
Abstract
The discovery of the Cystic fibrosis (CF) gene in 1989 has paved the way for incredible progress in treating the disease such that the mean survival age of individuals living with CF is now ~58 years in Canada. Recent developments in gene targeting tools and new cell and animal models have re-ignited the search for a permanent genetic cure for all CF. In this review, we highlight some of the more recent gene therapy approaches as well as new models that will provide insight into personalized therapies for CF.
Similar content being viewed by others

Introduction
Cystic fibrosis
Cystic fibrosis (CF) is the most common life-limiting fatal genetic disorder, affecting approximately 90,000 individuals worldwide [1]. It is an autosomal recessive disorder that requires mutations in the CF gene in both genetic alleles [2]. The CF gene encodes for a protein the cystic fibrosis transmembrance conductance regulator (CFTR) which is a protein chloride channel that belongs to the family of adenosine triphosphate (ATP)-binding cassette (ABC) transporters. It consists of two membrane-spanning domains (MSD1, MSD2), two nucleotide-binding domains (NBD1, NBD2) and the functional regulatory domain (R) with multiple phosphorylation consensus sites, which when phosphorylated, undergoes conformational change and opening of the chloride channel [3]. Mutations in the CF gene affecting CFTR expression, protein levels or function, now known as CFTR variants, affect multiple organ systems including the lung, pancreas, liver, gut and reproductive organs. Changes in chloride and bicarbonate transportation across this channel impairs epithelial cell functions including mucociliary transport of foreign agents out of the airways, elevated concentrations sweat chloride, impairment in pancreatic hormone regulation, and intestinal obstruction [4,5,6]. In the lungs, CFTR-mediated export of chloride and bicarbonate ions across the epithelium into airway surface liquid (ASL) plays a vital role in maintaining the ASL pH and airway secreted protein composition (i.e. mucins). Dehydration of the ASL thickens mucus secretions and impairs mucociliary clearance, antimicrobial enzyme activity and promotes a pro-inflammatory environment mediated by recurrent infections leading to lung damage [7].
Classes of CFTR variants
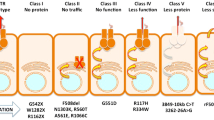
In 1989, CFTR was identified and localized on the long arm of chromosome 7 (1q.31.2), consisting of 27 exons spanning about 215 kb of the genomic sequence [8,9,10,11]. While there have been > 2000 CF mutations identified to date (http://www.genet.sickkids.on.ca/cftr/), over 360 are CF disease-causing variants (www.cftr2.org). Recently, these variants have been categorized into 7 classes based on CFTR protein dysfunction and/or gene expression [12] (Fig. 1): Class I are protein production variants that result in no functional CFTR protein with roughly 22% of CF patients harboring at least 1 mutant allele; Class II are protein processing variants that create misfolded CFTR protein and reduced expression on the cell membrane to function. Approximately 88% of CF patients have at least 1 mutant allele and the main variant p.Phe508del (F508del)-CFTR caused by a mutational deletion of the amino acid phenylalanine at the position 508 of the protein; Class III are gating variants that impair CFTR gate opening and encompasses roughly 6% of CF patients; Class IV result in defective ion channel conduction and approximately 6% of CF patients harbour this variant; Class V are insufficient protein variants and results in a reduced amount of CFTR at the surface membrane capturing 5% of CF patients; Class VI affects the stability of CFTR that causes a reduction in membrane retention and 5% of CF patients harbor at least one allele of this variant, and finally Class VII affects CFTR mRNA expression resulting in no mRNA and includes large deletions such as the dele2,3(21 Kb) mutation.

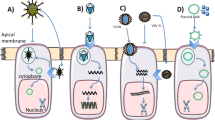
Classes of CFTR variants and CFTR modulators and the impact it has in CFTR expression and processing
Advantages and disadvantages of CFTR modulator therapy
Over the past 30 years, tremendous advances in clinical interventions and CF research have allowed for transformative advances in CF therapy. Prior to the development of small molecules targeting the CFTR protein (CFTR modulators), treatment of CF was solely aimed at alleviating the symptoms associated with the disease [13]. In recent years, CFTR modulators capable of directly correcting the genetic defect are paving the way for a cure for CF [14]. Here, we briefly touch on some current CFTR modulators that have been approved or are currently in clinical trials.
CFTR modulators are classified into 4 groups (Fig. 1): correctors, potentiators, stabilizers and amplifiers. Small molecules aimed at stabilizing the misfolded protein in the cytosol to prevent degradation are known as correctors (examples include lumacaftor (VX-809), tezacaftor (VX-661), and elexacaftor (VX-445) from Vertex Pharmaceuticals and posenacaftor (PTI-801) from Proteostasis). Small molecules that bind to the NBD domain of the CFTR channel to facilitate its opening are known as potentiators, (examples include ivacaftor (VX770) and dirocaftor (PTI-808)). Stabilizers such as cavosonstat (N91115 from Nivalis) rescues the protein stability on the plasma membrane, promotes CFTR maturation and is currently in phase II clinical trials. Amplifiers increase the amount of CFTR production and include nesolicaftor (PTI-428), a current candidate in phase III clinical trials in combination with PTI-801 and PTI-808. Finally, for CF-causing variants where in-frame nonsense, frameshift, and splicing variants that introduce a premature termination codon (PTC) into the CFTR mRNA (i.e. W1282X and G542X), read through agents such as ELX-02 developed by Eloxx Pharmaceuticals and Ataluren PTC-124 by PTC Therapeutics were designed to restore functional protein production by overriding PTC signals [15]. However, early clinical trials currently underway for ELX-02 and PTC-124 failed to show significant improvement in FEV1 measurements in patients with at least 1 mutant allele in a phase III clinical trial [16]. The number of transcripts differ considerably depending on the site of the PTC, the cell type and the patient’s genetic background [15, 17]. Other small molecule inhibitors of the nonsense mediated decay (NMD) pathway such as SMG1 inhibitor (SMGi) can restore CFTR expression and function in cells harboring W1282X CFTR [18]. Therefore, combining small molecules to improve CFTR transcript production and/or stability with CFTR modulators may provide better clinical outcomes.
The approved CFTR modulator therapies ORKAMBI™ (a combination of VX-770 and VX-809) and SYMDEKO™ (a combination of VX-661 and VX-809) are combination treatments that has shown improved clinical benefits for some patients harboring F508del-CFTR. However, there are wide variations in responses to the drugs which suggest while the drugs may be used to treat the same genetic defect, other factors such as environmental [19,20,21] and gene modifiers [22,23,24,25] may influence therapy response. A recently approved drug, TRIKAFTA™ is a combination of 2 correctors (VX445 and VX661) and 1 potentiator (VX770) drugs that have shown incredible promise in improving lung function, sweat chloride conductance and lowering pulmonary exacerbations in F508del-CFTR individuals [26]. The short-term effectiveness of these modulators offer hope for restoring basic lung functions. However, the efficacy of this drug in effectively curing all CF individuals harboring at least 1 F508del allele remains unknown. Many rare CF variants are not eligible for current modulator treatment as these drugs are not expected to work such as for Class I production variants. Moreover, the long-term potential side effects of modulator treatment remain unclear [27] and with the costs for CFTR modulator therapy averaging over $300,000/year/patient [28], many CF individuals will not receive potential life-saving therapies without financial support or reimbursements. Therefore, new therapy approaches are still needed to treat all CF.
