
Abstract
Background
Huntington’s disease (HD) is a neurodegenerative disorder characterized by psychiatric symptoms, serious motor and cognitive deficits. Certain pathological changes can already be observed in pre-symptomatic HD (pre-HD) patients; however, the underlying molecular pathogenesis is still uncertain and no effective treatments are available until now. Here, we reanalyzed HD-related differentially expressed genes from the GEO database between symptomatic HD patients, pre-HD individuals, and healthy controls using bioinformatics analysis, hoping to get more insight in the pathogenesis of both pre-HD and HD, and shed a light in the potential therapeutic targets of the disease.
Methods
Pre-HD and symptomatic HD differentially expressed genes (DEGs) were screened by bioinformatics analysis Gene Expression Omnibus (GEO) dataset GSE1751. A protein–protein interaction (PPI) network was used to select hub genes. Subsequently, Gene Ontology (GO) enrichment analysis of DEGs and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis of hub genes were applied. Dataset GSE24250 was downloaded to verify our hub genes by the Kaplan–Meier method using Graphpad Prism 5.0. Finally, target miRNAs of intersected hub genes involved in pre-HD and symptomatic HD were predicted.
Results
A total of 37 and 985 DEGs were identified in pre-HD and symptomatic HD, respectively. The hub genes, SIRT1, SUZ12, and PSMC6, may be implicated in pre-HD, and the hub genes, FIS1, SIRT1, CCNH, SUZ12, and 10 others, may be implicated in symptomatic HD. The intersected hub genes, SIRT1 and SUZ12, and their predicted target miRNAs, in particular miR-22-3p and miR-19b, may be significantly associated with pre-HD.
Conclusion
The PSMC6, SIRT1, and SUZ12 genes and their related ubiquitin-mediated proteolysis, transcriptional dysregulation, and histone metabolism are significantly associated with pre-HD. FIS1, CCNH, and their related mitochondrial disruption and transcriptional dysregulation processes are related to symptomatic HD, which might shed a light on the elucidation of potential therapeutic targets in HD.
Similar content being viewed by others


Background
Huntington’s disease (HD) is an inherited neurodegenerative disorder characterized by progressive motor and cognitive deficits [1]. The pathogenesis of HD is associated with the abnormal accumulation of mutant huntingtin (mHtt), a protein with expansion of CAG repeats at the amino-terminus of wild-type huntingtin (Htt) [2]. Currently, effective therapies are lacking for HD [3]. In addition, neuronal degeneration can be observed in pre-symptomatic patients as early as 20 years prior to diagnosis [4, 5], indicating that therapeutic intervention could occur several years before the manifestation of clinical symptoms.
Prediction results of gene expression profiling and bioinformatics analysis have become increasingly useful in the diagnosis and therapy of various diseases [6,7,8], including those of the nervous system [9]. In addition to being used to identify the functional connections among genes in an unbiased manner for the in-depth study of biological processes, it can also be used to predict up- and downstream genes and to explore the relationship between gene expression and disease phenotypes [10, 11].
MicroRNAs (miRNAs, 19–24 nucleotides in length) are the most abundant and representative small non-coding RNAs, which negatively regulate messenger RNA (mRNA) levels by binding to the 3′-untranslated region [12, 13]. Numerous studies have described dysregulation of miRNAs in neurodegenerative diseases, indicating that miRNAs may useful in many diseases, including HD [14, 15].
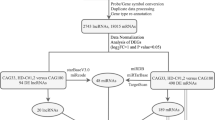
In the present study, the gene expression profile, GSE1751, from the GEO database was reanalyzed with respect to HD-related differentially expressed genes (DEGs) in whole blood samples between symptomatic HD patients, pre-symptomatic HD (pre-HD) individuals, and healthy controls using bioinformatics analysis. Hub genes were selected for deeper functional analysis by constructing a protein–protein interaction (PPI) network and used to predict their related miRNAs. As a result, sets of genes and miRNAs relating to the pathogenesis of HD were identified, increasing our understanding of the disease process and shedding light on markers for the treatment of HD.
