
Abstract
Programmed cell death (PCD) plays an important role in many aspects of individual development, maintenance of body homeostasis and pathological processes. Ferroptosis is a novel form of PCD characterized by the accumulation of iron-dependent lipid peroxides resulting in lethal cell damage. It contributes to tumor progression in an apoptosis-independent manner. In recent years, an increasing number of non-coding RNAs (ncRNAs) have been demonstrated to mediate the biological process of ferroptosis, hence impacting carcinogenesis, progression, drug resistance, and prognosis. However, the clear regulatory mechanism for this phenomenon remains poorly understood. Moreover, ferroptosis does not usually exist independently. Its interaction with PCD, like apoptosis, necroptosis, autophagy, pyroptosis, and cuproptosis, to destroy cells appears to exist. Furthermore, ncRNA seems to be involved. Here, we review the mechanisms by which ferroptosis occurs, dissect its relationship with other forms of death, summarize the key regulatory roles played by ncRNAs, raise relevant questions and predict possible barriers to its application in the clinic, offering new ideas for targeted tumour therapy.
Similar content being viewed by others

Introduction
Ferroptosis is a novel model of cell death, as defined in 2012 [1]. It is distinguished from other types of deaths by apoptosis, necroptosis, autophagy, pyroptosis, and cuproptosis [1, 2]. Its main morphological manifestations are shrinking mitochondria, increased membrane density, and fewer cristae. In recent years, research into ferroptosis has expanded tremendously. Numerous scientific breakthroughs have been gained in oncology, and targeting ferroptosis has become a potential cancer therapy.
Although each programmed cell death (PCD) has a unique mechanism of occurrence and cellular and biochemical properties, mixed types of cell death seem more prevalent than single types of death in most cells. Some of their components and factors are synergistic. Exploring how ferroptosis interacts with other PCDs at the molecular level and identifying and integrating shared pathways will open new areas for systematic research [3].
Ninety-eight percent of the human genome is transcribed into RNAs that do not encode proteins, known as non-coding RNAs (ncRNAs) [4]. Evidence suggests they are vital in basic biological processes like growth and development and almost every human disease, particularly cancer [5, 6]. At the same time, ncRNAs have been shown to be involved in the biology of ferroptosis and, in turn, influence tumour progression. This implies that ncRNA-based targeted iron death therapy is a promising novel anti-cancer therapy. However, the mechanisms by which ncRNAs regulate ferroptosis are still poorly understood. Furthermore, the role of ncRNAs in ferroptosis has not been fully defined.
In this review, we provide new ideas for targeting ncRNAs in ferroptosis-related therapeutic strategies by systematically summarizing ferroptosis mechanisms and the progress of ncRNA targeting of ferroptosis signaling pathways in tumors, paying particular attention to the interactions between ferroptosis and other PCDs.
Mechanism of ferroptosis
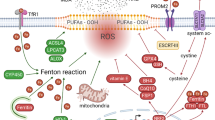
Ferroptosis is a novel form of cell death regulation that relies on iron ion-mediated oxidative damage. Ferroptosis may be triggered when intracellular iron ion-dependent reactive oxygen species (ROS) accumulate in excess and glutathione peroxidase 4 (GPX4) scavenging is diminished, resulting in an imbalance in the homeostasis of ROS production and degradation, i.e. a redox imbalance between intracellular oxidants and antioxidants [7]. Current molecular mechanisms of ferroptosis include glutathione (GSH) depletion, lipid peroxidation, and impaired iron metabolism (Fig. 1). The various molecules and signals involved in iron metabolism and lipid peroxidation will be discussed below.

The core molecular mechanisms of ferroptosis. The regulatory pathways of ferroptosis are divided into iron metabolism, lipid metabolism and the system xc-/GSH/GPX4 axis. Iron metabolism: Transferrin (TF); Transferrin receptor 1 (TFRC); ferroportin (FPN); Ferritin heavy chain 1 (FTH1); Ferritin light chain (FTL); solute carrier family 39 member 14 (SLC39A14); Six transmembrane epithelial antigen of protein 3 (STEAP3); Poly (RC) binding protein 1/2 (PCBP1/2); Reactive oxygen species (ROS); Lipid metabolism: Polyunsaturated fatty acid (PUFA); Long chain acyl CoA synthetase 4 (ACSL4); Lysophosphatidylcholine acyltransferase 3 (LPCAT3); Phosphatidylethanolamine (PE); arachidonic acid (AA); adrenic acid (AdA), coenzyme A (CoA); system xc-/GSH/GPX4 axis: Solute carrier family member 7A11 (SLC7A11); Solute carrier family member 3A2 (SLC3A2); Glutathione (GSH); glutathione-disulfide reductase (GSR); glucose 6-phosphate dehydrogenase (G6PD); Glutathione peroxidase 4 (GPX4); oxidized glutathione (GSSG); nicotinamide adenosine dinucleotide hydrogen phosphate (NADPH); Nuclear factor E2 related factor 2 (NRF2)
The canonical system XC-/GSH/GPX4 pathway
Amino acid metabolism is an important part of the metabolic cycle of organisms, and abnormal amino acid metabolism is closely related to ferroptosis. Cystine/glutamic acid reverse transporter (system Xc-) plays an important role in maintaining the balance and distribution of amino acids and is a very important antioxidant system in cells. Its inactivation of the cellular antioxidant system by downregulation or inhibition of the Cystine/glutamic acid reverse transporter (system Xc-) is a major determinant of the suceptibility to ferroptosis. System XC- consists of the light chain xCT/solute carrier family 7 member 11 (SLC7A11) and the heavy chain 4F2hc/solute carrier family 3 member 2 (SLC3A2), and SLC3A2 is a chaperone that facilitates momemnt of SLS7A11 to the plasma surface and SLC7A11 forms the transport channel in its oxidated form [8]. Cystine is transported intracellularly by system XC- then transformed into cysteine. Cysteine is the rate-limiting amino acid for GSH (a vital intracellular antioxidant) production. Moreover, GPX4, a member of the selenium family containing GPXs, is a recognized negative regulator of ferroptosis. It is an enzyme for the reduction of toxic peroxides (L-OOH) to non-toxic lipid alcohols (L-OH) [9, 10]. It was shown that GSH is an essential cofactor of GPX4 and can influence the GPX4 function [11]. Therefore, system XC-mediated cysteine can also indirectly affect GPX4 activity. Furthermore, GSH synthesis requires the nicotinamide adenosine dinucleotide hydrogen phosphate (NADPH) cycle to supply ATP.
Lipid metabolism pathway
Lipids are important regulators of cell death, and the accumulation of lipid peroxides is thought to be an important driver of ferroptosis [12]. Although the exact source of lipid peroxides is unknown, polyunsaturated fatty acids (PUFAs) have been identified as an important source. PUFAs are an important component of cell membranes and they can perform many cellular functions by enhancing cell mobility. However, they contain unstable carbon–carbon double bonds that can generate lipid reactive oxygen species, which can cause ferroptosis when accumulated in excess [13]. Among PUFAs, arachidonic acid (AA) and adrenoic acid (ADA) are the 203 main substrates causing lipid peroxidation during ferroptosis [14]. In contrast, acyl-coenzyme A synthase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are required for the biosynthesis and remodeling of AA/AdA derivatives. Both can catalyze the formation of AA/AdA-CoA derivatives and AA/AdA-phosphatidylethanolamine (AA/AdA-PE) from free AA/AdA. AA/AdA-PE then synthesizes lipid peroxides AA/AdA-hydroperoxide-PE (AA/AdA-OOH-PE) through enzymatic and non-enzymatic reactions [15]. Lipid peroxides themselves and their degradation products (malondialdehyde (MDA) and 4-hydroxynonenal (4-HNEs)) produce cytotoxicity and cause cell death [16]. Moreover, the degradation process involves cyclooxygenase-2 (COX2) and nicotinamide adenine dinucleotide phosphate oxidases 2 (NOX2), among others [17].
Iron metabolism pathway
Iron has a dual role in cell growth. Although iron is a trace element essential for cell proliferation, its excessive accumulation can cause cell damage and increase the risk of diseases such as tumors [7]. Iron ions are also an important component in the accumulation of lipid peroxides and the initiation of iron death. The key to iron metabolism is the regulation of iron pool capacity, which mainly includes iron uptake, storage and export.
-
(1)
Iron ions are transferred into the cytosol through multiple pathways. In one respect, transferrin (Tf) and lactotransferrin (LTF) store extracellular iron as Fe3+, which is then bound to the transferrin receptor (TfR) and another unknown receptor on the cell membrane, and Fe3+ is endocytosed to form endo nucleosomes [18, 19]. In the endosome, the metal reductase six transmembrane epithelial antigen of protein 3 (STEAP3) reduces Fe3+ to Fe2+. On the contrary, solute carrier family 39-member 14 (SLC39A14/ZIP14) and solute carrier family 39-member 8 (SLC39A8/ZIP8) transfer Fe2+ directly into the intracellular compartment by transporting non-transferrin-bound iron (NTBI) to the cell membrane [20].
-
(2)
Multiple mechanisms maintain the equilibrium of Fe2+ in the cytoplasm. Poly C-binding protein 1/2 (PCBP1/2) oxidizes most Fe2+ to Fe3+, which is stored in ferritin (composed of light chain (FTL) and heavy chain 1 (FTH1)), which itself can be degraded to increase free iron levels; iron regulatory protein (IRP1/2) promotes the free iron utilization in cells in multiple pathways; and heme oxygenase 1 (HO-1), regulated by the nuclear factor E2-related factor 2 (Nrf2 / NFE2L2) gene, catalyzes the degradation of heme to produce Fe2+ [21].
-
(3)
Iron efflux protein solute carrier family 40 member 1 (SLC40A1/ferroportin1/FPN) and ferritin transfer out protein Prominin2 can facilitate the export of intracellular ferric ions and ferritin [22]. When the intracellular iron metabolic pathway is abnormal, and an unstable iron pool is formed, Fe2+ then generates ROS through the Fenton reaction [1] or participates in the iron-containing lipoxygenase activation [23], triggering lipid peroxidation, leading to cell damage. This process is known as ferroptosis.