Gene therapy approaches for CF
Gene therapy offers great hope for the treatment of genetic diseases/disorders. By replacing the genetic mutation with a “correct version” of the CFTR gene, this method offers a potentially permanent cure. Indeed, since the discovery of the CF gene, many studies have attempted to correct the CFTR mutations through gene therapy approaches. While gene correction showed limited success in both cell and animal models [29,30,31], therapy for patients had proven to be more difficult. In-vitro studies have suggested that not all cells need to express normal CFTR to effect normal epithelial functions. In a mixing experiment where normal cells were mixed with CF mutant cells, only 6–10% of the epithelium needed to contain epithelial cells expressing normal CFTR to restore chloride transport similar to normal epithelia [32]. Conversely, in a gene targeting study, up to 25% gene correction could restore mucus transport in homozygous F508del human airway epithelial cells [33]. The number of cells harboring wild-type CFTR that is needed to translate into clinical benefit in patience remains unknown. However, theoretically correcting a stem cell population within the airways may provide a renewable and long-term source of endogenous cells capable of renewing the damaged epithelia with cells that express wild-type CFTR. Yet surprisingly, with the exception of a Phase I and II clinical trial for MRT5005 [https://www.cff.org/Trials/Pipeline/details/10157/MRT5005], a drug that delivers CFTR-encoded mRNA to the lungs (RESTORE-CF), there are no other clinical trials for CF gene therapy. This may largely be due to several reasons: 1. The need for repeated delivery due to the inability to target stem/progenitor cells of the airways to sustain expression during cell turnover, 2. Suboptimal delivery or low efficiency of targeting of the donor plasmid/gene to the CF airways due to the highly inflammatory microenvironment, 3. The inability to deliver large DNA fragments of the CFTR gene effectively with current delivery methods, 4. Concerns of off-target safety that can result in insertional mutagenesis, and 5. Immune barriers limiting effective delivery of viral vectors. In this review, we briefly touch on some of the more recent genetic approaches that can rejuvenate CF gene therapy and touch on new cell and animal models that are enabling the testing of current gene targeting strategies and providing insight into personalized approaches for CF therapy.
Gene editing approaches
Gene editing tools can provide new gene therapy strategies to achieve permanent correction. Here we list a few editing tools used to date to test the efficacy of genetic correction for CF in-vitro.
Zinc Finger Nucleases (ZFNs) and transcription activator-like effector nucleases (TALEN)
Early developments of gene editing approaches included use of artificial restriction enzymes, Zinc Finger Nucleases (ZFNs) and transcription activator-like effector nucleases (TALEN) [34, 35], (Table 1; Fig. 2). These gene modification tools enabled precise genome editing through targeted nucleases cleavages and renewed hope for gene therapy. ZFNs are composed of specific pairs of oligos attached to a FokI restriction enzyme that facilitate a precise double-strand break (DSB) at the target site [36]. TALENs are composed of TALE repeats that bind and recognize extended DNA sequences and are also attached with a FoKI restriction enzyme to create a DSB [37, 38]. In both instances, the DSB induces DNA repair mechanisms by either non homologous end joining (NHEJ), or homology-directed repair (HDR) [39, 40]. Neither ZFN and TALENs technology have been used in CF gene therapies and in the advent of CRISPR-Cas systems, gene editing using the latter tool is more flexible making it the editing tool of choice for many researchers. The specific requirement of a pair of ZFNs reduces the number of target sites that can be identified for gene correction. Moreover, the low binding affinity of the ZFN creates undesirable off-target mutations in the genome [41]. TALEN has shown less off-target and better binding affinity than ZFN, however, the size for cDNA encoding a TALEN (3 kb) can be an issue for delivery into cells with a limited cargo size [42].

Graphics of gene editing technologies
CRISPR gene editing
In 2013, a new gene-editing tool used by bacteria to fend off bacteriophages by called clustered regularly interspaced short palindromic repeats (CRISPR) and it’s enzyme CRISPR associated protein 9 (Cas9) [43] was shown to be useful in editing the genomes of cultured mammalian cells [44]. The precise editing of the CRISPR-Cas9 system along with the versatile use of the system to silence genes by removing part of the gene or substituting the gene with desired ones has made the CRISPR-Cas9 system the preferred editing tool for gene editing. Moreover, the relative ease in designing a specific target site and low cost allows efficient gene editing to be done within a relatively short period of time [45]. The CRISPR-Cas9 is composed of two main modules: the guide RNA (gRNA), and the Cas9 protein enzyme. The gRNA is designed to recognize a specific sequence motif near the target site and recruits the Cas9 protein to cut and create a double-stranded DNA break (DSB). The cell’s natural DNA repair mechanisms are then activated to repair the cleaved DNA through NHEJ or HDR [39, 40]. NHEJ directly ligates the broken ends, and can create “indels” or insertion or deletions of genes effectively creating mutants [46]. However, with a repair template, the HDR response will enable homologous recombination. This method is useful for introducing a desired gene (or a wild-type version of a gene). However, the frequency of HDR is very low [47] and therefore efficiency of “repairing” or replacing a mutant gene remains a challenge.
(i) Base editing: The CRIPSR-Cas9 system’s classical reliance on introducing DSBs poses an efficiency problem since undesirable random insertions or deletions (indels) occur more often at DNA cleavage sites than HDR. Base editing was thus pioneered to increase the efficiency of the CRISPR-Cas9 system by circumventing the need for DSBs altogether, allowing for the direct conversion of a DNA base to another without DSBs at a target locus [48]. Cytosine base editors (CBEs) facilitate the permanent conversion of C-G to T-A base pairs, while adenine base editors (ABEs) enzymatically convert A-T base pairs into G-C base pairs [49]. In the contexts of CF, base editing could then be an attractive new tool in treating CF, since many CFTR variants could be rescued with a single base pair change. Accordingly, Geurts et al. recently provided support to the efficacy and feasibility of utilizing such base editing tools safely within human cells to potentially treat CF with two respective ABEs [50]. A caveat of base editing is the limitation of only 4 possible base-to-base conversions and is too large for certain gene delivery vectors.
(ii) Prime editing: Prime editing has recently become an attractive advancement in the CRISPR toolbox [51]. This gene editing technology makes it possible to edit a specified DNA sequence, of variable lengths at a target site, with a fusion complex composed of a catalytically impaired Cas9 protein and an engineered reverse transcriptase [51]. A prime editing guide RNA (pegRNA) encodes the desired gene edit and directs the fusion complex to the target site [51]. As a possible gene replacement therapeutic technology, prime editing is very promising in the context of CF, given the most common CFTR variant (CFTR-F508del) has been repaired by prime editing in patient-derived intestinal organoids [52]. However, prime editing did result in varying degrees of targeting efficiency and undesired off-target mutations were also observed [52]. Nevertheless, since the CFTR gene is large, and a complete replacement of a mutant gene with wild-type CFTR would likely be inefficient, prime editing is leading method to address the vast number of CF disease-causing variants.
Gene delivery
There are several gene delivery methods to introduce a therapeutic gene or gene targeting. Both non-viral and viral delivery vectors have been tested in CF gene therapy research.
(i) Non-viral vectors: Non-viral vectors were developed as a strategy to deliver the CFTR gene. These non-integrating gene delivery methods do not disrupt the host genome and thus the risk of causing mutagenesis are low. Non-viral vectors are not restricted in the cargo load enabling larger donor DNA fragments to be used for gene repair. However, the efficacy of gene delivery is comparatively lower than viral methods. To enhance gene transfer into the nucleus, a cationic lipid is used to formulate the plasmid DNA [53] complexed with CFTR enhanced chloride transport by 20% in CF patients compared to non-CF levels [54]. Using a nebulized cationic lipid pGM169/GL67A to deliver the donor DNA, up to 3.7% increase in CFTR function in the lungs of CF patients was observed [55, 56]. The drawback of the cationic liposome-mediated approach is the need for repeated delivery as transient expression of CFTR did not have a lasting effect [57]. Despite these efforts, non-viral based methods of gene delivery cannot permanently restore lung functions.
(ii) Viral vectors: To improve efficacy of targeting the cells, several viral based delivery methods have been tested to including adenovirus (Ad), adeno-associated virus (AAV), and retroviral vector in pre-clinical and clinical trials to deliver the corrected CFTR gene.
Adenovirus (Ad)
Based vectors were once the preferred delivery vectors for gene delivery [58, 59]. Mutational deletion of viral replication genes and host immune cell evasion genes early region 1 and 3 (E1/E3) respectively, removed the ability of the virus to self-replicate making these viral vectors attractive for gene therapy. However, leaky expression of viral genes from E1 deleted vectors, in addition to capsid proteins, could elicit host immune responses to the Ad vectors [60,61,62]. The first clinical trial (in 1993) for CF gene therapy using an adenovirus vector failed to restore CFTR expression in CF patient’s nasal epithelia [63, 64]. This led to the identification and testing of other adenovirus serotypes 2 and 5 in CF clinical trials which resulted in transient restoration of chloride transport in the nasal and bronchial epithelium [65, 66]. However, evidence of a pro-inflammatory response was found with these Ad vectors which required repeated administration for effective gene delivery [63, 65]. Even so, the trials have only demonstrated limited clinical benefits in CF patients [66].