Methods
Data acquisition
The gene expression profile, GSE1751, from the GEO database was downloaded (https://www.ncbi.nlm.nih.gov/geo, May 18, 2017). GSE1751 is based on the GPL96 platform (Affymetrix Human Genome U133 Plus 2.0 Array) and contains a total of 31 peripheral whole blood samples, including 12 symptomatic HD cases, 5 pre-symptomatic HD cases, and 14 healthy controls [16]. People who are carriers of the HD mutation but have no clinically present signs or symptoms are considered pre-symptomatic cases [3].
Screening of differentially expressed genes (DEGs)
The R package, “limma” (https://cran.r-project.org/), obtained from https://www.bioconductor.org [17], was used to analyze the GSE1751 raw expression data. Background correction, quantile normalization, and log2-transformation were performed to create a robust multi-array average (RMA) and a log-transformed perfect match. The Benjamini–Hochberg method was used to adjust original p-values, and the false discovery rate (FDR) procedure was used to calculate fold changes (FC). The DEGs between symptomatic HD (GSM30530 to GSM30541) and healthy control (GSM30580 to GSM30593) groups and between pre-symptomatic HD (GSM30542 to GSM30546) and healthy control groups were evaluated. The differential gene expression threshold was log2 fold change > 2 and p-value < 0.05.
Integration of the protein–protein interaction (PPI) network and screening of the hub genes
The Search Tool for the Retrieval of Interacting Gene (STRING) database [18] was used to construct the PPI network of the DEGs in the two groups. Subsequently, the Cytoscape [19] software was used to build a PPI network, employing the Molecular Complex Detection (MCODE) plug-in to screen PPI network modules for these two groups of DEGs. The cluster method (No. 14) was used to cluster the modules, and the genes with an MCODE score ≥ 2 were selected as hub genes.
Gene Ontology and KEGG pathway analysis of DEGs
Database for Annotation, Visualization, and Integrated Discovery (DAVID) (https://david.abcc.ncifcrf.gov/) [20] is a gene functional annotation tool that is helpful for understanding biological functions. GO (Gene Ontology) function enrichment analysis of the DEGs in the two groups was performed, and those with a p-value < 0.05 were considered significantly enriched. Subsequently, the Kyoto Encyclopedia of Genes and Genomes (KEGG) database was employed to elucidate the KEGG pathways of the hub genes in the two groups.
Prediction of the miRNAs of the intersected hub genes and their validation
Finally, these hub genes were input into Targetscan [21] (https://www.targetscan.org/vert_71/) and miRDB [22] (https://www.mirdb.org/) to predict their possible miRNAs. The intersection portion of the two databases was selected as our predicted miRNAs. Moreover, based on the information in the individual MCODE modules, the node with the highest score was selected as the hub gene in GSE1751. Each hub gene was also found in the independent datasets (dataset GSE24250, HD samples n = 8, and HC samples n = 6) based on the downloaded raw data files, including the gene expression level, survival time, and survival state. Expression levels were divided into two groups, the HD group and the control group, according to X-tile [23]. The Kaplan–Meier method was used to determine the probability of survival in Graphpad Prism 5.0 for Windows.
Results
Differentially expressed genes (DEGs) and the heatmap
Based on the analysis of GSE1751 by R and the cut-off criteria, 37 differentially expressed genes were found between the pre-HD individuals and the healthy controls (Group 1) and 985 between the HD patients and the healthy controls (Group 2). There were 35 upregulated DEGs and 2 downregulated DEGs in group 1, and 756 upregulated DEGs and 229 downregulated DEGs in group 2. Additional file 1: Table S1 shows the gene expression level of all DEGs, indicating up- and downregulation as well as the expression differences between the two groups.