In conclusion, iron is crucial to the physiological functioning of cells. A lack of iron can cause cells to malfunction, whereas an abundance of iron can cause oxidative stress on cells and ferroptosis.
Other metabolic pathways
P53, the “star molecule” of oncology, is a double-edged sword in ferroptosis. P53 is a SLC7A11 transcriptional repressor, which increases cellular sensitivity to ferroptosis through SLC7A11 in a GPX4-dependent or non-dependent pathway [24]. Additionally, P53 negatively regulates ferroptosis by acting on dipeptidyl peptidase 4 (DPP4) or by inducing cell cycle protein-dependent kinase inhibitor 1A (CDKN1A/p21) [25].
The transcription factor Nrf2 is involved in antioxidant responses, and various iron and lipid metabolism factors are among its target genes [26]. Thus, Nrf2 can counteract ferroptosis by regulating intracellular iron ion content [27], GPX4 levels [28], and the NAPDH cycle [29].
The flavin protein apoptosis-inducing factor mitochondrial-associated 2 (AIFM2), subsequently renamed ferroptosis inhibitory protein 1 (FSP1) [30], regulates ferroptosis negatively. Interestingly, its function is independent of cellular GSH levels and GPX4 activity. FSP1 catalyzes CoQ10 regeneration with NAD(P)H and influences ferroptosis progression by an independent pathway FSP1-CoQ10-NAD(P)H [31].
Effect of ncRNA-mediated ferroptosis on tumor progression
ncRNAs are a unique class of RNAs transcribed from genes that do not encode proteins [32]. In addition to playing significant functions at the transcriptional and post-transcriptional levels, they can also govern the course of human disease through epigenetic alterations. The involvement of ncRNAs in regulating the progression of various cancer types has been well documented, and targeting ncRNAs has shown promising clinical therapeutic effects, which we will not repeat here. Recent studies have revealed that ncRNAs play an important role in regulating the progression of various cancer types through the iron death pathway, which can regulate iron death-related gene expression through epigenetic, transcriptional and translational modalities. They play a role in tumorigenesis, progression, treatment and prognosis. Although the role of ncRNAs in iron death is not yet fully defined, it has an invaluable role in the targeting of cancer therapy [33, 34]. The main relevant ncRNAs identified so far are microRNA (miRNA), long ncRNA (lncRNA) and circular RNA (circRNA).
miRNAs and ferroptosis
miRNAs exhibit function primarily by binding to and regulating the expression of the 3′-untranslated region of the target mRNA [35]. Since more than 60% of coding genes are potential targets of miRNAs [5], miRNAs among ncRNAs are the most widely studied. miRNAs can regulate ferroptosis key molecules in various cancer cells and participate in tumor progression in numerous ways, which we have sorted it out in detail (Table 1).
Previous studies have shown that a single miRNA can be involved in ferroptosis by regulating iron death-related genes in multiple cancers simultaneously, such as miR-324-3p, miR-200a and miR-7-5p. miR-324-3p was reported to be significantly downregulated in cis-diamminedichloroplatinum II (DDP, aka cisplatin)-resistant lung adenocarcinoma cells and increased the resistant cells' sensitivity to cisplatin by targeting GPX4 [36]. Meanwhile, metformin could promote ferroptosis by the miR-324-3p/GPX4 axis in breast cancer [37]. Additionally, the miR-200 family is known for its down-regulation in human tumor cells. By targeting important mRNAs involved in epithelial mesenchymal transition (EMT) (ZEB1 and ZEB2), -catenin/Wnt signaling (-catenin), EGFR inhibitor resistance (ERRFI-1), and chemoresistance to therapeutic drugs, it plays a critical role in reducing EMT, tumor cell adhesion, migration, invasion, and metastasis. As a ferroptosis regulator, NRF2 has antioxidant properties, and its levels are regulated by Keap1. It has been reported that miR-200a regulates the Keap1/Nrf2 pathway in the mammary epithelium [38], and methylseleninic acid (MSA) can act as a chemopreventive agent for oesophageal squamous cell carcinoma (ESCC) cells by the KLF4/miR-200a/Keap1/Nrf2 axis [39]. Although miR-200a can regulate essential ferroptosis components, its involvement in ferroptosis has not been experimentally confirmed. Moreover, miR-7-5p was highly expressed in radiation-resistant ovarian, oral squamous cell carcinoma, and hepatocellular carcinoma cell lines and affected ferroptosis by downregulating the mitochondrial iron transporter protein Mitoferrin and decreasing Fe2+ [40]; and later, Kazuo et al. demonstrated that miR-7-5p was upregulated in radiation-resistant cells of cervical cancer and was involved in the cellular regulation of ROS, mitochondrial membrane potential, and Fe2+ level regulation and affects the ALOX12 and HIF1α expression [41].
miRNA is an important exosome component, and it has been detected in exosomes of several cell types [42]. 15-lipoxygenase (ALOX15) is closely associated with the accumulation of lipid ROS in cancer cells [43]. Cisplatin and paclitaxel promote miR-522 secretion by cancer-associated fibroblasts (CAFs) through the USP522/hnRNPA7 axis, thereby downregulating ALOX15 and reducing ROS production in cancer cells, ultimately leading to chemoresistance [44]. This study confirms the occurrence of ferroptosis in tumor microenvironment-associated exosomes for the first time. Moreover, exosomal miR-4443 was highly expressed in cisplatin-resistant non-small cell lung cancer (NSCLC) cells. Further studies revealed that miR-4443 could target methyltransferase-like 3 (METTL3), thereby reducing the N6 methyladenosine (m6A) level in cells, while the FSP1 expression is regulated by m6A modifications. Overall, miR-4443 regulates the FSP1 expression by METTL3 in an m6A-like manner, which in turn is involved in ferroptosis and confers cisplatin resistance to NSCLC cells [45].
To summarize Table 1 we found that different miRNAs can regulate iron ion levels through different pathways, and an imbalance of iron ions can lead to uncontrolled miRNA expression. Also, miRNAs and NRF2 exist to regulate each other. In conclusion, miRNAs are involved in potential regulatory mechanisms of ferroptosis, including various pathways such as mitochondria-associated proteins, iron metabolism, glutathione metabolism and lipid peroxidation, and in turn, miRNAs and ROS can regulate each other in various pathways.
lncRNA and ferroptosis
lncRNA has a longer sequence than miRNA. It mainly acts as a regulator of transcription factors in the nucleus or as a sponge for miRNAs in the cytoplasm [46].
Unlike miRNAs, lincRNAs can operate as miRNA sponges to indirectly regulate the cell death process and act directly on ferroptosis key genes and proteins. The most recent research on the role of lincRNAs in ferroptosis is described in Table 2.
Stearoyl coenzyme A desaturase 1 (SCD1) is a mechano reactive enzyme that reprograms lipid metabolism in gastric cancer stem cells (GCSC) and participates in ferroptosis. In contrast, exosomal lncFERO (exo-lncFERO) regulates SCD1 mRNA levels, causing PUFA dysregulation and subsequent ferroptosis inhibition. This enhances dryness and regulates chemosensitivity in the body [47].
lncPVT1 is upregulated in various cancers [48,49,50]. It is involved in tumor cell proliferation, migration, autophagy, apoptosis, and EMT. It promotes the malignant progression of tumors through physiological or pathological mechanisms like hypoxia and exosomes [50, 51], which are potential therapeutic targets for human cancers. According to studies, the therapeutic anesthetic ketamine can limit hepatocarcinoma viability and induce ferroptosis. Moreover, lncPVT1 can interact with miR-214-3p and hinder it from acting as a sponge for GPX4, effectively responding to ketamine-induced ferroptosis [52].
Cancer genomic databases and bioinformatics analysis have identified many differentially expressed IncRNAs with prognostic value associated with ferroptosis [53,54,55]. However, these IncRNAs still lack experimental confirmation of their potential as ferroptosis markers.
Overall, lncRNAs can affect ROS metabolism directly or indirectly through a variety of mechanisms including GPX4, ferric ions, SLC7A11 and, conversely, lncRNAs are regulated by them.
circRNA and ferroptosis
CircRNA is a single-stranded RNA molecule in a covalently closed loop. Therefore, it is nucleic acid exonuclease resistant and exhibits high stability in the body [56]. Simultaneously, its high abundance is tissue- and stage-specific [57]. This provides an advantage for circRNAs to act as biomarkers and targets for cancer therapy.
Several studies have revealed a relationship between circRNA and ferroptosis. circRNAs can mediate ferroptosis through multiple mechanisms in many tumor types (Table 3). Compared to the nucleus, circRNAs are more often found in the cytoplasm and act as sponges for miRNAs that regulate the target genes' expression [58].
Tumor resistance can significantly compromise clinical efficacy. circ-BGN was first found to be highly expressed in trastuzumab-resistant HER2-positive breast cancer. Further studies revealed that circ-BGN could act directly on SLC7A11, a core molecule of ferroptosis, and enhanced OTUB1-mediated deubiquitination of SLC7A11, thereby inhibiting ferroptosis. The conclusion was also confirmed by in vivo experiments [59]. hsa_circ_0000745 has the potential to act as a diagnostic marker for cervical cancer, gastric cancer, and other cancers [60, 61]. Yanbi et al. recently found that circ_0000745 involves cell cycle progression, glycolytic metabolism, apoptosis, and ferroptosis in acute lymphoblastic leukemia. Furthermore, this role is accomplished through the circ_0000745/miR-494-3p/NET1 axis [62]. It has been reported that circKIF4A can promote numerous tumor progressions and mediate glycolytic metabolism and drug resistance through competitive endogenous RNA mechanism mechanism [63,64,65]. In papillary thyroid cancer, circKIF4A negatively regulates ferroptosis and promotes tumor proliferation in vitro and in vivo. In essence, circKIF4A can absorb miR-1231 to increase GPX4 levels [66].
In general, circRNAs could be potential therapeutic targets for the treatment of cancer through the ferroptosis pathway.
In this section, we systematically summarize the ncRNAs associated with ferroptosis in cancer to date and explore the regulatory role of ncRNAs in cancer progression and iron death, which implies that ncRNAs have great potential as anti-cancer therapeutic targets through regulation of ferroptosis. Moreover, ferroptosis-related ncRNAs are individually heterogeneous across tumors, which has significant implications for personalised tumor therapy.