Adeno-associated virus
AAV-based vectors have been tested as another gene delivery tool. With the ability to transduce terminally differentiated and non-dividing cells, AAV can also persist longer in-vivo [67] compared to its Ad counterpart. Transient immunosuppression can improve re-administration of AAV vectors in mouse lungs up to 8 months [68]. In 1998, the first successful human clinical trial with repeated delivery of AAV2-CFTR into the maxillary sinuses [69] demonstrated restoration of CFTR function without noticeable toxicity or an elevated immune response after 2 weeks of delivery. However, other clinical trial studies performed years later failed to show sufficient CFTR functional correction by AAV-CFTR [70, 71]. One caveat of the AAV vectors is the limited target gene size (less than 4.6 kb) that can be inserted into the viral vector for efficient expression.
Helper-dependent adenoviruses (Hd-Ad)
To avoid the harmful immune response of Ad, the Helper-dependent Adenovirus (Hd-Ad) was developed [72]. Deletion of all viral coding sequences allows Hd-Ad to deliver large DNA cargo (to 37 kb) without eliciting host immune responses [73, 74]. One unique feature of the Hd-Ad vectors is that they can be used to deliver both a gene editing endonuclease system and donor DNA in a single vector to achieve site-specific gene integration without expressing the endonuclease following gene correction [75,76,77]. Gene correction using Hd-Ad in CF mouse and pig airway basal cells can restore CFTR function similar to levels observed in normal wild-type cells as measured by fluorescence imaging plate reader (FLiPR) assay [30, 72, 78,79,80,81]. HD-Ad vectors have also been shown to be effective in correcting the CFTR gene in the lungs of CF knockout mice [82]. However, a major challenge remains for in-vivo gene therapy as the ability to sustain therapeutic effects is lost due to airway cell turnover. Therefore, targeting a stem cell compartment within the airways has become an attractive goal for permanent CF gene correction.
Retroviruses and lentiviruses
Retroviral and lentiviral vectors have been used for gene delivery methods as early as the late 1990s. Retroviruses harboring human CFTR gene transduced into rabbit tracheal epithelial cells showed persistent expression in the airways for up to 3 weeks. However, the transduced capacity by retroviruses were low and transduction occurred only in wounded areas [83]. Lentiviral vectors have been effective in delivering CFTR transgene into the airway epithelium [84] with potential to target the lung stem cell population for sustained and persistent CFTR expression [85]. While both retroviruses and lentiviruses can efficiently target host cells and integrate into the host genome, there remains significant concerns over their use as a delivery vector for gene therapy. The host immune responses remain a significant barrier in efficacious delivery of exogenous genetic materials by viral methods. In the context of CF airway disease, the proinflammatory milieu of the diseased airways compounded by the mucosal obstructions poses a challenge for any gene delivery methods. Second, there are concerns of insertional mutagenesis, epigenetic silencing, and secondary impact of altered expression levels derived by using viral promoters to drive the un-regulated expression of the transgene [86, 87].
Therefore, while new gene editing approaches may increase the targeting efficiency of gene correction, precise and efficient delivery of the genetic tools to the right cell type for permanent gene correction remains a barrier to clinical use. To study this, new animal and advanced stem cell-based models may enable research into cell delivery and targeting strategies.
Animal models of CF
Animal models of CF are valuable tools that may be utilized to further understand disease pathogenesis and test new therapeutics. There are two fundamental issues that remain to be resolved before gene therapy can become viable for patients, and animal models provide a relevant platform through which these obstacles may be safely addressed. First, in-vivo efficiencies of gene targeting need to achieve a level that will translate to therapeutic outcome. Second, the efficacy of gene targeting must outweigh concerns of off-target mutagenesis from the gene editing tools. Animal models have traditionally been useful models to understand basic mechanisms of disease pathogenesis. Recent animal models for CF, especially those harboring human CF variants offer opportunities to test new emerging CFTR modulators for which these modulators are designed to specifically target the specific functional outcome. Here we briefly touch on several of these animal models and their use in CF therapy discovery (Fig. 3).

CF animal models compared to human disease phenotypes
(i) Mouse model: With a 78% amino acid sequence conservation between mouse and human CFTR (hCFTR) [88], the use of mice for disease modelling comes as no surprise when also considering practical factors like costs, breeding time, and ease of maintenance. However, CF mouse models only exhibit mild pancreatic disease [89, 90] if any, present variable gallbladder abnormalities [90,91,92], and liver pathologies are largely only observed in mice studied later in life [89]. While new humanized mouse models have become available, and can be used to study CFTR modulator efficacies, they possess a major limitation in harboring ~ 6 copies of the hCFTR gene [93–95]. Therefore, it remains unclear how effective these humanized models are for gene therapy testing but may be a good model for CFTR modulator testing.
(ii) Rat model: CF rat models present similar phenotypes with CF mice. Like the CF mice, the rat models do not recapitulate spontaneous lung infection or pancreatic and liver disease [96, 97] though some models have displayed exocrine pancreas histopathology [98]. Nevertheless, rats possess a 76% amino acid sequence identity to hCFTR [99] and have submucosal glands in the large airways [97, 100]. Rat models have also provided the groundwork for exploring new genetic advancements in CF modelling, like the generation of the first G542X CF nonsense mutation rat model with CRISPR-Cas9 [101], and a new F508del rat models that may be invaluable in the development of therapeutics [97].
(iii) Rabbit model: Rabbit models of CF are rather new to the field [102, 103] thus the relevance to human CF disease remains to be seen. However, rabbits present as a very promising model for the study of lung diseases in general, due to their airway anatomy and inflammatory responses [104]. Further, there is a 92% amino acid sequence conservation between rabbit and human CFTR [88]. A caveat of the rabbit model is they lack submucosal glands within their airways [100, 104] which contain CFTR-expressing cells in human airways.
(iv) Ferret model: Due to the highly conserved anatomy between human and ferret lungs [105, 106], ferret CF models accurately mirror the key disease phenotypes of CF, including those unable to be recapitulated in other models [107,108,109] With a sequence homology of 92% with hCFTR [88], and an abundance of submucosal glands throughout their airways [110], ferrets are an attractive translational model of CF [111]. A caveat of the ferret model is the costs associated with maintaining these animal colonies and current CF ferret models require CFTR modulators to survive, making long-term study of the disease pathogenesis difficult.
(v) Pig model: Pig models share a 92% amino acid sequence identity with hCFTR [88], and arguably offer the highest translational potential for CF research due to their comparable genetics, physiology, and anatomy to humans [112,113,114]. However, porcine CF models present an even larger practical and cost challenge than ferrets. Their sheer size, while beneficially comparable to humans, calls for much consideration regarding labor costs and maintenance. For testing new drugs, the pig model can become astronomically expensive. Nevertheless, CF pig models recapitulate all key CF disease phenotypes, though notably with more severe manifestations than in humans [113,114,115,116,117].
Cell models for studying CF disease pathogenesis and therapy.
