Lactate promotes glioma migration by TGF-beta2-dependent regulation of matrix metalloproteinase-2. Neuro Oncol. 2009;11:4.
Wang H, Zhou R, Sun L, Xia J, Yang X, Pan C, et al. TOP1MT deficiency promotes GC invasion and migration via the enhancements of LDHA expression and aerobic glycolysis. Endocr Relat Cancer. 2017;24:11.
Chen M, Cen K, Song Y, Zhang X, Liou YC, Liu P, et al. NUSAP1-LDHA-Glycolysis-Lactate feedforward loop promotes Warburg effect and metastasis in pancreatic ductal adenocarcinoma. Cancer Lett. 2023;567:216285.
Shi Q, Le X, Wang B, Abbruzzese JL, Xiong Q, He Y, et al. Regulation of vascular endothelial growth factor expression by acidosis in human cancer cells. Oncogene. 2001;20:28.
Arseneault R, Chien A, Newington JT, Rappon T, Harris R, Cumming RC. Attenuation of LDHA expression in cancer cells leads to redox-dependent alterations in cytoskeletal structure and cell migration. Cancer Lett. 2013;338:2.
Govoni M, Rossi V, Di Stefano G, Manerba M. Lactate upregulates the expression of DNA repair genes, causing intrinsic resistance of cancer cells to cisplatin. Pathol Oncol Res. 2021;27:1609951.
Zhang K, Zhang T, Yang Y, Tu W, Huang H, Wang Y, et al. N(6)-methyladenosine-mediated LDHA induction potentiates chemoresistance of colorectal cancer cells through metabolic reprogramming. Theranostics. 2022;12:10.
Amrutkar M, Berg K, Balto A, Skilbrei MG, Finstadsveen AV, Aasrum M, et al. Pancreatic stellate cell-induced gemcitabine resistance in pancreatic cancer is associated with LDHA- and MCT4-mediated enhanced glycolysis. Cancer Cell Int. 2023;23:1.
Shi L, Duan R, Sun Z, Jia Q, Wu W, Wang F, et al. LncRNA GLTC targets LDHA for succinylation and enzymatic activity to promote progression and radioiodine resistance in papillary thyroid cancer. Cell Death Differ. 2023;30:6.
Das CK, Parekh A, Parida PK, Bhutia SK, Mandal M. Lactate dehydrogenase A regulates autophagy and tamoxifen resistance in breast cancer. Biochim Biophys Acta. 2019;1866:6.
Koukourakis M, Tsolou A, Pouliliou S, Lamprou I, Papadopoulou M, Ilemosoglou M, et al. Blocking LDHA glycolytic pathway sensitizes glioblastoma cells to radiation and temozolomide. Biochem Biophys Res Commun. 2017;491:4.
Claps G, Faouzi S, Quidville V, Chehade F, Shen S, Vagner S, et al. The multiple roles of LDH in cancer. Nat Rev Clin Oncol. 2022;19:12.
Mishra D, Banerjee D. Lactate dehydrogenases as metabolic links between tumor and stroma in the tumor microenvironment. Cancers. 2019;11:6.
Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA-Associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. 2016;24:5.
Harmon C, Robinson MW, Hand F, Almuaili D, Mentor K, Houlihan DD, et al. Lactate-mediated acidification of tumor microenvironment induces apoptosis of liver-resident NK cells in colorectal liver metastasis. Cancer Immunol Res. 2019;7:2.
Ge W, Meng L, Cao S, Hou C, Zhu X, Huang D, et al. The SIX1/LDHA axis promotes lactate accumulation and leads to NK cell dysfunction in pancreatic cancer. J Immunol Res. 2023. https://doi.org/10.1155/2023/6891636.
Lemos H, Huang L, Prendergast GC, Mellor AL. Immune control by amino acid catabolism during tumorigenesis and therapy. Nat Rev Cancer. 2019;19:3.
Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer. 2017;5:1.
Munder M, Schneider H, Luckner C, Giese T, Langhans CD, Fuentes JM, et al. Suppression of T-cell functions by human granulocyte arginase. Blood. 2006;108:5.
Lamas B, Vergnaud-Gauduchon J, Goncalves-Mendes N, Perche O, Rossary A, Vasson MP, et al. Altered functions of natural killer cells in response to L-Arginine availability. Cell Immunol. 2012;280:2.