Despite the full potential of ferroptosis-related ncRNAs, there are still many unanswered questions. Although a clear regulatory role for ncRNAs in the development of ferroptosis in tumors has been identified, little is still known about the in-depth mechanisms underlying this component. This makes the clinical application of ncRNA-dependent approaches to ferroptosis a major obstacle. Furthermore, to translate basic research into clinical trials, the construction of additional animal models to validate the role of ncRNAs in ferroptosis is a must. In addition, given the shortcomings of conventional treatment options for tumors, research on the application of biomaterials such as molecular nanomaterials for targeted tumor ferroptosis therapy is urgently needed. Besides, due to the diversity of ncRNA biological functions, targeting ncRNA therapy is likely to cause some complications and cause damage to non-tumor organs. For example, miR-375-3p and miR-214-3p, which have the potential to both promote ferroptosis in tumor cells of cervical cancer and HCC, may also cause fibrosis of cardiomyocytes and acute renal impairment[67, 68]. It is therefore important to achieve tumor-targeted metastasis of ncRNAs, and multidisciplinary cross-fertilisation will facilitate this process.
Relationship between ferroptosis and other PCDs
Abnormal cell death regulation is an important feature of cancer. PCDs are highly involved in tumor development, including apoptosis, necroptosis, autophagy, pyroptosis, ferroptosis, and cuproptosis. Therefore, exploring the mechanisms of different types of cell death is of great importance in cancer. Researchers have discovered that ferroptosis is independent and connected to other types of cell death and that its essential regulators are also involved in regulating other types of cell death [69]. These death types usually share a common pathway [70]. Consequently, further investigation of the inter regulation of ferroptosis with other types of programmed cell death and developing strategies that can trigger numerous planned cell deaths are extremely promising cancer treatment strategies.
Apoptosis and ferroptosis
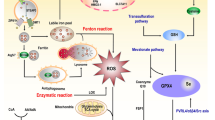
Apoptosis is a form of cellular suicide induced by the activation of intracellular death programs and was initially thought to be the only way of PCD. It is an intrinsic tumor suppressor mechanism that physically displays cellular crumpling, chromatin aggregation, and the production of apoptotic vesicles followed by phagocytosis [2]. Mechanistically, apoptosis consists of three main aspects: oxidative damage, imbalance of calcium homeostasis and mitochondrial damage. Apoptosis can be initiated by ncRNAs through regulation of the relevant receptors or as cerRNAs.
Death structural domain-associated protein (Daxx) mediates apoptosis through the Fas-Daxx-ASK1-JNK1 axis, while the ferritin FTH1 inhibits the action of Daxx [71]. Ferroptosis inducer erastin activates the C/EBP homogenic protein (CHOP) signal pathway, affecting the expression of p53 non-dependent PUMA and increasing sensitivity to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induced cell death [72]. Furthermore, apoptosis may be directly transformed into ferroptosis [73].
Necroptosis and ferroptosis
Necroptosis is an alternate cell death mechanism triggered when apoptosis is blocked and is a degenerative pathology caused by damaging factors. Morphological features include cell swelling, membrane rupture, release of cytoplasmic contents and chromosome condensation. The basic molecular mechanism consists of receptor-interacting kinases (RIPK1 and RIPK3) and mixed-spectrum kinase structural domain-like pseudokinases (MLKL). The RIPK1/RIPK3 complex recruits and phosphorylates MLKL translocates to the plasma membrane, and forms channels, releasing damage-associated molecular patterns (DAMPs), permeabilization of the plasma membrane, and release of contents [74].
By activating the mitochondrial permeability transition pore (MPTP) and phosphorylating RIPK1, iron excess induces necrotic apoptosis in ischemic stroke. Heat shock protein 90 (HSP90) is an evolutionarily conserved and commonly expressed molecular chaperone. It intensifies RIPK1 phosphorylation, inhibits GPX4 activity, and can induce necroptosis and ferroptosis [75]. Thus, HSP90 acts as a co-regulatory node for necroptosis and iron sagging. ferroptosis and necroptosis are known to be positively regulated by ACSL4 and MLKL, respectively. In a mouse model of renal ischemia–reperfusion injury, ACSL4 and MLKL knockdown modulate the sensitivity of necroptosis and ferroptosis, respectively [76]. This led us to wonder if ferroptosis and necroptosis have complementing processes reasonably. Therefore, it is essential to continue to explore the relationship between ferroptosis and necroptosis.
Autophagy and ferroptosis
Autophagy is a process by which cells ‘self-feed’. Under physiological conditions, basal autophagy is a cellular self-protection mechanism, while induced autophagy under stressful conditions may cause cell death. Morphologically, it is characterised by the accumulation of autophagic vesicles and cytoplasmic vesiculation without chromatin condensation [77]. There are three main forms of autophagy: microautophagy, macroautophagy, and chaperone-mediated autophagy (CMA). Autophagy begins mechanistically with pre-autophagic structures in the cytoplasm, which create autophagosomes after phagocytosis of damaged organelles and denatured macromolecules. Subsequently, autophagosomes combine with lysosomes to generate autolysosomes, which destroy the contents of autophagosomes [77].
In exploring the relationship between autophagy and ferroptosis, we once again identified HSP90. HSP90 increases the protein stability of CMA receptor lysosome-associated membrane protein 2A (LAMP2A) to accelerate GPX4 degradation and enhance ferroptosis [78]. Zili et al. found that increased BECN1 mRNA stability with the involvement of ELAVL1 caused ferritin phagocytosis and subsequent ferroptosis [79]. While in Parkinson's disease (PD), FTH1 overexpression inhibits ferritin phagocytosis and, ultimately, ferroptosis [80]. We, therefore, hypothesize that ferritin phagocytosis (a sort of selective autophagy) may have a good connection with ferroptosis. Nuclear receptor coactivator 4 (NCOA4) has been reported to be involved in autophagy-dependent ferritin degradation [81], and NCOA4 overexpression can contribute to ferritin degradation and promote increased free iron and subsequent ferroptosis [82]. Interestingly, intracellular free iron regulates NCOA4 levels [81]. Moreover, RAB7A and SQSTM1 are regulators of lipophagy and clockophagy, respectively, and their downregulation prevents lipid peroxidation-dependent ferroptosis [83, 84]. High mobility group box-1 protein (HMGB1) is a DAMP, and its relationship with autophagy and ferroptosis is more complex. On one side, autophagy-dependent ferroptosis can increase the HMGB1 release [85], whereas HMGB1 can be engaged in the advancement of autophagy and ferroptosis [86, 87]. Recent studies have revealed that hippocampal calmodulin-like 1 (HPCAL1) is an autophagy receptor that affects membrane tension by regulating CDH2, which further affects lipid peroxidation and ultimately inhibits ferroptosis in vitro and in vivo [88]. Another autophagy receptor, Tax1 (human T cell leukemia virus type I) binding protein 1 (TAX1BP1), promotes GPX4 degradation and subsequent ferroptosis in response to copper stress [89]. The above studies suggest a close association between autophagy and ferroptosis.
Pyroptosis and ferroptosis
Programmed cell death induced by inflammatory vesicles mediated by gasdermins is known as cell scorch death and can amplify local or systemic inflammatory effects [90]. Unique to cell death by scorch is the formation of many bubble-like protrusions, known as scorch vesicles, within the cell. Mechanistically, inflammatory vesicles sense danger and recruit and activate caspase 1, which stimulates inflammatory proteins that cleave gastrin D (GSDMD), causing it to attach to the cell membrane and generate pores, which is the conventional mechanism of scorch death. The non-classical pathway of scorch death is mainly mediated by cystatase-4, caspase-5, and caspase-11 [91].
We found that there are multiple co-stimulatory factors for scorch death and ferroptosis. Transcription factor P53 is an important regulatory molecule of ferroptosis. Moreover, in NSCLC, P53 can directly increase scorch death and inhibit tumor growth [92]. In a myocardial fibrosis model, MLK3 regulates ferroptosis and scorch death through the JNK/p53 pathway and the NF-κB/NLRP3 pathway, while miR-351 can inhibit MLK3 expression [93]. Additionally, elevated ferric ions and ROS levels can induce scorch death and ferroptosis. Rui et al. found synergistic effects of scorch death and ferroptosis using dual-induced nano drugs [94]. Furthermore, iron-activated ROS can induce scorch death in melanoma through the Tom20-Bax-caspase-GSDME axis [95]. Another study found that in macrophages, GPX4, a core regulatory protein of ferroptosis, can block GSDMD activity and trigger scorch death by reducing lipid peroxidation. Interestingly, HMGB1 levels were thus altered, eventually leading to sepsis [96]. In conclusion, the regulatory relationship between scorch death and ferroptosis should be explored in depth.
Cuproptosis and ferroptosis
Copper is a key factor in cell signaling, and cell death induced by copper overload was found to be a new form of cell death called cuproptosis. The main targets of copper death are the mitochondria, which are morphologically characterised by mitochondrial wrinkling and mitochondrial membrane rupture. Both copper ion carrier induction and dysregulation of copper homeostasis lead to copper death. Copper binds to lipases in the tricarboxylic acid (TCA) cycle, leading to protein aggregation, proteotoxic stress, and cell death [97].
Elesclomol (ES) is a copper ion carrier. In CRC cells, ES allows copper ions to be retained in mitochondria, leading to ROS accumulation, promoting SLC7A11 degradation, and increasing susceptibility to ferroptosis [98]. Given the novelty of cuproptosis, its relationship with ferroptosis has not been extensively studied.
Based on the initial investigation, we have generated Fig. 2, in which molecules such as HSP90, HMGB1, and P53 show multiple times. Thus, are there shared regulatory proteins and signaling pathways between ferroptosis and other PCDs? Is this sharing related to the positive correlation between ferroptosis and other forms of death? Can we suppress multiple death pathways through this sharing? Hopefully, these questions can be addressed in subsequent studies. Although many of the study subjects are non-tumor disorders, this suggests the complexity of the relationships between ferroptosis and other PCDs, hence pointing the way for future tumor-related research.