(i) Current gold-standard lung cell models: Cell models have played instrumental roles in understanding the biophysical properties of CFTR, the mechanistic cause of the defects and evaluating novel therapeutic strategies (Fig. 4). Human primary epithelial cell lines have been the main tool for assessing ion channel functions and for drug development [118–121]. While recent improvements in culture conditions have improved the expansion potential of primary cells, this expansive ability is limited [122] and primary cells enter senescence shortly in culture. To circumvent this, immortalized epithelial cell lines, such as A549, BEAS-2B, Calu-3 and 16HBE14o, are commonly used to study drug transport, metabolism, and epithelial integrity [123–127]. However, these immortalized cell lines are derived from lung tumour cells or have been transformed, and thus do not show original lung cell characteristics or reflect the repertoire of epithelial cell types found in the native lungs. Primary nasal cells are an alternative cell type to study CF airway disease due to the ease of generating nasal epithelial cultures from patients. The pros of these cells are the relative ease of obtaining samples from patients and they can be sampled several times (if needed). Studies have suggested nasal epithelial cells are a good surrogate of airway bronchial epithelial cells [128, 129]. However, like primary bronchial cells, the ability to expand these cells in culture for sufficient use without re-sampling remains a problem. In addition, sampling variability can impact CFTR protein expression and function of the epithelium. Recently, lung stem cells isolated from bronchoalveolar lavage fluid can generate renewable airway organoids for multiple passages in cultures [130]. It remains to be seen whether a method of airway organoid generation can be achieved from individuals with airway diseases for disease modeling. Nonetheless, generation of a renewable source of patient-specific lung airway cells is a key enabler for identifying patient-specific therapies for lung diseases.

Cell models to study CF disease and therapies. For gene editing approaches, green “✓” indicates research data supporting the use of these approaches in the cell models for CF gene correction. Red “X” indicates no information available. For advantages/limitations section, green “✓” indicates possible and red “X” indicates not possible
(ii) Human pluripotent stem cell (PSC) models for personalized medicine: Human embryonic stem (hES) cells were discovered in 1998 and hold enormous promise to repair disease and regenerate tissues [131]. With the ability to self-renew and differentiate into cells of all three embryonic germ layers endoderm, ectoderm and mesoderm, hES became an intriguing source of cells for regenerative medicine. However, research in the use of hES for regeneration faced paucity due to the growing ethical concerns associated with the use of “embryonic/fetal” tissue. In 2006, the first discovery of induced pluripotent stem cells (iPSC) was made and revealed these cells shared similar characteristics to mouse ES [132]. By 2007, the first human iPSC was made by introducing four transcription factors associated with pluripotency to fibroblasts [133]. Since this discovery, therapeutic applications of human iPSC have led to > 65 market competitors offering iPSC-based products. Indeed, iPSC are a great source of cells for patient-specific disease modeling, drug discovery and personalized regenerative medicine. Biobanks of iPSC from individuals with various genetic mutations have become a useful resource for disease modeling. The Hospital for Sick Children in Toronto has now acquired over 100 CF patient cells harboring various CFTR variants and generated iPSC from each individual including some gene-corrected isogenic iPSC lines for benchmarking patient-specific “normal” responses [134]. This will undoubtedly enable research in modelling CF organ and patient-specific disease and therapy discoveries.
Differentiation of human iPSC into multiple tissue cell types has now been achieved albeit with varying efficiencies. Most directed differentiation methods use a stepwise approach of activating and/or inhibiting pathways known to affect developmental growth in animal models, especially the mouse. Indeed, we and others have identified key developmental pathways required to generate lung epithelial cells from human iPSC [135–138]. Moreover, airway and intestinal cells derived from homozygous F508del CF iPSC model CF phenotype (lack of CFTR membrane expression) can be used to screen for CFTR small molecule correctors [136, 139, 140]. We have shown that CF iPSC-derived airway cells are amenable to high throughput CFTR functional screens—a step towards using these cells for personalized medicine [139–143]. Recently, we have improved the generation of lung cells from human PSC and demonstrate the utility of capturing CFTR expression and function in the differentiated cells modeling development [136, 144]. Understanding the impact of mutant CFTR during development remains poorly understood and these new PSC models will advance our understanding of the prenatal origins of disease mechanisms.
Another benefit of using iPSC models is the ability to determine both patient and tissue-specific responses. This is important as CFTR expression and activity levels differ in different tissues. Correction of CF mutations have been tested in iPSC, however the efficacy of these gene-editing strategies in-vivo remains to be seen [141–143]. Ultimately, establishing predictive patient and tissue specific models to predict patient outcome is key to advancing precision medicine.
New models, new gene editing tools, new targets?
One of the biggest challenges in generating treatment strategies for CF is the sheer number of CF-causing variants. Even among patients with the same variant, there are vast differences in severity of symptoms and responses to treatments. To date, treatment options for CF are mutation-dependent, and no viable options exist to universally address all CF patients. Though recent advancements in gene editing have fostered hope for personalized treatments, this is neither viable nor practical for treating all CF.
Recently, Kemaladewi et al. demonstrated a novel mutant-independent therapeutic approach to treat congenital muscular dystrophy type 1A (MDC1A) [145]. Using CRISPR, the feasibility of treating inherited diseases by looking beyond the singular disease-causing gene, and instead targeting compensatory modifier genes, was illustrated. In the context of CF, ion channels aside from CFTR have been implicated in CF disease severity and responses to modulator therapy. Therefore, targeting other ion channels known to also affect CF disease severity such as the sodium channel ENaC [146] or alternative ion channels TMEM16A (ANO1 [147, 148]) and SLC26A9 [149, 150] may need to be assessed to find effective therapies for all individuals with CF.
Conclusion
Since the discovery of the CF gene over 30 years ago, it has become apparent that finding an effective therapy to treat all CF remains a challenge. While the discoveries of new small molecule modulators have greatly advanced treatment for some CF, the effectiveness of these lifesaving drugs have not been universally effective and rather limited to specific classes of mutations. Rare CFTR variants remain uncured. Now, with recent advances in new gene editing tools coupled with both iPSC-derived tissue models and new animal models, new precise gene targeting methods to treat CF disease will emerge and lead to potential effective personalized therapies. Classical approaches of targeting the disease-causing variant may also be replaced or coupled with mutation-agnostic approaches to treat complex CF phenotypes and with improved pre-clinical models, this can now be tested. With new advancements in gene editing technologies coupled with advanced cell models to test gene engineering approaches, this will lead to rapid developments of new therapies for all CF.
Availability of data and materials
Not applicable.
Abbreviations
- CF:
-
Cystic fibrosis
- CFTR:
-
Cystic fibrosis transmembrane conductance regulator
- F598del:
-
P.Phe508del
- ABC transporters:
-
Adenosine triphosphate (ATP)-binding cassette transporters
- MSD1 and MSD2:
-
Membrane-spanning domain 1 and 2
- NBD1 and MBD2:
-
Nucleotide-binding domain 1 and 2
- R domain:
-
Regulatory domain
- ASL:
-
Airway surface liquid
- PTC:
-
Premature termination codon
- NMD:
-
Nonsense mediated decay
- ZFNs:
-
Zinc Finger Nucleases
- TALEN:
-
Transcription activator-like effector nucleases
- DSB:
-
Double-strand break
- NHEJ:
-
Non homologous end joining
- HDR:
-
Homology-directed repair
- CRISPR:
-
Clustered regularly interspaced short palindromic repeats
- Cas9:
-
CRISPR associated protein 9
- gRNA:
-
Guide RNA
- indels:
-
Insertions or deletions
- CBEs:
-
Cytosine base editors
- ABEs:
-
Adenine base editors
- pegRNA:
-
Prime editing guide RNA
- Ad:
-
Adenovirus
- AAV:
-
Adeno-associated virus
- Ad:
-
Adenovirus
- E1:
-
Early region 1
- E3:
-
Early region 3
- AAV:
-
Adeno-associated virus
- Hd-Ad:
-
Helper-dependent adenoviruses
- FLIPR:
-
Fluorescence imaging plate reader
- hCFTR :
-
Human cystic fibrosis transmembrane conductance regulator
- hES:
-
Human embryonic stem
- iPSC:
-
Induced pluripotent stem cells
- MDC1A:
-
Muscular dystrophy type 1A
- ENaC:
-
Epithelial sodium channel
- TMEM16A:
-
Transmembrane member 16A
- ANO1:
-
Anoctamin-1
- SLC26A9:
-
Solute carrier family 26 member 9
References
Schmidt BZ, Haaf JB, Leal T, Noel S. Cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis: current perspectives. Clin Pharmacol. 2016;8:127–40.
Ratjen F, Bell SC, Rowe SM, Goss CH, Quittner AL, Bush A. Cystic fibrosis. Nat Rev Dis Primers. 2015;1:15010.
Moran O. On the structural organization of the intracellular domains of CFTR. Int J Biochem Cell Biol. 2014;52:7–14.