Functional GO terms and pathway enrichment analysis
The top 3 GO terms related to the biological processes (BP) of the DEGs in Group 1 were regulation of sequestering of zinc ionic (fold enrichment: 94.78; p-value: 0.0201), response to zinc ion (Fold Enrichment: 72.91; p-value: 0.026), and retrograde transport, endosome to Golgi (Fold Enrichment: 19.34; p-value: 0.095). In Group 2, these BPs were nuclear-transcribed mRNA catabolic process, nonsense-mediated decay (Fold Enrichment: 3.56; p-value: 7.60E-07), SRP-dependent cotranslational protein targeting to membrane (Fold Enrichment: 3.68; p-value: 6.01E−06), and viral transcription (Fold Enrichment: 3.09; p-value: 6.38E−05). Moreover, the top 3 molecular function (MF) terms in Group 1 were chromatin DNA binding (Fold Enrichment: 30.31; p-value: 0.004) and zinc ion transmembrane transporter activity (Fold Enrichment: 44.26; p-value: 0.032). These MFs in Group 2 were protein binding (Fold Enrichment: 1.28; p-value: 2.49E−19), poly(A) RNA binding (fold enrichment: 1.54; p-value: 4.22E−5), and structural constituent of ribosome (Fold Enrichment: 2.34; p-value: 8.66E−5). These results show that zinc ion and chromatin DNA binding are involved in the pre-symptomatic stage and transcription and translation processes are important in the symptomatic stage (Figs. 1 and 2).

Gene ontology (GO) KEGG analysis of Group 1 (pre-HD & HC). The length of the orange bar represents the negative Log10 p value

Gene ontology (GO) KEGG analysis of Group 2 (HD & HC). The length of the orange bar represents the negative Log10 p value
Hub genes selected by PPI network analysis of DEGs
A total of 10 and 5394 nodes were identified from PPI network analysis of groups 1 and 2, respectively. Here, 3 hub genes were found in group 1 [sirtuin 1 (SIRT1), suppressor of zeste 12 (SUZ12), and proteasome 26S subunit, ATPase 6 (PSMC6)] and 14 were found in group 2 [fission mitochondrial 1 (FIS1), chromobox 1 (CBX1), zinc finger protein 217 (ZNF217), TATA-box binding protein-associated factor, RNA polymerase I subunit D (TAF1D) DEK, family with sequence similarity 60 member A(FAM60A), GABA type A receptor-associated protein like 2 (GABARAPL2), SIRT1, histone acetyltransferase 1 (HAT1), cyclin H (CCNH), histone deacetylase 2 (HDAC2), SUZ12, RRN3 homolog, RNA polymerase I transcription factor (RRN3), and PEST proteolytic signal-containing nuclear protein (PCNP)]. The genes and their scores are shown in Additional file 2: Table S2. Importantly, SIRT1 and SUZ12 are unregulated hub genes in both groups, but their expression levels were higher in group 2. The FC values of PSMC6 and SIRT1 in Group 2 were approximately twice those in Group 1 and 1.25 times that of the SUZ12 gene. The expression levels of these three hub genes in HD patients, pre-HD individuals, and healthy controls are shown in Fig. 3.

Box diagram showing the levels of the PSMC6 (a), SIRT1 (b), and SUZ12 (c) genes. The gene expression levels of PSMC6, SIRT1 and SUZ12 decreased on sequence HD group, preHD group and HC group
GO and KEGG analysis results of hub genes in two groups
The functional enrichment results of the hub genes in the two groups are shown in Additional file 3: Table S3. Those in Group 1 mainly participate in the chromatin DNA binding process, and those in Group 2 play roles in the regulation of transcription, chromatin remodeling, cell cycle, proteasome-mediated ubiquitin-dependent protein catabolic processes, and histone-related metabolism processes.
The KEGG database was used for pathway analysis of the hub genes in the two groups. Those in Group 1 participate in 11 pathways, and those in Group 2 participate in 17 pathways. Among them, 9 pathways were intersected in the two groups (Fig. 4). All KEGG pathway results are shown in Additional file 4: Table S4.