The mutual regulatory mechanisms between ferroptosis and other forms of death. The various initiators and effector molecules involved in ferroptosis, apoptosis, necroptosis, autophagy, pyroptosis and cuproptosis can interact to promote cell death
Role of ncRNA in crosstalk between ferroptosis and other PCDs in tumors
ncRNAs are important regulators of eukaryotic gene expression, and many ncRNAs have been found to mediate PCD to influence tumor malignant progression. The data above demonstrate the relationship and similarities between ferroptosis and numerous forms of cell death. Without a doubt, ncRNAs participate in regulating crosstalk between these PCDs. This section provides a summary of relevant studies (Table 4).
Zuli et al. found that LINC00618 promotes apoptosis by increasing BCL2-related X (BAX) levels and cleaved caspase-3 and by repressing SLC7A11 transcription through lymphatic-specific decapping enzymes (LSH) to promote ferroptosis. However, ferroptosis initiated by LINC00618 depends on vincristine (VCR)-triggered apoptosis. Thus, LINC00618 promotes ferroptosis in an apoptosis-dependent manner [99]. Additionally, many ncRNAs are involved in cancer progression by simultaneously regulating apoptosis and ferroptosis. For example, the methylation-modified lncRNA P53RRA is down-regulated in lung cancer and promotes nucleoplasmic translocation of p53 by interacting with G3BP1, ultimately leading to cell cycle arrest, apoptosis, and ferroptosis [99]. Another study found that the oncogenic factor circDTL upregulates GPX4 by acting as a ceRNA competing for binding with miR-1287-5p, ultimately inhibiting ferroptosis and apoptosis [100].
The link between ferroptosis and autophagy appears to be closer. ALKBH5 is a negative regulator of autophagic flux, and cIARS decreases ferroptosis via inhibiting ALKBH5-mediated autophagy, which increases sorafenib (SF) resistance in HCC cells [101]. Oncology studies have shown that LINC00551 inhibits cell viability in lung adenocarcinoma (LUAD). Mechanistically, LINC00551 inhibits mTOR activity through the miR-4/DDIT4 signaling pathway, upregulates autophagy levels, and then promotes ferroptosis in an autophagy-dependent manner [102]. Recent studies have found that lincRNA NEAT1 is involved in ferroptosis and autophagy induced by gambogenic acid (GNA), a natural anticancer compound, through SLC7A11 / GPX4 and AMPK / mTOR axis in melanoma [103].
With the preceding data, we hypothesize that ferroptosis, apoptosis, and autophagy have synergistic effects. However, there are few reports on the ncRNAs regulation in tumors in the crosstalk between ferroptosis and other PCDs, and the corresponding regulatory relationships still need further study.
Conclusion
Recently, there has been considerable interest in developing cancer drugs targeting the PCD pathway. Besides, ferroptosis has attracted much attention as a newly discovered form of cell death. Although ferroptosis research has surged in recent years, many questions remain unresolved. To address the direction of this review, the following questions and perspectives are presented.
First and foremost, the ultimate triggering cause for ferroptosis is unknown. Although iron and lipid peroxide accumulation are critical stages, not all lipid peroxidation damage leads to cellular ferroptosis. Then, it remains to be investigated whether lipid peroxidation reaches a certain threshold to cause plasma membrane rupture directly; or needs to be activated by some unknown molecule to cause the final effect phase.
Although a growing number of ncRNAs have been linked to the regulation of ferroptosis, the regulatory mechanisms remain poorly understood. Furthermore, there is still a lack of ferroptosis-specific markers for clinical diagnosis. Notably, novel small ncRNAs such as PIWI-interacting RNA (piRNA) and tRNA-derived small RNA (tsRNA) have been shown to have biological functions in cancer. What role do they play in ferroptosis?
Endoplasmic reticulum (ER) stress, redox stress, and mitochondrial dysfunction appear to be common pathways for multiple death types [104]. Investigating the biological relevance of ferroptosis to other PCDs is of great interest. Nevertheless, the findings discussed in Part V indicate the complexity of this relationship. Furthermore, there are limited investigations on the role of ncRNAs in the crosstalk between ferroptosis and other forms of crosstalk. Future research may reveal if we may adversely regulate many death pathways through a single target.
The advantages of ncRNA as tumour prevention, monitoring treatment response and prognosis have been illustrated in the literature and have yielded some promising results in the clinic [105]. However, the clinical application of ferroptosis and thus tumour suppression through an ncRNA-dependent approach faces significant obstacles. On the one hand, the lack of understanding of specific mechanisms has led to limited application of ncRNA modifying agents in ferroptosis. On the other hand, although promoting cellular ferroptosis can inhibit tumour progression, will it be accompanied by damage to other non-tumour organs or fibrosis? In addition, ncRNA-based therapies inherently have many limitations, such as instability and tolerability [106]. Due to the instability of ncRNAs, the mode of transport has a significant impact on the efficiency of transport. Currently, nanoparticle-based, phage-based and other delivery methods are being optimized. Also, ncRNAs, being RNAs, are likely to be recognized and cleared by the immune system. It is hoped that the next generation of ncRNA therapies will overcome these drawbacks and allow for real clinical applications.
Availability of data and materials
Not applicable.
Abbreviations
- ncRNAs:
-
Non-coding RNAs
- PCD:
-
Programmed cell death
- GSH:
-
Glutathione
- Tf:
-
Transferrin
- LTF:
-
Lactotransferrin
- TfR:
-
Transferrin receptor
- STEAP3:
-
Six transmembrane epithelial antigen of protein 3
- SLC39A14/ZIP14:
-
Solute carrier family 39 member 14
- SLC39A8/ZIP8:
-
Solute carrier family 39 member 8
- NTBI:
-
Non-transferrin-bound iron
- PCBP1/2:
-
Poly C-binding protein 1/2
- FTL:
-
Light chain
- FTH1:
-
Heavy chain 1
- IRP1/2:
-
Iron regulatory protein
- HO-1:
-
Heme oxygenase 1
- Nrf2 / NFE2L2:
-
Transcription factor nuclear factor E2-related factor 2
- SLC40A1 / ferroportin1 / FPN:
-
Iron efflux protein solute carrier family 40 member 1
- ROS:
-
Reactive oxygen species
- PUFAs:
-
Polyunsaturated fatty acids
- AA:
-
Arachidonic acid
- AdA:
-
Adrenoic acid
- ACSL4:
-
Acyl coenzyme A synthase long chain family member 4
- LPCAT3:
-
Lysophosphatidylcholine acyltransferase 3
- AA / AdA-PE:
-
AA/AdA-phosphatidylethanolamine
- AA / AdA-OOH-PE:
-
AA/AdA-hydroperoxide-PE
- MDA:
-
Malondialdehyde
- 4-HNEs:
-
4-Hydroxynonenal
- COX2:
-
Cyclooxygenase-2
- NOX2:
-
Nicotinamide adenine dinucleotide phosphate oxidases 2
- SLC7A11:
-
Solute carrier family 7 member 11
- SLC3A2:
-
Solute carrier family 3 member 2
- GPX4:
-
Glutathione peroxidase 4
- L-OOH:
-
Peroxides
- L-OH:
-
Alcohols
- NADPH:
-
Nicotinamide adenosine dinucleotide hydrogen phosphate
- RSL3:
-
RAS selective lethal small molecule 3
- DPP4:
-
Dipeptidyl peptidase 4
- CDKN1A/p21:
-
Cell cycle protein-dependent kinase inhibitor 1A
- AIFM2:
-
Apoptosis-inducing factor mitochondrial-associated 2
- FSP1:
-
Iron death inhibitory protein 1
- miRNA:
-
MicroRNA
- lncRNA:
-
Long ncRNA
- circRNA:
-
Circular RNA
- DDP, aka cisplatin:
-
Cis-diamminedichloroplatinum II
- MSA:
-
Methylseleninic acid
- ESCC:
-
Oesophageal squamous cell
- ALOX15:
-
15-Lipoxygenase
- CAFs:
-
Cancer-associated fibroblasts
- METTL3:
-
Methyltransferase-like 3
- m6A:
-
Methyladenosine
- CSCs:
-
Cancer stem cells
- SCD1:
-
Stearoyl coenzyme A desaturase 1
- GCSC:
-
Gastric cancer stem cells
- exo-lncFERO:
-
Exosomal lncFERO
- EMT:
-
Epithelial mesenchymal transition
- Daxx:
-
Death structural domain-associated protein
- CHOP C/EBP:
-
Homogenic protein
- TRAIL:
-
Tumor necrosis factor-related apoptosis-inducing ligand
- RIPK1 and RIPK3:
-
Receptor-interacting kinases
- MLKL:
-
Mixed-spectrum kinase structural domain-like pseudokinases
- DAMPs:
-
Damage-associated molecular patterns
- MPTP:
-
Mitochondrial permeability transition pore
- HSP90:
-
Heat shock protein 90
- CMA:
-
Chaperone-mediated autophagy
- LAMP2A:
-
Lysosome-associated membrane protein 2A
- PD:
-
Parkinson’s disease
- NCOA4:
-
Nuclear receptor coactivator 4
- HMGB1:
-
High mobility group box-1 protein
- HPCAL1:
-
Hippocampal calmodulin-like 1
- TAX1BP1:
-
Tax1 (human T cell leukemia virus type I) binding protein 1
- GSDMD:
-
Gastrin d
- TCA:
-
Tricarboxylic acid
- ES:
-
Elesclomol
- BAX:
-
BCL2-related X
- LSH:
-
Lymphatic-specific decapping enzymes
- VCR:
-
Vincristine
- SF:
-
Sorafenib
- LUAD:
-
Lung adenocarcinoma
- GNA:
-
Gambogenic acid
- piRNA:
-
PIWI-interacting RNA
- tsRNA:
-
TRNA-derived small RNA
- ER:
-
Endoplasmic reticulum
- COAD:
-
Colorectal cancer
- GBM:
-
Glioblastoma
- GC:
-
Gastric cancer
- OSCC:
-
Oral squamous cell carcinomas
- CRC:
-
Colorectal cancer
- OC:
-
Ovarian cancer
- CCRCC:
-
Clear cell renal cell carcinoma
- UGC:
-
Upper gastrointestinal adenocarcinoma
- TPC:
-
Papillary thyroid cancer
- PCa:
-
Prostate cancer
- NPC:
-
Nasopharyngeal carcinoma
- ALL:
-
Acute lymphoblastic leukemia
- AML:
-
Acute myeloid leukemia
References
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25:486–541.