Estivill X, Bancells C, Ramos C. Geographic distribution and regional origin of 272 cystic fibrosis mutations in European populations. The Biomed CF Mutation Analysis Consortium. Hum Mutat. 1997;10(2):135–154.
Gadsby DC, Vergani P, Csanady L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature. 2006;440(7083):477–83.
Kopito RR. Biosynthesis and degradation of CFTR. Physiol Rev. 1999;79(1 Suppl):S167–73.
Stoltz DA, Meyerholz DK, Welsh MJ. Origins of cystic fibrosis lung disease. N Engl J Med. 2015;372(4):351–62.
Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245(4922):1066–73.
Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245(4922):1073–80.
Tsui LC, Buchwald M, Barker D, Braman JC, Knowlton R, Schumm JW, et al. Cystic fibrosis locus defined by a genetically linked polymorphic DNA marker. Science. 1985;230(4729):1054–7.
Zengerling S, Tsui LC, Grzeschik KH, Olek K, Riordan JR, Buchwald M. Mapping of DNA markers linked to the cystic fibrosis locus on the long arm of chromosome 7. Am J Hum Genet. 1987;40(3):228–36.
De Boeck K, Amaral MD. Progress in therapies for cystic fibrosis. Lancet Respir Med. 2016;4(8):662–74.
Clancy JP, Cotton CU, Donaldson SH, Solomon GM, VanDevanter DR, Boyle MP, et al. CFTR modulator theratyping: current status, gaps and future directions. J Cyst Fibros. 2019;18(1):22–34.
Patel SD, Bono TR, Rowe SM, Solomon GM. CFTR targeted therapies: recent advances in cystic fibrosis and possibilities in other diseases of the airways. Eur Respir Rev. 2020;29(156):190068.
Pranke I, Bidou L, Martin N, Blanchet S, Hatton A, Karri S, et al. Factors influencing readthrough therapy for frequent cystic fibrosis premature termination codons. ERJ Open Res. 2018;4(1):00080–2017.
Kerem E, Konstan MW, De Boeck K, Accurso FJ, Sermet-Gaudelus I, Wilschanski M, et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir Med. 2014;2(7):539–47.
Nguyen LS, Wilkinson MF, Gecz J. Nonsense-mediated mRNA decay: inter-individual variability and human disease. Neurosci Biobehav Rev. 2014;46(Pt 2):175–86.
Keenan MM, Huang L, Jordan NJ, Wong E, Cheng Y, Valley HC, et al. Nonsense-mediated RNA decay pathway inhibition restores expression and function of W1282X CFTR. Am J Respir Cell Mol Biol. 2019;61(3):290–300.
Ramsay KA, Stockwell RE, Bell SC, Kidd TJ. Infection in cystic fibrosis: impact of the environment and climate. Expert Rev Respir Med. 2016;10(5):505–19.
Szczesniak R, Rice JL, Brokamp C, Ryan P, Pestian T, Ni Y, et al. Influences of environmental exposures on individuals living with cystic fibrosis. Expert Rev Respir Med. 2020;14(7):737–48.
Oates GR, Schechter MS. Socioeconomic status and health outcomes: cystic fibrosis as a model. Expert Rev Respir Med. 2016;10(9):967–77.
O’Neal WK, Knowles MR. Cystic fibrosis disease modifiers: complex genetics defines the phenotypic diversity in a monogenic disease. Annu Rev Genomics Hum Genet. 2018;19:201–22.
Strug LJ, Gonska T, He G, Keenan K, Ip W, Boelle PY, et al. Cystic fibrosis gene modifier SLC26A9 modulates airway response to CFTR-directed therapeutics. Hum Mol Genet. 2016;25(20):4590–600.
Gong J, Wang F, Xiao B, Panjwani N, Lin F, Keenan K, et al. Genetic association and transcriptome integration identify contributing genes and tissues at cystic fibrosis modifier loci. PLoS Genet. 2019;15(2):e1008007.
Corvol H, Mesinele J, Douksieh IH, Strug LJ, Boelle PY, Guillot L. SLC26A9 Gene is associated with lung function response to ivacaftor in patients with cystic fibrosis. Front Pharmacol. 2018;9:828.
Middleton PG, Mall MA, Drevinek P, Lands LC, McKone EF, Polineni D, et al. Elexacaftor-tezacaftor-ivacaftor for cystic fibrosis with a single Phe508del allele. N Engl J Med. 2019;381(19):1809–19.
Lopes-Pacheco M. CFTR modulators: the changing face of cystic fibrosis in the era of precision medicine. Front Pharmacol. 2019;10:1662.
Vadagam P, Kamal KM, Covvey JR, Giannetti V, Mukherjee K. Cost-effectiveness and budget impact of lumacaftor/ivacaftor in the treatment of cystic fibrosis. J Manag Care Spec Pharm. 2018;24(10):987–97.
Cao H, Ouyang H, Grasemann H, Bartlett C, Du K, Duan R, et al. Transducing airway basal cells with a helper-dependent adenoviral vector for lung gene therapy. Hum Gene Ther. 2018;29(6):643–52.
Cao H, Machuca TN, Yeung JC, Wu J, Du K, Duan C, et al. Efficient gene delivery to pig airway epithelia and submucosal glands using helper-dependent adenoviral vectors. Mol Ther Nucleic Acids. 2013;2:e127.
Molinski SV, Ahmadi S, Ip W, Ouyang H, Villella A, Miller JP, et al. Orkambi(R) and amplifier co-therapy improves function from a rare CFTR mutation in gene-edited cells and patient tissue. EMBO Mol Med. 2017;9(9):1224–43.
Johnson LG, Olsen JC, Sarkadi B, Moore KL, Swanstrom R, Boucher RC. Efficiency of gene transfer for restoration of normal airway epithelial function in cystic fibrosis. Nat Genet. 1992;2(1):21–5.
Zhang L, Button B, Gabriel SE, Burkett S, Yan Y, Skiadopoulos MH, et al. CFTR delivery to 25% of surface epithelial cells restores normal rates of mucus transport to human cystic fibrosis airway epithelium. PLoS Biol. 2009;7(7):e1000155.
Carroll D. Genome engineering with zinc-finger nucleases. Genetics. 2011;188(4):773–82.
Liu J, Gaj T, Yang Y, Wang N, Shui S, Kim S, et al. Efficient delivery of nuclease proteins for genome editing in human stem cells and primary cells. Nat Protoc. 2015;10(11):1842–59.
Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11(9):636–46.
Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, et al. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010;186(2):757–61.
Li T, Huang S, Jiang WZ, Wright D, Spalding MH, Weeks DP, et al. TAL nucleases (TALNs): hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucleic Acids Res. 2011;39(1):359–72.
Lukacsovich T, Yang D, Waldman AS. Repair of a specific double-strand break generated within a mammalian chromosome by yeast endonuclease I-SceI. Nucleic Acids Res. 1994;22(25):5649–57.
Ceccaldi R, Rondinelli B, D’Andrea AD. Repair pathway choices and consequences at the double-strand break. Trends Cell Biol. 2016;26(1):52–64.
Pattanayak V, Ramirez CL, Joung JK, Liu DR. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods. 2011;8(9):765–70.
Holkers M, Maggio I, Liu J, Janssen JM, Miselli F, Mussolino C, et al. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013;41(5):e63.
Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–6.
Lee JK, Jeong E, Lee J, Jung M, Shin E, Kim YH, et al. Directed evolution of CRISPR-Cas9 to increase its specificity. Nat Commun. 2018;9(1):3048.
Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NT, Zheng Z, et al. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529(7587):490–5.
Mann CM, Martinez-Galvez G, Welker JM, Wierson WA, Ata H, Almeida MP, et al. The Gene Sculpt Suite: a set of tools for genome editing. Nucleic Acids Res. 2019;47(W1):W175–82.
Capecchi MR. Altering the genome by homologous recombination. Science. 1989;244(4910):1288–92.
Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature. 2016;533(7603):420–4.
Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature. 2017;551(7681):464–71.