Venn diagram of the KEGG pathway results of the hub genes in the two groups. The blue cylinder represents group 1 and the green cylinder represents group 2. The intersection pathways are shown
Prediction of miRNAs of the intersected hub genes and validation of hub genes
Targetscan and miRDB were used to predict the miRNAs of the target genes, SIRT1 and SUZ12, and the intersected prediction results of these two databases became the final predicted miRNAs; 15 for SIRT1 and 13 for SUZ12. For the SIRT1 gene, 6 predicted miRNAs have previously been reported to have a clear relationship with HD (miR-22-3p, miR-138-5p, miR-9-5p, miR-132-3p, miR-135b-5p, and miR-135a-5p) [24, 25], and 1 miRNA for the SUZ12 gene (miR-19b-3p) [26]. All these results are shown in Table 1. To verify the 5 hub genes, the survival rate was calculated for the two groups in the validation dataset (GSE24250) through Kaplan–Meier analysis. We can see clearly that the HD patients with high expression levels of SIRT1, FIS1, and CCNH have a decreased overall survival time compared to those with low expression levels. Additionally, the Kaplan–Meier analysis curves of the high-expression group do not intersect with those of the low-expression group, indicating that the survival time of the high-expression group is always lower than that of the low-expression group. Figure 5a–c shows that the median survival time (dotted line) of the high-expression group is lower than that of the low-expression group. However, the P values of the three genes in the verification dataset are not statistically significant (0.2 for SIRT1, 0.1 for FIS1, and 0.2 for CCNH). This finding might be due to the small size of the validation dataset. The remaining 2 genes showed no statistical significance between gene expression and clinical outcome of HD in the validation dataset.

Kaplan–Meier analysis of the survival time for SIRT1 (a), FIS1 (b), CCNH (c), PSMC6 (d), and SUZ12 (e) in the validation dataset of 85 cases. The results show that the HD patients with high expression levels (blue line) of SIRT1, FIS1, and CCNH have a decreased overall survival time compared to those with low expression levels (red line).The remaining 2 genes showed no statistical significance between gene expression and clinical outcome of HD in the validation dataset. The dotted line represents the median survival time of those genes with different expression levels
Discussion
HD is a neurodegenerative disorder with a long symptomatic phase [27]. Although mHtt is expressed during embryonic development, clinical HD manifests in adulthood [28]. Various studies focusing on the early molecular pathogenesis in the pre-symptomatic stage may provide new insight into the therapeutic treatment of HD [29]; therefore, we attempted to elucidate hub genes, miRNAs, of pre-HD and HD. In recent years, bioinformatics has been increasingly applied to many fields including neurology, laying the foundation for further experimental verification [11, 30, 31]. In the present study, bioinformatics methods were used to predict hub genes and miRNAs associated with HD and pre-HD in both generation and validation datasets with the hope of providing insight into the pathogenesis in both HD and pre-HD patients. The results indicate that SIRT1, SUZ12, and PSMC6 may be involved in the pathogenesis of pre-HD, and FIS1, SIRT1, CCNH, and SUZ12 may play roles in symptomatic HD. GO term enrichment and KEGG analysis demonstrates that PSMC6 and its ubiquitin-mediated proteolysis pathway play an important role in pre-HD. Moreover, CCNH and transcriptional dysregulation, in addition to FIS1 and mitochondrial disruption, may participate in the pathogenesis of HD in the late stage. Interestingly, the intersected hub genes, SIRT1 and SUZ12, and their transcriptional regulation and histone-related metabolism pathways may play important roles in both pre-HD and HD. MiR-22-3p and miR-19b may be involved in the pathogenesis through these intersected hub genes, respectively.
Since no survival data regarding GSE1751 are available, an independent validated dataset, GSE24250, was employed to elucidate whether the up- or downregulation of hub genes could affect the survival time of HD patients. According to the Kaplan–Meier analysis results, the upregulation of SIRT1, FIS1, and CCNH has a negative correlation with survival time in HD patients, which is consistent with our results. However, the association of SUZ12 and PSMC6 with survival time was not statistically significant, which may be due to the low incidence of HD and the small sample size of our datasets.