Mou Y, Wang J, Wu J, He D, Zhang C, Duan C, Li B. Ferroptosis, a new form of cell death: opportunities and challenges in cancer. J Hematol Oncol. 2019;12:34.
Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. Landscape of transcription in human cells. Nature. 2012;489:101–8.
Adams BD, Parsons C, Walker L, Zhang WC, Slack FJ. Targeting noncoding RNAs in disease. J Clin Invest. 2017;127:761–71.
Slack FJ, Chinnaiyan AM. The role of non-coding RNAs in oncology. Cell. 2019;179:1033–55.
Kuang F, Liu J, Tang D, Kang R. Oxidative damage and antioxidant defense in ferroptosis. Front Cell Dev Biol. 2020;8: 586578.
Lin CH, Lin PP, Lin CY, Lin CH, Huang CH, Huang YJ, Lane HY. Decreased mRNA expression for the two subunits of system xc(-), SLC3A2 and SLC7A11, in WBC in patients with schizophrenia: evidence in support of the hypo-glutamatergic hypothesis of schizophrenia. J Psychiatr Res. 2016;72:58–63.
Qin D, Wang J, Le A, Wang TJ, Chen X, Wang J. Traumatic brain injury: ultrastructural features in neuronal ferroptosis, glial cell activation and polarization, and blood-brain barrier breakdown. Cells. 2021;10:1009.
Maiorino M, Conrad M, Ursini F. GPx4, lipid peroxidation, and cell death: discoveries, rediscoveries, and open issues. Antioxid Redox Signal. 2018;29:61–74.
Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29:347–64.
Rishi G, Huang G, Subramaniam VN. Cancer: the role of iron and ferroptosis. Int J Biochem Cell Biol. 2021;141: 106094.
Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, Superti-Furga G, Stockwell BR. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 2015;10:1604–9.
Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90.
Yin H, Xu L, Porter NA. Free radical lipid peroxidation: mechanisms and analysis. Chem Rev. 2011;111:5944–72.
Feng H, Stockwell BR. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018;16: e2006203.
Ayala A, Muñoz MF, Argüelles S. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev. 2014;2014: 360438.
Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15:234–45.
Wang Y, Liu Y, Liu J, Kang R, Tang D. NEDD4L-mediated LTF protein degradation limits ferroptosis. Biochem Biophys Res Commun. 2020;531:581–7.
Chen X, Yu C, Kang R, Tang D. Iron metabolism in ferroptosis. Front Cell Dev Biol. 2020;8: 590226.
Chang LC, Chiang SK, Chen SE, Yu YL, Chou RH, Chang WC. Heme oxygenase-1 mediates BAY 11–7085 induced ferroptosis. Cancer Lett. 2018;416:124–37.
Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, Zhu LJ, Baer CE, Dixon SJ, Mercurio AM. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev Cell. 2019;51:575-586.e574.
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113:E4966-4975.
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, Gu W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62.
Kang R, Kroemer G, Tang D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic Biol Med. 2019;133:162–8.
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, Tang D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology. 2016;63:173–84.
Agyeman AS, Chaerkady R, Shaw PG, Davidson NE, Visvanathan K, Pandey A, Kensler TW. Transcriptomic and proteomic profiling of KEAP1 disrupted and sulforaphane-treated human breast epithelial cells reveals common expression profiles. Breast Cancer Res Treat. 2012;132:175–87.
Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. 2014;39:199–218.
Abdalkader M, Lampinen R, Kanninen KM, Malm TM, Liddell JR. Targeting Nrf2 to suppress ferroptosis and mitochondrial dysfunction in neurodegeneration. Front Neurosci. 2018;12:466.
Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Goya Grocin A, da Xavier Silva TN, Panzilius E, Scheel CH, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–8.
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–92.
Zhang J, Liu X, Li X, Cai Y, Zhou Y, Wang Q, Xu Z, Xia P, Yang P, Jun L, et al. The emerging role of noncoding RNA regulation of the ferroptosis in cardiovascular diseases. Oxid Med Cell Longev. 2022;2022:3595745.
Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–74.
Kim HK, Yeom JH, Kay MA. Transfer RNA-derived small RNAs: another layer of gene regulation and novel targets for disease therapeutics. Mol Ther. 2020;28:2340–57.
Tan W, Liu B, Qu S, Liang G, Luo W, Gong C. MicroRNAs and cancer: Key paradigms in molecular therapy. Oncol Lett. 2018;15:2735–42.
Deng SH, Wu DM, Li L, Liu T, Zhang T, Li J, Yu Y, He M, Zhao YY, Han R, Xu Y. miR-324-3p reverses cisplatin resistance by inducing GPX4-mediated ferroptosis in lung adenocarcinoma cell line A549. Biochem Biophys Res Commun. 2021;549:54–60.
Hou Y, Cai S, Yu S, Lin H. Metformin induces ferroptosis by targeting miR-324-3p/GPX4 axis in breast cancer. Acta Biochim Biophys Sin. 2021;53:333–41.
Eades G, Yang M, Yao Y, Zhang Y, Zhou Q. miR-200a regulates Nrf2 activation by targeting Keap1 mRNA in breast cancer cells. J Biol Chem. 2011;286:40725–33.
Liu M, Hu C, Xu Q, Chen L, Ma K, Xu N, Zhu H. Methylseleninic acid activates Keap1/Nrf2 pathway via up-regulating miR-200a in human oesophageal squamous cell carcinoma cells. 2015. Biosci Rep. https://doi.org/10.1042/BSR20150092
Tomita K, Fukumoto M, Itoh K, Kuwahara Y, Igarashi K, Nagasawa T, Suzuki M, Kurimasa A, Sato T. MiR-7-5p is a key factor that controls radioresistance via intracellular Fe(2+) content in clinically relevant radioresistant cells. Biochem Biophys Res Commun. 2019;518:712–8.
Tomita K, Nagasawa T, Kuwahara Y, Torii S, Igarashi K, Roudkenar MH, Roushandeh AM, Kurimasa A, Sato T. MiR-7–5p is involved in ferroptosis signaling and radioresistance Thru the generation of ROS in radioresistant HeLa and SAS cell lines. Int J Mol Sci. 2021;22:830.
O’Brien K, Breyne K, Ughetto S, Laurent LC, Breakefield XO. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat Rev Mol Cell Biol. 2020;21:585–606.
Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Rådmark O, Wurst W, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8:237–48.
Zhang H, Deng T, Liu R, Ning T, Yang H, Liu D, Zhang Q, Lin D, Ge S, Bai M, et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol Cancer. 2020;19:43.
Song Z, Jia G, Ma P, Cang S. Exosomal miR-4443 promotes cisplatin resistance in non-small cell lung carcinoma by regulating FSP1 m6A modification-mediated ferroptosis. Life Sci. 2021;276: 119399.
Wu ZY, Trenner M, Boon RA, Spin JM, Maegdefessel L. Long noncoding RNAs in key cellular processes involved in aortic aneurysms. Atherosclerosis. 2020;292:112–8.
Zhang H, Wang M, He Y, Deng T, Liu R, Wang W, Zhu K, Bai M, Ning T, Yang H, et al. Chemotoxicity-induced exosomal lncFERO regulates ferroptosis and stemness in gastric cancer stem cells. Cell Death Dis. 2021;12:1116.
Boloix A, Masanas M, Jiménez C, Antonelli R, Soriano A, Roma J, Sánchez de Toledo J, Gallego S, Segura MF. Long non-coding RNA PVT1 as a prognostic and therapeutic target in pediatric cancer. Front Oncol. 2019;9:1173.
Ghafouri-Fard S, Omrani MD, Taheri M. Long noncoding RNA PVT1: a highly dysregulated gene in malignancy. J Cell Physiol. 2020;235:818–35.
Wu BQ, Jiang Y, Zhu F, Sun DL, He XZ. Long noncoding RNA PVT1 promotes EMT and cell proliferation and migration through downregulating p21 in pancreatic cancer cells. Technol Cancer Res Treat. 2017;16:819–27.
Lai SW, Chen MY, Bamodu OA, Hsieh MS, Huang TY, Yeh CT, Lee WH, Cherng YG. Exosomal lncRNA PVT1/VEGFA axis promotes colon cancer metastasis and stemness by downregulation of tumor suppressor miR-152-3p. Oxid Med Cell Longev. 2021;2021:9959807.
He GN, Bao NR, Wang S, Xi M, Zhang TH, Chen FS. Ketamine induces ferroptosis of liver cancer cells by targeting lncRNA PVT1/miR-214-3p/GPX4. Drug Des Devel Ther. 2021;15:3965–78.
Xiao S, Liu X, Yuan L, Wang F. A ferroptosis-related lncRNAs signature predicts prognosis and therapeutic response of gastric cancer. Front Cell Dev Biol. 2021;9: 736682.
Zheng Z, Zhang Q, Wu W, Xue Y, Liu S, Chen Q, Lin D. Identification and validation of a ferroptosis-related long non-coding RNA signature for predicting the outcome of lung adenocarcinoma. Front Genet. 2021;12: 690509.
Zheng Z, Wu W, Lin Z, Liu S, Chen Q, Jiang X, Xue Y, Lin D. Identification of seven novel ferroptosis-related long non-coding RNA signatures as a diagnostic biomarker for acute myeloid leukemia. BMC Med Genomics. 2021;14:236.
Bach DH, Lee SK, Sood AK. Circular RNAs in cancer. Mol Ther Nucleic Acids. 2019;16:118–29.
Jeck WR, Sorrentino JA, Wang K, Slevin MK, Burd CE, Liu J, Marzluff WF, Sharpless NE. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19:141–57.
Guria A, Sharma P, Natesan S, Pandi G. Circular RNAs-the road less traveled. Front Mol Biosci. 2019;6:146.
Wang S, Wang Y, Li Q, Li X, Feng X. A novel circular RNA confers trastuzumab resistance in human epidermal growth factor receptor 2-positive breast cancer through regulating ferroptosis. Environ Toxicol. 2022;37:1597–607.
Wang YW, Xu Y, Wang YY, Zhu J, Gao HD, Ma R, Zhang K. Elevated circRNAs circ_0000745, circ_0001531 and circ_0001640 in human whole blood: potential novel diagnostic biomarkers for breast cancer. Exp Mol Pathol. 2021;121: 104661.