Geurts MH, de Poel E, Amatngalim GD, Oka R, Meijers FM, Kruisselbrink E, et al. CRISPR-based adenine editors correct nonsense mutations in a cystic fibrosis organoid biobank. Cell Stem Cell. 2020;26(4):503-10e7.
Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576(7785):149–57.
Maarten H. Geurts EdP, Cayetano Pleguezuelos-Manzano, Léo Carrillo, Amanda Andersson-Rolf, Matteo Boretto, Jeffrey M. Beekman, View ORCID ProfileHans Clevers. Evaluating CRISPR-based Prime Editing for cancer modeling and CFTR repair in intestinal organoids. biorxiv. 2020.
Zabner J, Fasbender AJ, Moninger T, Poellinger KA, Welsh MJ. Cellular and molecular barriers to gene transfer by a cationic lipid. J Biol Chem. 1995;270(32):18997–9007.
Caplen NJ, Alton EW, Middleton PG, Dorin JR, Stevenson BJ, Gao X, et al. Liposome-mediated CFTR gene transfer to the nasal epithelium of patients with cystic fibrosis. Nat Med. 1995;1(1):39–46.
Alton EW, Boyd AC, Cheng SH, Cunningham S, Davies JC, Gill DR, et al. A randomised, double-blind, placebo-controlled phase IIB clinical trial of repeated application of gene therapy in patients with cystic fibrosis. Thorax. 2013;68(11):1075–7.
Alton E, Armstrong DK, Ashby D, Bayfield KJ, Bilton D, Bloomfield EV, et al. A randomised, double-blind, placebo-controlled trial of repeated nebulisation of non-viral cystic fibrosis transmembrane conductance regulator (CFTR) gene therapy in patients with cystic fibrosis. Efficacy and Mechanism Evaluation. Southampton (UK) 2016.
Hyde SC, Southern KW, Gileadi U, Fitzjohn EM, Mofford KA, Waddell BE, et al. Repeat administration of DNA/liposomes to the nasal epithelium of patients with cystic fibrosis. Gene Ther. 2000;7(13):1156–65.
Zabner J, Petersen DM, Puga AP, Graham SM, Couture LA, Keyes LD, et al. Safety and efficacy of repetitive adenovirus-mediated transfer of CFTR cDNA to airway epithelia of primates and cotton rats. Nat Genet. 1994;6(1):75–83.
Yei S, Mittereder N, Tang K, O’Sullivan C, Trapnell BC. Adenovirus-mediated gene transfer for cystic fibrosis: quantitative evaluation of repeated in vivo vector administration to the lung. Gene Ther. 1994;1(3):192–200.
Sheets RL, Stein J, Bailer RT, Koup RA, Andrews C, Nason M, et al. Biodistribution and toxicological safety of adenovirus type 5 and type 35 vectored vaccines against human immunodeficiency virus-1 (HIV-1), Ebola, or Marburg are similar despite differing adenovirus serotype vector, manufacturer’s construct, or gene inserts. J Immunotoxicol. 2008;5(3):315–35.
Shirley JL, de Jong YP, Terhorst C, Herzog RW. Immune responses to viral gene therapy vectors. Mol Ther. 2020;28(3):709–22.
Flotte TR, Ng P, Dylla DE, McCray PB Jr, Wang G, Kolls JK, et al. Viral vector-mediated and cell-based therapies for treatment of cystic fibrosis. Mol Ther. 2007;15(2):229–41.
Zabner J, Couture LA, Gregory RJ, Graham SM, Smith AE, Welsh MJ. Adenovirus-mediated gene transfer transiently corrects the chloride transport defect in nasal epithelia of patients with cystic fibrosis. Cell. 1993;75(2):207–16.
Rich DP, Couture LA, Cardoza LM, Guiggio VM, Armentano D, Espino PC, et al. Development and analysis of recombinant adenoviruses for gene therapy of cystic fibrosis. Hum Gene Ther. 1993;4(4):461–76.
Alton E, Armstrong DK, Ashby D, Bayfield KJ, Bilton D, Bloomfield EV, et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: a randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir Med. 2015;3(9):684–91.
Simon RH, Engelhardt JF, Yang Y, Zepeda M, Weber-Pendleton S, Grossman M, et al. Adenovirus-mediated transfer of the CFTR gene to lung of nonhuman primates: toxicity study. Hum Gene Ther. 1993;4(6):771–80.
Hallek M, Wendtner CM. Recombinant adeno-associated virus (rAAV) vectors for somatic gene therapy: recent advances and potential clinical applications. Cytokines Mol Ther. 1996;2(2):69–79.
Halbert CL, Standaert TA, Wilson CB, Miller AD. Successful readministration of adeno-associated virus vectors to the mouse lung requires transient immunosuppression during the initial exposure. J Virol. 1998;72(12):9795–805.
Wagner JA, Reynolds T, Moran ML, Moss RB, Wine JJ, Flotte TR, et al. Efficient and persistent gene transfer of AAV-CFTR in maxillary sinus. Lancet. 1998;351(9117):1702–3.
Moss RB, Rodman D, Spencer LT, Aitken ML, Zeitlin PL, Waltz D, et al. Repeated adeno-associated virus serotype 2 aerosol-mediated cystic fibrosis transmembrane regulator gene transfer to the lungs of patients with cystic fibrosis: a multicenter, double-blind, placebo-controlled trial. Chest. 2004;125(2):509–21.
Moss RB, Milla C, Colombo J, Accurso F, Zeitlin PL, Clancy JP, et al. Repeated aerosolized AAV-CFTR for treatment of cystic fibrosis: a randomized placebo-controlled phase 2B trial. Hum Gene Ther. 2007;18(8):726–32.
O’Neal WK, Zhou H, Morral N, Langston C, Parks RJ, Graham FL, et al. Toxicity associated with repeated administration of first-generation adenovirus vectors does not occur with a helper-dependent vector. Mol Med. 2000;6(3):179–95.
Parks RJ, Chen L, Anton M, Sankar U, Rudnicki MA, Graham FL. A helper-dependent adenovirus vector system: removal of helper virus by Cre-mediated excision of the viral packaging signal. Proc Natl Acad Sci USA. 1996;93(24):13565–70.
Toietta G, Koehler DR, Finegold MJ, Lee B, Hu J, Beaudet AL. Reduced inflammation and improved airway expression using helper-dependent adenoviral vectors with a K18 promoter. Mol Ther. 2003;7(5 Pt 1):649–58.
Xia E, Duan R, Shi F, Seigel KE, Grasemann H, Hu J. Overcoming the undesirable CRISPR-Cas9 expression in gene correction. Mol Ther Nucleic Acids. 2018;13:699–709.
Zhou ZP, Yang LL, Cao H, Chen ZR, Zhang Y, Wen XY, et al. In vitro validation of a CRISPR-mediated CFTR correction strategy for preclinical translation in pigs. Hum Gene Ther. 2019;30(9):1101–16.
Xia E, Zhang Y, Cao H, Li J, Duan R, Hu J. TALEN-mediated gene targeting for cystic fibrosis-gene therapy. Genes (Basel). 2019;10(1):39.
Cao H, Yang T, Li XF, Wu J, Duan C, Coates AL, et al. Readministration of helper-dependent adenoviral vectors to mouse airway mediated via transient immunosuppression. Gene Ther. 2011;18(2):173–81.
Koehler DR, Frndova H, Leung K, Louca E, Palmer D, Ng P, et al. Aerosol delivery of an enhanced helper-dependent adenovirus formulation to rabbit lung using an intratracheal catheter. J Gene Med. 2005;7(11):1409–20.
Cao H, Ouyang H, Ip W, Du K, Duan W, Avolio J, et al. Testing gene therapy vectors in human primary nasal epithelial cultures. Mol Ther Methods Clin Dev. 2015;2:15034.
Cao H, Ouyang H, Laselva O, Bartlett C, Zhou ZP, Duan C, et al. A helper-dependent adenoviral vector rescues CFTR to wild-type functional levels in cystic fibrosis epithelial cells harbouring class I mutations. Eur Respir J. 2020;56(5):2000205.
Koehler DR, Sajjan U, Chow YH, Martin B, Kent G, Tanswell AK, et al. Protection of Cftr knockout mice from acute lung infection by a helper-dependent adenoviral vector expressing Cftr in airway epithelia. Proc Natl Acad Sci USA. 2003;100(26):15364–9.