In the present study, the PSMC6 gene was highly associated with the pre-symptomatic phase of HD. PSMC6, also called proteasome regulatory subunit 4 (RPT4), is a subunit of the 19S proteasome regulatory protein, which can participate in ubiquitin-mediated proteolysis [32, 33]. Disassembly of the proteasome by Htt aggregates has been reported in various studies [34], and overexpression of RPT4 has been shown to facilitate aggregation of mHtt in a cellular model of HD [35]. PSMC6 and the related ubiquitin-mediated proteolysis were shown to be involved in the pre-symptomatic stage. Interestingly, the expression level of PSMC6 in pre-HD patients was 74 as compared with 123 in HD patients and 20 in healthy controls, indicating that PSMC6 is already involved in the pre-symptomatic stage of HD and continues to play a role in the symptomatic stage.
Moreover, it was found that CCNH and FIS1 were highly associated with late-stage HD rather than the pre-symptomatic stage. CCNH is assembled with TFIIH core proteins and phosphorylates the C-terminal domain of RNA polymerase II to facilitate promoter clearance [36]. RNA polymerase II has been shown to increase in the postmortem HD brain [37] and in vitro [25]. We hypothesize that CCNH may play a role in the pathogenesis of HD. FIS1 could interact with dynamin-related protein 1 (DRP1), which is the primary component of mitochondrial fission [38]. An inhibitor of the DRP1–FIS1 interaction was protective in a mouse model of HD [39]. Therefore, CCNH and the related transcriptional dysregulation and FIS1 and the related mitochondrial disruption may be involved in the pathogenesis of HD during the symptomatic stage.
The SIRT1 and SUZ12 genes may play important roles during both the pre-symptomatic and symptomatic stage in HD patients. The FC values of SIRT1 and SUZ12 in Group 2 were approximately 2- and 1.25-times those in Group 1, respectively. SIRT1 belongs to a highly conserved family of sirtuins, the overexpression of which exerts neuroprotection through deacetylation of several transcription factors such as PCG-1α, p53, and FOXO3a [40,41,42]. The Sirt1 activator, resveratrol (RESV), decreases histone H3 acetylation at lysine 9 and improves motor coordination in the YAC128 and N171-82Q HD mouse model [43, 44]. SUZ12 is the core subunit of the polycomb repressive complex 2 (PRC2), which can implement gene silencing through methylation and ubiquitylation of histones [45,46,47]. There exists some evidence showing inhibition of PRC2 in HD through the upregulation of histone methylation with the participation of SUZ12 [48, 49]. Interestingly, another study also found that full-length huntingtin can stimulate the histone methylation of PRC2 [50]. Moreover, SUZ12 histone ubiquitylation and SIRT1-mediated deacetylation promote ubiquitin-dependent degradation [47, 51,52,53]. According to our functional enrichment results, SIRT1 and SUZ12 are involved in the pathogenesis of HD through several pathways, including transcriptional regulation, histone-related metabolism, and proteasome-mediated ubiquitin-dependent protein catabolic processes.
MiR-22-3p had the highest ranking among the predicted results for the SIRT1 gene, and has previously been reported to be directly related to HD in a mouse model [24]. Various studies have confirmed that the miR-22-3p/SIRT1 pathway plays an important role in the development of HD [42, 54]. As one of the predicted miRNAs of SUZ12, miR-19b has been reported previously. Although we failed to find an association between miR-19b and SUZ12, we still think that a correlation between miR-19b and SUZ12 may exist in HD [26]. More intriguingly, another 26 predicted miRNAs of SUZ12 and SIRT1 may also be related to the pathogenesis of HD, but further experiments are required.
Conclusion
In our studies, PSMC6 and related ubiquitin-mediated proteolysis may participate in the pre-symptomatic phase of HD, while CCNH and related transcriptional dysregulation and FIS1 and related mitochondrial disruption may be involved in late-stage HD. SIRT1 and SUZ12 have been confirmed to play crucial roles from the pre-symptomatic to the symptomatic stage, and their associated transcriptional dysregulation, histone metabolism, and proteasome-mediated ubiquitin-dependent protein catabolic processes may be important. However, few studies have focused on the role of SUZ12 in HD to date, and further experiments are still required.