Huang M, He YR, Liang LC, Huang Q, Zhu ZQ. Circular RNA hsa_circ_0000745 may serve as a diagnostic marker for gastric cancer. World J Gastroenterol. 2017;23:6330–8.
Yang X, Li Y, Zhang Y, Liu J. Circ_0000745 promotes acute lymphoblastic leukemia progression through mediating miR-494-3p/NET1 axis. Hematology. 2022;27:11–22.
Luo K, Liu A, Wu H, Liu Q, Dai J, Liu Y, Wang Z. CircKIF4A promotes glioma growth and temozolomide resistance by accelerating glycolysis. Cell Death Dis. 2022;13:740.
Huang J, Deng X, Chen X, Chang Z, Lu Q, Tang A, Liu P. Circular RNA KIF4A promotes liver metastasis of breast cancer by reprogramming glucose metabolism. J Oncol. 2022;2022:8035083.
Shi YR, Wu Z, Xiong K, Liao QJ, Ye X, Yang P, Zu XB. Circular RNA circKIF4A sponges miR-375/1231 to promote bladder cancer progression by upregulating NOTCH2 expression. Front Pharmacol. 2020;11:605.
Chen W, Fu J, Chen Y, Li Y, Ning L, Huang D, Yan S, Zhang Q. Circular RNA circKIF4A facilitates the malignant progression and suppresses ferroptosis by sponging miR-1231 and upregulating GPX4 in papillary thyroid cancer. Aging. 2021;13:16500–12.
Zhuang Y, Yang D, Shi S, Wang L, Yu M, Meng X, Fan Y, Zhou R, Wang F. MiR-375-3p promotes cardiac fibrosis by regulating the ferroptosis mediated by GPX4. Comput Intell Neurosci. 2022;2022:9629158.
Zhou J, Xiao C, Zheng S, Wang Q, Zhu H, Zhang Y, Wang R. MicroRNA-214-3p aggravates ferroptosis by targeting GPX4 in cisplatin-induced acute kidney injury. Cell Stress Chaperones. 2022;27:325–36.
Wang D, Tang L, Zhang Y, Ge G, Jiang X, Mo Y, Wu P, Deng X, Li L, Zuo S, et al. Regulatory pathways and drugs associated with ferroptosis in tumors. Cell Death Dis. 2022;13:544.
Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N. Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta. 2013;1833:3448–59.
Liu F, Du ZY, He JL, Liu XQ, Yu QB, Wang YX. FTH1 binds to Daxx and inhibits Daxx-mediated cell apoptosis. Mol Biol Rep. 2012;39:873–9.
Hong SH, Lee DH, Lee YS, Jo MJ, Jeong YA, Kwon WT, Choudry HA, Bartlett DL, Lee YJ. Molecular crosstalk between ferroptosis and apoptosis: emerging role of ER stress-induced p53-independent PUMA expression. Oncotarget. 2017;8:115164–78.
Zheng DW, Lei Q, Zhu JY, Fan JX, Li CX, Li C, Xu Z, Cheng SX, Zhang XZ. Switching apoptosis to ferroptosis: metal-organic network for high-efficiency anticancer therapy. Nano Lett. 2017;17:284–91.
Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, Liu ZG. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci USA. 2012;109:5322–7.
Zhou Y, Liao J, Mei Z, Liu X, Ge J. Insight into crosstalk between ferroptosis and necroptosis: novel therapeutics in ischemic stroke. Oxid Med Cell Longev. 2021;2021:9991001.
Müller T, Dewitz C, Schmitz J, Schröder AS, Bräsen JH, Stockwell BR, Murphy JM, Kunzendorf U, Krautwald S. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell Mol Life Sci. 2017;74:3631–45.
Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–21.
Wu Z, Geng Y, Lu X, Shi Y, Wu G, Zhang M, Shan B, Pan H, Yuan J. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci USA. 2019;116:2996–3005.
Zhang Z, Yao Z, Wang L, Ding H, Shao J, Chen A, Zhang F, Zheng S. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy. 2018;14:2083–103.
Tian Y, Lu J, Hao X, Li H, Zhang G, Liu X, Li X, Zhao C, Kuang W, Chen D, Zhu M. FTH1 inhibits ferroptosis through ferritinophagy in the 6-OHDA model of parkinson’s disease. Neurotherapeutics. 2020;17:1796–812.
Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509:105–9.
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, Kang R, Tang D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12:1425–8.
Bai Y, Meng L, Han L, Jia Y, Zhao Y, Gao H, Kang R, Wang X, Tang D, Dai E. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun. 2019;508:997–1003.
Yang M, Chen P, Liu J, Zhu S, Kroemer G, Klionsky DJ, Lotze MT, Zeh HJ, Kang R, Tang D. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv. 2019;5:eaaw2238.
Wen Q, Liu J, Kang R, Zhou B, Tang D. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun. 2019;510:278–83.
Tang D, Kang R, Livesey KM, Cheh CW, Farkas A, Loughran P, Hoppe G, Bianchi ME, Tracey KJ, Zeh HJ 3rd, Lotze MT. Endogenous HMGB1 regulates autophagy. J Cell Biol. 2010;190:881–92.
Ye F, Chai W, Xie M, Yang M, Yu Y, Cao L, Yang L. HMGB1 regulates erastin-induced ferroptosis via RAS-JNK/p38 signaling in HL-60/NRAS(Q61L) cells. Am J Cancer Res. 2019;9:730–9.
Chen X, Song X, Li J, Zhang R, Yu C, Zhou Z, Liu J, Liao S, Klionsky DJ, Kroemer G, et al. Identification of HPCAL1 as a specific autophagy receptor involved in ferroptosis. Autophagy. 2023;19:54–74.
Tang W, Zhu S, Liang X, Liu C, Song L. The crosstalk between long non-coding RNAs and various types of death in cancer cells. Technol Cancer Res Treat. 2021;20:15330338211033044.
Wang YY, Liu XL, Zhao R. Induction of pyroptosis and its implications in cancer management. Front Oncol. 2019;9:971.
Zhang C, Liu N. Ferroptosis, necroptosis, and pyroptosis in the occurrence and development of ovarian cancer. Front Immunol. 2022;13: 920059.
Zhang T, Li Y, Zhu R, Song P, Wei Y, Liang T, Xu G. Transcription factor p53 suppresses tumor growth by prompting pyroptosis in non-small-cell lung cancer. Oxid Med Cell Longev. 2019;2019:8746895.
Wang J, Deng B, Liu Q, Huang Y, Chen W, Li J, Zhou Z, Zhang L, Liang B, He J, et al. Pyroptosis and ferroptosis induced by mixed lineage kinase 3 (MLK3) signaling in cardiomyocytes are essential for myocardial fibrosis in response to pressure overload. Cell Death Dis. 2020;11:574.
Xu R, Yang J, Qian Y, Deng H, Wang Z, Ma S, Wei Y, Yang N, Shen Q. Ferroptosis/pyroptosis dual-inductive combinational anti-cancer therapy achieved by transferrin decorated nanoMOF. Nanoscale Horiz. 2021;6:348–56.
Zhou B, Zhang JY, Liu XS, Chen HZ, Ai YL, Cheng K, Sun RY, Zhou D, Han J, Wu Q. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018;28:1171–85.
Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen Q, Cao L, Xie M, Ran Q, Kroemer G, et al. Lipid peroxidation drives gasdermin d-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe. 2018;24:97-108.e104.
Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, Rossen J, Joesch-Cohen L, Humeidi R, Spangler RD, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375:1254–61.
Gao W, Huang Z, Duan J, Nice EC, Lin J, Huang C. Elesclomol induces copper-dependent ferroptosis in colorectal cancer cells via degradation of ATP7A. Mol Oncol. 2021;15:3527–44.
Mao C, Wang X, Liu Y, Wang M, Yan B, Jiang Y, Shi Y, Shen Y, Liu X, Lai W, et al. A G3BP1-interacting lncRNA promotes ferroptosis and apoptosis in cancer via nuclear sequestration of p53. Cancer Res. 2018;78:3484–96.
Shanshan W, Hongying M, Jingjing F, Yiming Y, Yu R, Rui Y. CircDTL functions as an oncogene and regulates both apoptosis and ferroptosis in non-small cell lung cancer cells. Front Genet. 2021;12: 743505.
Liu Z, Wang Q, Wang X, Xu Z, Wei X, Li J. Circular RNA cIARS regulates ferroptosis in HCC cells through interacting with RNA binding protein ALKBH5. Cell Death Discov. 2020;6:72.
Peng X, Yang R, Peng W, Zhao Z, Tu G, He B, Cai Q, Shi S, Yin W, Yu F, et al. Overexpression of LINC00551 promotes autophagy-dependent ferroptosis of lung adenocarcinoma via upregulating DDIT4 by sponging miR-4328. PeerJ. 2022;10: e14180.
Wang M, Cheng H, Wu H, Liu C, Li S, Li B, Su J, Luo S, Li Q. Gambogenic acid antagonizes the expression and effects of long non-coding RNA NEAT1 and triggers autophagy and ferroptosis in melanoma. Biomed Pharmacother. 2022;154: 113636.
Tang D, Chen X, Kang R, Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31:107–25.
Winkle M, El-Daly SM, Fabbri M, Calin GA. Noncoding RNA therapeutics - challenges and potential solutions. Nat Rev Drug Discov. 2021;20:629–51.
Balihodzic A, Prinz F, Dengler MA, Calin GA, Jost PJ, Pichler M. Non-coding RNAs and ferroptosis: potential implications for cancer therapy. Cell Death Differ. 2022;29:1094–106.
Bao C, Zhang J, Xian SY, Chen F. MicroRNA-670-3p suppresses ferroptosis of human glioblastoma cells through targeting ACSL4. Free Radic Res. 2021;55:853–64.
Lu Y, Chan YT, Tan HY, Zhang C, Guo W, Xu Y, Sharma R, Chen ZS, Zheng YC, Wang N, Feng Y. Epigenetic regulation of ferroptosis via ETS1/miR-23a-3p/ACSL4 axis mediates sorafenib resistance in human hepatocellular carcinoma. J Exp Clin Cancer Res. 2022;41:3.