Westhaver LP, Nersesian S, Nelson A, MacLean LK, Carter EB, Rowter D, et al. Mitochondrial damage-associated molecular patterns trigger arginase-dependent lymphocyte immunoregulation. Cell Rep. 2022;39:8.
Oberlies J, Watzl C, Giese T, Luckner C, Kropf P, Müller I, et al. Regulation of NK cell function by human granulocyte arginase. J Immunol. 2009;182:9.
Nachef M, Ali AK, Almutairi SM, Lee SH. Targeting SLC1A5 and SLC3A2/SLC7A5 as a potential strategy to strengthen anti-tumor immunity in the tumor microenvironment. Front Immunol. 2021. https://doi.org/10.3389/fimmu.2021.624324.
Sinclair LV, Neyens D, Ramsay G, Taylor PM, Cantrell DA. Single cell analysis of kynurenine and system L amino acid transport in T cells. Nat Commun. 2018;9:1.
Della Chiesa M, Carlomagno S, Frumento G, Balsamo M, Cantoni C, Conte R, et al. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood. 2006;108:13.
Song H, Park H, Kim YS, Kim KD, Lee HK, Cho DH, et al. L-kynurenine-induced apoptosis in human NK cells is mediated by reactive oxygen species. Int Immunopharmacol. 2011;11:8.
Wang D, Saga Y, Mizukami H, Sato N, Nonaka H, Fujiwara H, et al. Indoleamine-2,3-dioxygenase, an immunosuppressive enzyme that inhibits natural killer cell function, as a useful target for ovarian cancer therapy. Int J Oncol. 2012;40:4.
Ma G, Zhang Z, Li P, Zhang Z, Zeng M, Liang Z, et al. Reprogramming of glutamine metabolism and its impact on immune response in the tumor microenvironment. Cell Commun Signal. 2022;20:1.
Kuo CL, Chou HY, Lien HW, Yeh CA, Wang JR, Chen CH, et al. A Fc-VEGF chimeric fusion enhances PD-L1 immunotherapy via inducing immune reprogramming and infiltration in the immunosuppressive tumor microenvironment. Cancer Immunol Immunother. 2023;72:2.
Smolarczyk R, Cichoń T, Pilny E, Jarosz-Biej M, Poczkaj A, Kułach N, et al. Combination of anti-vascular agent - DMXAA and HIF-1α inhibitor - digoxin inhibits the growth of melanoma tumors. Sci Rep. 2018;8:1.
Noman MZ, Desantis G, Janji B, Hasmim M, Karray S, Dessen P, et al. PD-L1 is a novel direct target of HIF-1α, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J Exp Med. 2014;211:5.
Xu LJ, Ma Q, Zhu J, Li J, Xue BX, Gao J, et al. Combined inhibition of JAK1,2/Stat3-PD-L1 signaling pathway suppresses the immune escape of castration-resistant prostate cancer to NK cells in hypoxia. Mol Med Rep. 2018;17:6.
Bendell J, LoRusso P, Overman M, Noonan AM, Kim DW, Strickler JH, et al. First-in-human study of oleclumab, a potent, selective anti-CD73 monoclonal antibody, alone or in combination with durvalumab in patients with advanced solid tumors. Cancer Immunol Immunother. 2023;72:7.
Hay CM, Sult E, Huang Q, Mulgrew K, Fuhrmann SR, McGlinchey KA, et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology. 2016;5:8.
Schäkel L, Schmies CC, Idris RM, Luo X, Lee SY, Lopez V, et al. Nucleotide Analog ARL67156 as a lead structure for the development of CD39 and dual CD39/CD73 ectonucleotidase inhibitors. Front Pharmacol. 2020. https://doi.org/10.3389/fphar.2020.01294.
Leone RD, Lo YC, Powell JD. A2aR antagonists: next generation checkpoint blockade for cancer immunotherapy. Comput Struct Biotechnol J. 2015;13:265.
Kamai T, Kijima T, Tsuzuki T, Nukui A, Abe H, Arai K, et al. Increased expression of adenosine 2A receptors in metastatic renal cell carcinoma is associated with poorer response to anti-vascular endothelial growth factor agents and anti-PD-1/Anti-CTLA4 antibodies and shorter survival. Cancer Immunol Immunother. 2021;70:7.