Halbert CL, Aitken ML, Miller AD. Retroviral vectors efficiently transduce basal and secretory airway epithelial cells in vitro resulting in persistent gene expression in organotypic culture. Hum Gene Ther. 1996;7(15):1871–81.
Castellani S, Conese M. Lentiviral vectors and cystic fibrosis gene therapy. Viruses. 2010;2(2):395–412.
Cooney ALT, McCray Jr PB, Pezzulo AA, Sinn PL. Lentiviral vectors transduce lung stem cells without disrupting plasticity. 2020.
Bushman FD. Retroviral integration and human gene therapy. J Clin Invest. 2007;117(8):2083–6.
Ichise H, Ichise T, Sasanuma H, Yoshida N. The Cd6 gene as a permissive locus for targeted transgenesis in the mouse. Genesis. 2014;52(5):440–50.
Liu F, Zhang Z, Csanady L, Gadsby DC, Chen J. Molecular structure of the human CFTR ion channel. Cell. 2017;169(1):85-95e8.
Durie PR, Kent G, Phillips MJ, Ackerley CA. Characteristic multiorgan pathology of cystic fibrosis in a long-living cystic fibrosis transmembrane regulator knockout murine model. Am J Pathol. 2004;164(4):1481–93.
Guilbault C, Saeed Z, Downey GP, Radzioch D. Cystic fibrosis mouse models. Am J Respir Cell Mol Biol. 2007;36(1):1–7.
Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, et al. An animal model for cystic fibrosis made by gene targeting. Science. 1992;257(5073):1083–8.
Delaney SJ, Alton EW, Smith SN, Lunn DP, Farley R, Lovelock PK, et al. Cystic fibrosis mice carrying the missense mutation G551D replicate human genotype–phenotype correlations. EMBO J. 1996;15(5):955–63.
Gawenis LR, Hodges CA, McHugh DR, Valerio DM, Miron A, Cotton CU, et al. A BAC transgene expressing human CFTR under control of its regulatory elements rescues Cftr knockout mice. Sci Rep. 2019;9(1):11828.
McHugh DR, Steele MS, Valerio DM, Miron A, Mann RJ, LePage DF, et al. A G542X cystic fibrosis mouse model for examining nonsense mutation directed therapies. PLoS ONE. 2018;13(6):e0199573.
McCarron A, Parsons D, Donnelley M. Animal and cell culture models for cystic fibrosis: which model is right for your application? Am J Pathol. 2021;191(2):228–42.
Tuggle KL, Birket SE, Cui X, Hong J, Warren J, Reid L, et al. Characterization of defects in ion transport and tissue development in cystic fibrosis transmembrane conductance regulator (CFTR)-knockout rats. PLoS ONE. 2014;9(3):e91253.
Dreano E, Bacchetta M, Simonin J, Galmiche L, Usal C, Slimani L, et al. Characterization of two rat models of cystic fibrosis-KO and F508del CFTR-Generated by Crispr-Cas9. Animal Model Exp Med. 2019;2(4):297–311.
McCarron A, Cmielewski P, Reyne N, McIntyre C, Finnie J, Craig F, et al. Phenotypic characterization and comparison of cystic fibrosis rat models generated using CRISPR/Cas9 gene editing. Am J Pathol. 2020;190(5):977–93.
Trezise AE, Szpirer C, Buchwald M. Localization of the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) in the rat to chromosome 4 and implications for the evolution of mammalian chromosomes. Genomics. 1992;14(4):869–74.
Widdicombe JH, Chen LL, Sporer H, Choi HK, Pecson IS, Bastacky SJ. Distribution of tracheal and laryngeal mucous glands in some rodents and the rabbit. J Anat. 2001;198(Pt 2):207–21.
Sharma J, Abbott J, Klaskala L, Zhao G, Birket SE, Rowe SM. A novel G542X CFTR rat model of cystic fibrosis is sensitive to nonsense mediated decay. Front Physiol. 2020;11:611294.
Yang D, Liang X, Pallas B, Hoenerhoff M, Ren Z, Han R, et al. Production of CFTR-DeltaF508 rabbits. Front Genet. 2020;11:627666.
Livraghi-Butrico ART, Wilkinson MH, Terrell K, Pickles RJ, O’Neal WK, Sun F, Boucher R, Grubb BR. CF rabbits: the UNC experience. Pediatr Pulmonol. 2018;53:S148S456.
Kamaruzaman NA, Kardia E, Kamaldin N, Latahir AZ, Yahaya BH. The rabbit as a model for studying lung disease and stem cell therapy. Biomed Res Int. 2013;2013:691830.
Johnson-Delaney CA, Orosz SE. Ferret respiratory system: clinical anatomy, physiology, and disease. Vet Clin North Am Exot Anim Pract. 2011;14(2):357–67 (vii).
Liu X, Luo M, Zhang L, Ding W, Yan Z, Engelhardt JF. Bioelectric properties of chloride channels in human, pig, ferret, and mouse airway epithelia. Am J Respir Cell Mol Biol. 2007;36(3):313–23.
Sun X, Olivier AK, Liang B, Yi Y, Sui H, Evans TI, et al. Lung phenotype of juvenile and adult cystic fibrosis transmembrane conductance regulator-knockout ferrets. Am J Respir Cell Mol Biol. 2014;50(3):502–12.
Sun X, Olivier AK, Yi Y, Pope CE, Hayden HS, Liang B, et al. Gastrointestinal pathology in juvenile and adult CFTR-knockout ferrets. Am J Pathol. 2014;184(5):1309–22.
Olivier AK, Yi Y, Sun X, Sui H, Liang B, Hu S, et al. Abnormal endocrine pancreas function at birth in cystic fibrosis ferrets. J Clin Invest. 2012;122(10):3755–68.
Rosen BH, Chanson M, Gawenis LR, Liu J, Sofoluwe A, Zoso A, et al. Animal and model systems for studying cystic fibrosis. J Cyst Fibros. 2018;17(2S):S28–34.
Sun X, Yi Y, Yan Z, Rosen BH, Liang B, Winter MC, et al. In utero and postnatal VX-770 administration rescues multiorgan disease in a ferret model of cystic fibrosis. Sci Transl Med. 2019;11(485).
Rogers CS, Abraham WM, Brogden KA, Engelhardt JF, Fisher JT, McCray PB Jr, et al. The porcine lung as a potential model for cystic fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;295(2):L240–63.
Meyerholz DK, Stoltz DA, Namati E, Ramachandran S, Pezzulo AA, Smith AR, et al. Loss of cystic fibrosis transmembrane conductance regulator function produces abnormalities in tracheal development in neonatal pigs and young children. Am J Respir Crit Care Med. 2010;182(10):1251–61.
Ostedgaard LS, Meyerholz DK, Chen JH, Pezzulo AA, Karp PH, Rokhlina T, et al. The DeltaF508 mutation causes CFTR misprocessing and cystic fibrosis-like disease in pigs. Sci Transl Med. 2011;3(74):74ra24.
Rogers CS, Hao Y, Rokhlina T, Samuel M, Stoltz DA, Li Y, et al. Production of CFTR-null and CFTR-DeltaF508 heterozygous pigs by adeno-associated virus-mediated gene targeting and somatic cell nuclear transfer. J Clin Invest. 2008;118(4):1571–7.
Uc A, Olivier AK, Griffin MA, Meyerholz DK, Yao J, Abu-El-Haija M, et al. Glycaemic regulation and insulin secretion are abnormal in cystic fibrosis pigs despite sparing of islet cell mass. Clin Sci (Lond). 2015;128(2):131–42.
Uc A, Giriyappa R, Meyerholz DK, Griffin M, Ostedgaard LS, Tang XX, et al. Pancreatic and biliary secretion are both altered in cystic fibrosis pigs. Am J Physiol Gastrointest Liver Physiol. 2012;303(8):G961–8.
Awatade NT, Uliyakina I, Farinha CM, Clarke LA, Mendes K, Sole A, et al. Measurements of functional responses in human primary lung cells as a basis for personalized therapy for cystic fibrosis. EBioMedicine. 2015;2(2):147–53.