Yang X, Liu J, Wang C, Cheng KK, Xu H, Li Q, Hua T, Jiang X, Sheng L, Mao J, Liu Z. miR-18a promotes glioblastoma development by down-regulating ALOXE3-mediated ferroptotic and anti-migration activities. Oncogenesis. 2021;10:15.
Bai T, Liang R, Zhu R, Wang W, Zhou L, Sun Y. MicroRNA-214-3p enhances erastin-induced ferroptosis by targeting ATF4 in hepatoma cells. J Cell Physiol. 2020;235:5637–48.
Guan L, Wang F, Wang M, Han S, Cui Z, Xi S, Xu H, Li S. Downregulation of HULC induces ferroptosis in hepatocellular carcinoma via targeting of the miR-3200-5p/ATF4 Axis. Oxid Med Cell Longev. 2022;2022:9613095.
Wang P, Zhu CF, Ma MZ, Chen G, Song M, Zeng ZL, Lu WH, Yang J, Wen S, Chiao PJ, et al. Micro-RNA-155 is induced by K-Ras oncogenic signal and promotes ROS stress in pancreatic cancer. Oncotarget. 2015;6:21148–58.
Zhu C, Song Z, Chen Z, Lin T, Lin H, Xu Z, Ai F, Zheng S. MicroRNA-4735-3p facilitates ferroptosis in clear cell renal cell carcinoma by targeting SLC40A1. Anal Cell Pathol. 2022;2022:4213401.
Bazhabayi M, Qiu X, Li X, Yang A, Wen W, Zhang X, Xiao X, He R, Liu P. CircGFRA1 facilitates the malignant progression of HER-2-positive breast cancer via acting as a sponge of miR-1228 and enhancing AIFM2 expression. J Cell Mol Med. 2021;25:10248–56.
Gomaa A, Peng D, Chen Z, Soutto M, Abouelezz K, Corvalan A, El-Rifai W. Epigenetic regulation of AURKA by miR-4715-3p in upper gastrointestinal cancers. Sci Rep. 2019;9:16970.
Zhang K, Wu L, Zhang P, Luo M, Du J, Gao T, O’Connell D, Wang G, Wang H, Yang Y. miR-9 regulates ferroptosis by targeting glutamic-oxaloacetic transaminase GOT1 in melanoma. Mol Carcinog. 2018;57:1566–76.
Liu L, Yao H, Zhou X, Chen J, Chen G, Shi X, Wu G, Zhou G, He S. MiR-15a-3p regulates ferroptosis via targeting glutathione peroxidase GPX4 in colorectal cancer. Mol Carcinog. 2022;61:301–10.
Yang Y, Lin Z, Han Z, Wu Z, Hua J, Zhong R, Zhao R, Ran H, Qu K, Huang H, et al. miR-539 activates the SAPK/JNK signaling pathway to promote ferropotosis in colorectal cancer by directly targeting TIPE. Cell Death Discov. 2021;7:272.
Xu Q, Zhou L, Yang G, Meng F, Wan Y, Wang L, Zhang L. CircIL4R facilitates the tumorigenesis and inhibits ferroptosis in hepatocellular carcinoma by regulating the miR-541-3p/GPX4 axis. Cell Biol Int. 2020;44:2344–56.
Xu P, Wang Y, Deng Z, Tan Z, Pei X. MicroRNA-15a promotes prostate cancer cell ferroptosis by inhibiting GPX4 expression. Oncol Lett. 2022;23:67.
Xu Z, Chen L, Wang C, Zhang L, Xu W. MicroRNA-1287-5p promotes ferroptosis of osteosarcoma cells through inhibiting GPX4. Free Radic Res. 2021;55:1119–29.
Zhou Y, Wu H, Wang F, Xu L, Yan Y, Tong X, Yan H. GPX7 Is targeted by miR-29b and GPX7 knockdown enhances ferroptosis induced by erastin in glioma. Front Oncol. 2021;11: 802124.
Fan H, Ai R, Mu S, Niu X, Guo Z, Liu L. MiR-19a suppresses ferroptosis of colorectal cancer cells by targeting IREB2. Bioengineered. 2022;13:12021–9.
Liao Y, Jia X, Ren Y, Deji Z, Gesang Y, Ning N, Feng H, Yu H, Wei A. Suppressive role of microRNA-130b-3p in ferroptosis in melanoma cells correlates with DKK1 inhibition and Nrf2-HO-1 pathway activation. Hum Cell. 2021;34:1532–44.
Kabaria S, Choi DC, Chaudhuri AD, Jain MR, Li H, Junn E. MicroRNA-7 activates Nrf2 pathway by targeting Keap1 expression. Free Radic Biol Med. 2015;89:548–56.
Bi G, Liang J, Zhao M, Zhang H, Jin X, Lu T, Zheng Y, Bian Y, Chen Z, Huang Y, et al. miR-6077 promotes cisplatin/pemetrexed resistance in lung adenocarcinoma via CDKN1A/cell cycle arrest and KEAP1/ferroptosis pathways. Mol Ther Nucleic Acids. 2022;28:366–86.
Huang W, Shi G, Yong Z, Li J, Qiu J, Cao Y, Zhao Y, Yuan L. Downregulation of RKIP promotes radioresistance of nasopharyngeal carcinoma by activating NRF2/NQO1 axis via downregulating miR-450b-5p. Cell Death Dis. 2020;11:504.
Gai C, Liu C, Wu X, Yu M, Zheng J, Zhang W, Lv S, Li W. MT1DP loaded by folate-modified liposomes sensitizes erastin-induced ferroptosis via regulating miR-365a-3p/NRF2 axis in non-small cell lung cancer cells. Cell Death Dis. 2020;11:751.
Luo M, Wu L, Zhang K, Wang H, Zhang T, Gutierrez L, O’Connell D, Zhang P, Li Y, Gao T, et al. miR-137 regulates ferroptosis by targeting glutamine transporter SLC1A5 in melanoma. Cell Death Differ. 2018;25:1457–72.
Sun D, Li YC, Zhang XY. Lidocaine promoted ferroptosis by targeting miR-382-5p /SLC7A11 axis in ovarian and breast cancer. Front Pharmacol. 2021;12: 681223.
Mao SH, Zhu CH, Nie Y, Yu J, Wang L. Levobupivacaine induces ferroptosis by miR-489-3p/SLC7A11 signaling in gastric cancer. Front Pharmacol. 2021;12: 681338.
Yu Y, MohamedAl-Sharani H, Zhang B. EZH2-mediated SLC7A11 upregulation via miR-125b-5p represses ferroptosis of TSCC. Oral Dis. 2023;29:880–91.
Sun K, Ren W, Li S, Zheng J, Huang Y, Zhi K, Gao L. MiR-34c-3p upregulates erastin-induced ferroptosis to inhibit proliferation in oral squamous cell carcinomas by targeting SLC7A11. Pathol Res Pract. 2022;231: 153778.
Lyu N, Zeng Y, Kong Y, Chen Q, Deng H, Ou S, Bai Y, Tang H, Wang X, Zhao M. Ferroptosis is involved in the progression of hepatocellular carcinoma through the circ0097009/miR-1261/SLC7A11 axis. Ann Transl Med. 2021;9:675.
Drayton RM, Dudziec E, Peter S, Bertz S, Hartmann A, Bryant HE, Catto JW. Reduced expression of miRNA-27a modulates cisplatin resistance in bladder cancer by targeting the cystine/glutamate exchanger SLC7A11. Clin Cancer Res. 2014;20:1990–2000.
Ni H, Qin H, Sun C, Liu Y, Ruan G, Guo Q, Xi T, Xing Y, Zheng L. MiR-375 reduces the stemness of gastric cancer cells through triggering ferroptosis. Stem Cell Res Ther. 2021;12:325.
Yadav P, Sharma P, Sundaram S, Venkatraman G, Bera AK, Karunagaran D. SLC7A11/ xCT is a target of miR-5096 and its restoration partially rescues miR-5096-mediated ferroptosis and anti-tumor effects in human breast cancer cells. Cancer Lett. 2021;522:211–24.
Zhu JH, De Mello RA, Yan QL, Wang JW, Chen Y, Ye QH, Wang ZJ, Tang HJ, Huang T. MiR-139-5p/SLC7A11 inhibits the proliferation, invasion and metastasis of pancreatic carcinoma via PI3K/Akt signaling pathway. Biochim Biophys Acta Mol Basis Dis. 2020;1866: 165747.
Lu X, Kang N, Ling X, Pan M, Du W, Gao S. MiR-27a-3p promotes non-small cell lung cancer through SLC7A11-mediated-ferroptosis. Front Oncol. 2021;11: 759346.
Liu YP, Qiu ZZ, Li XH, Li EY. Propofol induces ferroptosis and inhibits malignant phenotypes of gastric cancer cells by regulating miR-125b-5p/STAT3 axis. World J Gastrointest Oncol. 2021;13:2114–28.
Luo Y, Niu G, Yi H, Li Q, Wu Z, Wang J, Yang J, Li B, Peng Y, Liang Y, et al. Nanomedicine promotes ferroptosis to inhibit tumour proliferation in vivo. Redox Biol. 2021;42: 101908.
Zheng S, Hu L, Song Q, Shan Y, Yin G, Zhu H, Kong W, Zhou C. miR-545 promotes colorectal cancer by inhibiting transferring in the non-normal ferroptosis signaling. Aging. 2021;13:26137–47.
Guo W, Wu Z, Chen J, Guo S, You W, Wang S, Ma J, Wang H, Wang X, Wang H, et al. Nanoparticle delivery of miR-21–3p sensitizes melanoma to anti-PD-1 immunotherapy by promoting ferroptosis. J Immunother Cancer. 2022. https://doi.org/10.1136/jitc-2021-004381.
Wu H, Liu A. Long non-coding RNA NEAT1 regulates ferroptosis sensitivity in non-small-cell lung cancer. J Int Med Res. 2021;49:300060521996183.
Sui X, Hu N, Zhang Z, Wang Y, Wang P, Xiu G. ASMTL-AS1 impedes the malignant progression of lung adenocarcinoma by regulating SAT1 to promote ferroptosis. Pathol Int. 2021;71:741–51.