Cekic C, Day YJ, Sag D, Linden J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Can Res. 2014;74:24.
Yan P, Luo Y, Li X, Li Y, Wang Y, Wu J, et al. A redox-responsive nanovaccine combined with A2A receptor antagonist for cancer immunotherapy. Adv Healthcare Mater. 2021;10:21.
Raez LE, Papadopoulos K, Ricart AD, Chiorean EG, Dipaola RS, Stein MN, et al. A phase I dose-escalation trial of 2-deoxy-D-glucose alone or combined with docetaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol. 2013;71:2.
Stein M, Lin H, Jeyamohan C, Dvorzhinski D, Gounder M, Bray K, et al. Targeting tumor metabolism with 2-deoxyglucose in patients with castrate-resistant prostate cancer and advanced malignancies. Prostate. 2010;70:13.
Wang Z, Guan D, Wang S, Chai LYA, Xu S, Lam KP. Glycolysis and oxidative phosphorylation play critical roles in natural killer cell receptor-mediated natural killer cell functions. Front Immunol. 2020;11:202.
Terrén I, Orrantia A, Mosteiro A, Vitallé J, Zenarruzabeitia O, Borrego F. Metabolic changes of Interleukin-12/15/18-stimulated human NK cells. Sci Rep. 2021;11:1.
Wu T, Ke Y, Tang H, Liao C, Li J, Wang L. Fidarestat induces glycolysis of NK cells through decreasing AKR1B10 expression to inhibit hepatocellular carcinoma. Mol Ther Oncolytics. 2021;23:420.
Cong J, Wang X, Zheng X, Wang D, Fu B, Sun R, et al. Dysfunction of natural killer cells by FBP1-induced inhibition of glycolysis during lung cancer progression. Cell Metab. 2018;28:2.
Zhao Z, Han F, Yang S, Wu J, Zhan W. Oxamate-mediated inhibition of lactate dehydrogenase induces protective autophagy in gastric cancer cells: involvement of the Akt-mTOR signaling pathway. Cancer Lett. 2015;358:1.
Billiard J, Dennison JB, Briand J, Annan RS, Chai D, Colón M, et al. Quinoline 3-sulfonamides inhibit lactate dehydrogenase A and reverse aerobic glycolysis in cancer cells. Cancer Metab. 2013;1:1.
Gupta GS, Kapur S, Kinsky RG. Inhibition kinetics of lactate dehydrogenase isoenzymes by gossypol acetic acid. Biochem Int. 1988;17:1.
Yu Y, Deck JA, Hunsaker LA, Deck LM, Royer RE, Goldberg E, et al. Selective active site inhibitors of human lactate dehydrogenases A4, B4, and C4. Biochem Pharmacol. 2001;62:1.
Granchi C, Roy S, Giacomelli C, Macchia M, Tuccinardi T, Martinelli A, et al. Discovery of N-hydroxyindole-based inhibitors of human lactate dehydrogenase isoform A (LDH-A) as starvation agents against cancer cells. J Med Chem. 2011;54:6.
Granchi C, Calvaresi EC, Tuccinardi T, Paterni I, Macchia M, Martinelli A, et al. Assessing the differential action on cancer cells of LDH-A inhibitors based on the N-hydroxyindole-2-carboxylate (NHI) and malonic (Mal) scaffolds. Org Biomol Chem. 2013;11:38.
Halford S, Veal GJ, Wedge SR, Payne GS, Bacon CM, Sloan P, et al. A phase I dose-escalation study of AZD3965, an oral monocarboxylate transporter 1 inhibitor, in patients with advanced cancer. Clin Cancer Res. 2023;29:8.
Beloueche-Babari M, Casals Galobart T, Delgado-Goni T, Wantuch S, Parkes HG, Tandy D, et al. Monocarboxylate transporter 1 blockade with AZD3965 inhibits lipid biosynthesis and increases tumour immune cell infiltration. Br J Cancer. 2020;122:6.