Forbes B, Ehrhardt C. Human respiratory epithelial cell culture for drug delivery applications. Eur J Pharm Biopharm. 2005;60(2):193–205.
Suzuki S, Crane AM, Anirudhan V, Barilla C, Matthias N, Randell SH, et al. Highly efficient gene editing of cystic fibrosis patient-derived airway basal cells results in functional CFTR correction. Mol Ther. 2020;28(7):1684–95.
Maule G, Casini A, Montagna C, Ramalho AS, De Boeck K, Debyser Z, et al. Allele specific repair of splicing mutations in cystic fibrosis through AsCas12a genome editing. Nat Commun. 2019;10(1):3556.
Rayner RE, Makena P, Prasad GL, Cormet-Boyaka E. Optimization of normal human bronchial epithelial (NHBE) cell 3D cultures for in vitro lung model studies. Sci Rep. 2019;9(1):500.
Crabbe A, Ledesma MA, Nickerson CA. Mimicking the host and its microenvironment in vitro for studying mucosal infections by Pseudomonas aeruginosa. Pathog Dis. 2014;71(1):1–19.
Haws C, Finkbeiner WE, Widdicombe JH, Wine JJ. CFTR in Calu-3 human airway cells: channel properties and role in cAMP-activated Cl− conductance. Am J Physiol. 1994;266(5 Pt 1):L502–12.
Cozens AL, Yezzi MJ, Kunzelmann K, Ohrui T, Chin L, Eng K, et al. CFTR expression and chloride secretion in polarized immortal human bronchial epithelial cells. Am J Respir Cell Mol Biol. 1994;10(1):38–47.
Bednarski C, Tomczak K, Vom Hovel B, Weber WM, Cathomen T. Targeted integration of a super-exon into the CFTR locus leads to functional correction of a cystic fibrosis cell line model. PLoS ONE. 2016;11(8):e0161072.
Bellec J, Bacchetta M, Losa D, Anegon I, Chanson M, Nguyen TH. CFTR inactivation by lentiviral vector-mediated RNA interference and CRISPR-Cas9 genome editing in human airway epithelial cells. Curr Gene Ther. 2015;15(5):447–59.
Brewington JJ, Filbrandt ET, LaRosa FJ, 3rd, Moncivaiz JD, Ostmann AJ, Strecker LM, et al. Brushed nasal epithelial cells are a surrogate for bronchial epithelial CFTR studies. JCI Insight. 2018;3(13).
Chu HW, Rios C, Huang C, Wesolowska-Andersen A, Burchard EG, O’Connor BP, et al. CRISPR-Cas9-mediated gene knockout in primary human airway epithelial cells reveals a proinflammatory role for MUC18. Gene Ther. 2015;22(10):822–9.
Sachs N, Papaspyropoulos A, Zomer-van Ommen DD, Heo I, Bottinger L, Klay D, et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019;38(4).
Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282(5391):1145–7.
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663–76.
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–72.
Eckford PDW, McCormack J, Munsie L, He G, Stanojevic S, Pereira SL, et al. The CF Canada-Sick Kids Program in individual CF therapy: a resource for the advancement of personalized medicine in CF. J Cyst Fibros. 2019;18(1):35–43.
Wong AP, Bear CE, Chin S, Pasceri P, Thompson TO, Huan LJ, et al. Directed differentiation of human pluripotent stem cells into mature airway epithelia expressing functional CFTR protein. Nat Biotechnol. 2012;30(9):876–82.
Wong AP, Chin S, Xia S, Garner J, Bear CE, Rossant J. Efficient generation of functional CFTR-expressing airway epithelial cells from human pluripotent stem cells. Nat Protoc. 2015;10(3):363–81.
McCauley KB, Hawkins F, Serra M, Thomas DC, Jacob A, Kotton DN. Efficient derivation of functional human airway epithelium from pluripotent stem cells via temporal regulation of Wnt signaling. Cell Stem Cell. 2017;20(6):844-57e6.
Miller AJ, Yu Q, Czerwinski M, Tsai YH, Conway RF, Wu A, et al. In vitro and in vivo development of the human airway at single-cell resolution. Dev Cell. 2020;54(6):818.
Ahmadi S, Bozoky Z, Di Paola M, Xia S, Li C, Wong AP, et al. Phenotypic profiling of CFTR modulators in patient-derived respiratory epithelia. NPJ Genom Med. 2017;2:12.
Merkert S, Schubert M, Olmer R, Engels L, Radetzki S, Veltman M, et al. High-throughput screening for modulators of CFTR activity based on genetically engineered cystic fibrosis disease-specific iPSCs. Stem cell reports. 2019;12:1389–403.
Crane AM, Kramer P, Bui JH, Chung WJ, Li XS, Gonzalez-Garay ML, et al. Targeted correction and restored function of the CFTR gene in cystic fibrosis induced pluripotent stem cells. Stem Cell Reports. 2015;4(4):569–77.
Suzuki S, Sargent RG, Illek B, Fischer H, Esmaeili-Shandiz A, Yezzi MJ, et al. TALENs facilitate single-step seamless SDF correction of F508del CFTR in airway epithelial submucosal gland cell-derived CF-iPSCs. Mol Ther Nucleic Acids. 2016;5:e273.
Smirnikhina SA, Kondrateva EV, Adilgereeva EP, Anuchina AA, Zaynitdinova MI, Slesarenko YS, et al. P.F508del editing in cells from cystic fibrosis patients. PLoS ONE. 2020;15(11):e0242094.
Shuk Yee Ngan HQ, Joshua Dierolf, Jin-A Lee, Elena Huang, Amy P. Wong. Modeling lung cell development using human pluripotent stem cells. bioRxiv. 2021.
Kemaladewi DU, Bassi PS, Erwood S, Al-Basha D, Gawlik KI, Lindsay K, et al. A mutation-independent approach for muscular dystrophy via upregulation of a modifier gene. Nature. 2019;572(7767):125–30.
Shei RJ, Peabody JE, Kaza N, Rowe SM. The epithelial sodium channel (ENaC) as a therapeutic target for cystic fibrosis. Curr Opin Pharmacol. 2018;43:152–65.
Quesada R, Dutzler R. Alternative chloride transport pathways as pharmacological targets for the treatment of cystic fibrosis. J Cyst Fibros. 2020;19(Suppl 1):S37–41.
Huang F, Rock JR, Harfe BD, Cheng T, Huang X, Jan YN, et al. Studies on expression and function of the TMEM16A calcium-activated chloride channel. Proc Natl Acad Sci USA. 2009;106(50):21413–8.
Xu J, Henriksnas J, Barone S, Witte D, Shull GE, Forte JG, et al. SLC26A9 is expressed in gastric surface epithelial cells, mediates Cl−/HCO3− exchange, and is inhibited by NH4+. Am J Physiol Cell Physiol. 2005;289(2):C493–505.
Liu X, Li T, Riederer B, Lenzen H, Ludolph L, Yeruva S, et al. Loss of Slc26a9 anion transporter alters intestinal electrolyte and HCO3(−) transport and reduces survival in CFTR-deficient mice. Pflugers Arch. 2015;467(6):1261–75.
Acknowledgements
This work was supported by the SickKids Foundation-CIHR IHDCYH NI20-1070 and Medicine by Design (University of Toronto) grants. AC is a recipient of the University of Toronto Summer Studentship Award (2021); All figures were prepared with BioRender.com.
Funding
Applicable.
Author information
Authors and Affiliations
Contributions
J-AL: Review of literature and manuscript preparation, Figure preparation. AC: Review of literature and manuscript preparation. EH: Manuscript and figure preparation. YX: Review of literature and manuscript preparation. HQ: Review of literature and manuscript preparation. JH: Review and editing of manuscript. APW: Review of literature and manuscript preparation, Review and editing of manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable
Competing interests
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Lee, JA., Cho, A., Huang, E.N. et al. Gene therapy for cystic fibrosis: new tools for precision medicine. J Transl Med 19, 452 (2021). https://doi.org/10.1186/s12967-021-03099-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-021-03099-4