Zhang Y, Luo M, Cui X, O’Connell D, Yang Y. Long noncoding RNA NEAT1 promotes ferroptosis by modulating the miR-362-3p/MIOX axis as a ceRNA. Cell Death Differ. 2022;29:1850–63.
Chen J, Qin C, Zhou Y, Chen Y, Mao M, Yang J. Metformin may induce ferroptosis by inhibiting autophagy via lncRNA H19 in breast cancer. FEBS Open Bio. 2022;12:146–53.
Zhang R, Pan T, Xiang Y, Zhang M, Xie H, Liang Z, Chen B, Xu C, Wang J, Huang X, et al. Curcumenol triggered ferroptosis in lung cancer cells via lncRNA H19/miR-19b-3p/FTH1 axis. Bioact Mater. 2022;13:23–36.
Gong H, Gao M, Lin Y, Liu J, Hu Z, Liu J. TUG1/MAZ/FTH1 axis attenuates the antiglioma effect of dihydroartemisinin by inhibiting ferroptosis. Oxid Med Cell Longev. 2022;2022:7843863.
Qi W, Li Z, Xia L, Dai J, Zhang Q, Wu C, Xu S. LncRNA GABPB1-AS1 and GABPB1 regulate oxidative stress during erastin-induced ferroptosis in HepG2 hepatocellular carcinoma cells. Sci Rep. 2019;9:16185.
Pan C, Chen G, Zhao X, Xu X, Liu J. lncRNA BBOX1-AS1 silencing inhibits esophageal squamous cell cancer progression by promoting ferroptosis via miR-513a-3p/SLC7A11 axis. Eur J Pharmacol. 2022;934: 175317.
Wang Z, Chen X, Liu N, Shi Y, Liu Y, Ouyang L, Tam S, Xiao D, Liu S, Wen F, Tao Y. A nuclear long non-coding RNA LINC00618 accelerates ferroptosis in a manner dependent upon apoptosis. Mol Ther. 2021;29:263–74.
Zhang Y, Guo S, Wang S, Li X, Hou D, Li H, Wang L, Xu Y, Ma B, Wang H, Jiang X. LncRNA OIP5-AS1 inhibits ferroptosis in prostate cancer with long-term cadmium exposure through miR-128-3p/SLC7A11 signaling. Ecotoxicol Environ Saf. 2021;220:112376.
Li YZ, Zhu HC, Du Y, Zhao HC, Wang L. Silencing lncRNA SLC16A1-AS1 induced ferroptosis in renal cell carcinoma through miR-143-3p/SLC7A11 signaling. Technol Cancer Res Treat. 2022;21:15330338221077804.
Zhang B, Bao W, Zhang S, Chen B, Zhou X, Zhao J, Shi Z, Zhang T, Chen Z, Wang L, et al. LncRNA HEPFAL accelerates ferroptosis in hepatocellular carcinoma by regulating SLC7A11 ubiquitination. Cell Death Dis. 2022;13:734.
Huang G, Xiang Z, Wu H, He Q, Dou R, Lin Z, Yang C, Huang S, Song J, Di Z, et al. The lncRNA BDNF-AS/WDR5/FBXW7 axis mediates ferroptosis in gastric cancer peritoneal metastasis by regulating VDAC3 ubiquitination. Int J Biol Sci. 2022;18:1415–33.
Luo W, Wang J, Xu W, Ma C, Wan F, Huang Y, Yao M, Zhang H, Qu Y, Ye D, Zhu Y. LncRNA RP11-89 facilitates tumorigenesis and ferroptosis resistance through PROM2-activated iron export by sponging miR-129-5p in bladder cancer. Cell Death Dis. 2021;12:1043.
Wang G, Sun L, Wang S, Guo J, Xiao R, Li W, Qi W, Qiu W. Ferroptosis-related long non-coding RNAs and the roles of LASTR in stomach adenocarcinoma. Mol Med Rep. 2022. https://doi.org/10.3892/mmr.2022.12634.
Li X, Li Y, Lian P, Lv Q, Liu F. Silencing lncRNA HCG18 regulates GPX4-inhibited ferroptosis by adsorbing miR-450b-5p to avert sorafenib resistance in hepatocellular carcinoma. Hum Exp Toxicol. 2023;42:9603271221142818.
Ma Q, Dai X, Lu W, Qu X, Liu N, Zhu C. Silencing long non-coding RNA MEG8 inhibits the proliferation and induces the ferroptosis of hemangioma endothelial cells by regulating miR-497-5p/NOTCH2 axis. Biochem Biophys Res Commun. 2021;556:72–8.
Chen Q, Wang W, Wu Z, Chen S, Chen X, Zhuang S, Song G, Lv Y, Lin Y. Over-expression of lncRNA TMEM161B-AS1 promotes the malignant biological behavior of glioma cells and the resistance to temozolomide via up-regulating the expression of multiple ferroptosis-related genes by sponging hsa-miR-27a-3p. Cell Death Discov. 2021;7:311.
Luo Y, Huang S, Wei J, Zhou H, Wang W, Yang J, Deng Q, Wang H, Fu Z. Long noncoding RNA LINC01606 protects colon cancer cells from ferroptotic cell death and promotes stemness by SCD1-Wnt/β-catenin-TFE3 feedback loop signalling. Clin Transl Med. 2022;12:e752.
Wang M, Mao C, Ouyang L, Liu Y, Lai W, Liu N, Shi Y, Chen L, Xiao D, Yu F, et al. Long noncoding RNA LINC00336 inhibits ferroptosis in lung cancer by functioning as a competing endogenous RNA. Cell Death Differ. 2019;26:2329–43.
Luo C, Nie C, Zeng Y, Qian K, Li X, Wang X. LINC01564 promotes the TMZ resistance of glioma cells by upregulating NFE2L2 expression to inhibit ferroptosis. Mol Neurobiol. 2022;59:3829–44.
Ou R, Lu S, Wang L, Wang Y, Lv M, Li T, Xu Y, Lu J, Ge RS. Circular RNA circLMO1 suppresses cervical cancer growth and metastasis by triggering miR-4291/ACSL4-mediated ferroptosis. Front Oncol. 2022;12: 858598.
Xian ZY, Hu B, Wang T, Cai JL, Zeng JY, Zou Q, Zhu PX. CircABCB10 silencing inhibits the cell ferroptosis and apoptosis by regulating the miR-326/CCL5 axis in rectal cancer. Neoplasma. 2020;67:1063–73.
Li C, Tian Y, Liang Y, Li Q. Circ_0008035 contributes to cell proliferation and inhibits apoptosis and ferroptosis in gastric cancer via miR-599/EIF4A1 axis. Cancer Cell Int. 2020;20:84.
Yao W, Wang J, Meng F, Zhu Z, Jia X, Xu L, Zhang Q, Wei L. Circular RNA CircPVT1 inhibits 5-fluorouracil chemosensitivity by regulating ferroptosis through MiR-30a-5p/FZD3 axis in esophageal cancer cells. Front Oncol. 2021;11: 780938.
Wang Y, Chen H, Wei X. Circ_0007142 downregulates miR-874-3p-mediated GDPD5 on colorectal cancer cells. Eur J Clin Invest. 2021;51: e13541.
Zhang HY, Zhang BW, Zhang ZB, Deng QJ. Circular RNA TTBK2 regulates cell proliferation, invasion and ferroptosis via miR-761/ITGB8 axis in glioma. Eur Rev Med Pharmacol Sci. 2020;24:2585–600.
Chen S, Zhang Z, Zhang B, Huang Q, Liu Y, Qiu Y, Long X, Wu M, Zhang Z. CircCDK14 promotes tumor progression and resists ferroptosis in glioma by regulating PDGFRA. Int J Biol Sci. 2022;18:841–57.
Dong LH, Huang JJ, Zu P, Liu J, Gao X, Du JW, Li YF. CircKDM4C upregulates P53 by sponging hsa-let-7b-5p to induce ferroptosis in acute myeloid leukemia. Environ Toxicol. 2021;36:1288–302.
Wu P, Li C, Ye DM, Yu K, Li Y, Tang H, Xu G, Yi S, Zhang Z. Circular RNA circEPSTI1 accelerates cervical cancer progression via miR-375/409-3P/515-5p-SLC7A11 axis. Aging. 2021;13:4663–73.
Yang J, Cao XH, Luan KF, Huang YD. Circular RNA FNDC3B protects oral squamous cell carcinoma cells from ferroptosis and contributes to the malignant progression by regulating miR-520d-5p/SLC7A11 axis. Front Oncol. 2021;11: 672724.
Wang HH, Ma JN, Zhan XR. Circular RNA Circ_0067934 attenuates ferroptosis of thyroid cancer cells by miR-545-3p/SLC7A11 signaling. Front Endocrinol. 2021;12:670031.
Wang W, Xie Y, Malhotra A. Potential of curcumin and quercetin in modulation of premature mitochondrial senescence and related changes during lung carcinogenesis. J Environ Pathol Toxicol Oncol. 2021;40:53–60.
Zhang H, Ge Z, Wang Z, Gao Y, Wang Y, Qu X. Circular RNA RHOT1 promotes progression and inhibits ferroptosis via mir-106a-5p/STAT3 axis in breast cancer. Aging. 2021;13:8115–26.
Jiang M, Mo R, Liu C, Wu H. Circ_0000190 sponges miR-382–5p to suppress cell proliferation and motility and promote cell death by targeting ZNRF3 in gastric cancer. J Biochem. 2022. https://doi.org/10.1093/jb/mvac003.
Acknowledgements
Not applicable.
Funding
This project was supported by grants from the National Natural Science Foundation of China (No. 82272411, No. 82072363) and Jiangsu Provincial Key Medical Discipline (Laboratory) (ZDXK202240).
Author information
Authors and Affiliations
Contributions
QZ conceived the structure of the manuscript and drafted the first manuscript, XF collected the related article. XZ checked and revised the manuscript. SJ gave constructive guidance and made critical revisions. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors have read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, Q., Fan, X., Zhang, X. et al. Ferroptosis in tumors and its relationship to other programmed cell death: role of non-coding RNAs. J Transl Med 21, 514 (2023). https://doi.org/10.1186/s12967-023-04370-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-023-04370-6