Pötzl J, Roser D, Bankel L, Hömberg N, Geishauser A, Brenner CD, et al. Reversal of tumor acidosis by systemic buffering reactivates NK cells to express IFN-γ and induces NK cell-dependent lymphoma control without other immunotherapies. Int J Cancer. 2017;140:9.
Gillies RJ, Ibrahim-Hashim A, Ordway B, Gatenby RA. Back to basic: trials and tribulations of alkalizing agents in cancer. Front Oncol. 2022;12:981718.
Heys SD, Segar A, Payne S, Bruce DM, Kernohan N, Eremin O. Dietary supplementation with L-arginine: modulation of tumour-infiltrating lymphocytes in patients with colorectal cancer. Br J Surg. 1997;84:2.
Du R, Zhang X, Lu X, Ma X, Guo X, Shi C, et al. PDPN positive CAFs contribute to HER2 positive breast cancer resistance to trastuzumab by inhibiting antibody-dependent NK cell-mediated cytotoxicity. Drug Resist Updat. 2023;68:100947.
Nafia I, Toulmonde M, Bortolotto D, Chaibi A, Bodet D, Rey C, et al. IDO targeting in sarcoma: biological and clinical implications. Front Immunol. 2020. https://doi.org/10.3389/fimmu.2020.00274.
Rautela J, Dagley LF, de Oliveira CC, Schuster IS, Hediyeh-Zadeh S, Delconte RB, et al. Therapeutic blockade of activin-A improves NK cell function and antitumor immunity. Science signaling. 2019;12:596.
Mei KC, Liao YP, Jiang J, Chiang M, Khazaieli M, Liu X, et al. Liposomal delivery of mitoxantrone and a cholesteryl indoximod prodrug provides effective chemo-immunotherapy in multiple solid tumors. ACS Nano. 2020;14:10.
Yan M, Liu Y, Zhu X, Wang X, Liu L, Sun H, et al. Nanoscale reduced graphene oxide-mediated photothermal therapy together with IDO inhibition and PD-L1 blockade synergistically promote antitumor immunity. ACS Appl Mater Interfaces. 2019;11:2.
Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science (New York, NY). 2019;366:6468.
Gray JE, Infante JR, Brail LH, Simon GR, Cooksey JF, Jones SF, et al. A first-in-human phase I dose-escalation, pharmacokinetic, and pharmacodynamic evaluation of intravenous LY2090314, a glycogen synthase kinase 3 inhibitor, administered in combination with pemetrexed and carboplatin. Invest New Drugs. 2015;33:6.
Rizzieri DA, Cooley S, Odenike O, Moonan L, Chow KH, Jackson K, et al. An open-label phase 2 study of glycogen synthase kinase-3 inhibitor LY2090314 in patients with acute leukemia. Leuk Lymphoma. 2016;57:8.
O’Brien KL, Assmann N, O’Connor E, Keane C, Walls J, Choi C, et al. De novo polyamine synthesis supports metabolic and functional responses in activated murine NK cells. Eur J Immunol. 2021;51:1.
Li X, Chen YT, Hu P, Huang WC. Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol Cancer Ther. 2014;13:4.
Król SK, Kiełbus M, Rivero-Müller A, Stepulak A. Comprehensive review on betulin as a potent anticancer agent. BioMed Res Int. 2015. https://doi.org/10.1155/2015/584189.
Acknowledgements
Not applicable.
Funding
This research was funded by Xiaonan Cui, the host of the Capacity Construction Project of Major Clinical (specialized) Departments of Traditional Chinese Medicine of Liaoning Province (No.LNZYXZK201909), and the host of the Distinguished Professor Program of Liaoning Province. And Ying Liu, the host of the Hospital Research Fund by Affiliated Zhongshan Hospital of Dalian University (No.20230320).
Author information
Authors and Affiliations
Contributions
LX-M wrote the manuscript. XN-C and YL provided financial and guidance of the manuscript. LX-M, CL-L, BZ, HL-L, XZ, HR-C, YL, XN-C proofread and revised the manuscript. All authors approved the final version to be published.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declared that no competing interest exists.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Miao, L., Lu, C., Zhang, B. et al. Advances in metabolic reprogramming of NK cells in the tumor microenvironment on the impact of NK therapy. J Transl Med 22, 229 (2024). https://doi.org/10.1186/s12967-024-05033-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-024-05033-w