Availability of data and materials
GSE1751 and GSE24250 datasets were downloaded from GEO (https://www.ncbi.nlm.nih.gov/geo/)) [16, 55] and expression profiling arrays were generated using GPL96 (Affymetrix Human Genome U133 Plus 2.0 Array). R packages of “limma” (https://cran.r-project.org/), provided by a bioconductor project ( https://www.bioconductor.org/) [17], were applied to assess GSE1751 and GSE24250 RAW datasets. We applied online prediction tools utilizing Targetscan [21] (https://www.targetscan.org/vert_71/) and miRDB [22] (https://www.mirdb.org/), to predict potential microRNA targeting.
Abbreviations
- HD:
-
Huntington’s disease
- pre-HD:
-
Pre-symptomatic HD
- mHtt:
-
Mutant huntingtin
- DEGs:
-
Differentially expressed genes
- GEO:
-
Gene expression omnibus
- PPI:
-
Protein–protein interaction
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- miRNA:
-
MicroRNA
- mRNA:
-
Messenger RNA
- RMA:
-
Robust multi-array average
- FDR:
-
False discovery rate
- FC:
-
Fold changes
- STRING:
-
Search Tool for the Retrieval of Interacting Gene
- MCODE:
-
Molecular Complex Detection
- DAVID:
-
Database for Annotation, Visualization, and Integrated Discovery
- BP:
-
Biological processes
- MF:
-
Molecular function
- SIRT1:
-
Sirtuin 1
- SUZ12:
-
Suppressor of zeste 12
- PSMC6:
-
Proteasome 26S subunit, ATPase 6
- FIS1:
-
Fission mitochondrial 1
- CCNH:
-
Cyclin H
- RPT4:
-
Proteasome regulatory subunit 4
- DRP1:
-
Dynamin-related protein 1
- RESV:
-
Resveratrol
- PRC2:
-
Polycomb repressive complex 2
References
MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, Barnes G, Taylor SA, James M, Groot N, MacFarlane H. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72(6):971–83.
Fusilli C, Migliore S, Mazza T, Consoli F, De Luca A, Barbagallo G, Ciammola A, Gatto EM, Cesarini M, Etcheverry JL, et al. Biological and clinical manifestations of juvenile Huntington's disease: a retrospective analysis. Lancet Neurol. 2018;17(11):986–93.
Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, Scahill RI, Leavitt BR, Stout JC, Paulsen JS, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. 2014;10(4):204–16.
Aylward EH, Sparks BF, Field KM, Yallapragada V, Shpritz BD, Rosenblatt A, Brandt J, Gourley LM, Liang K, Zhou H, et al. Onset and rate of striatal atrophy in preclinical Huntington disease. Neurology. 2004;63(1):66–72.
Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP Jr. Neuropathological classification of Huntington’s disease. J Neuropathol Exp Neurol. 1985;44(6):559–77.
Freije WA, Castro-Vargas FE, Fang Z, Horvath S, Cloughesy T, Liau LM, Mischel PS, Nelson SF. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 2004;64(18):6503–10.
De Preter K, Vermeulen J, Brors B, Delattre O, Eggert A, Fischer M, Janoueix-Lerosey I, Lavarino C, Maris JM, Mora J, et al. Accurate outcome prediction in neuroblastoma across independent data sets using a multigene signature. Clin Cancer Res. 2010;16(5):1532–41.
Kim YW, Liu TJ, Koul D, Tiao N, Feroze AH, Wang J, Powis G, Yung WK. Identification of novel synergistic targets for rational drug combinations with PI3 kinase inhibitors using siRNA synthetic lethality screening against GBM. Neuro-oncology. 2011;13(4):367–75.
Liang JW, Fang ZY, Huang Y, Liuyang ZY, Zhang XL, Wang JL, Wei H, Wang JZ, Wang XC, Zeng J, et al. Application of weighted gene co-expression network analysis to explore the key genes in Alzheimer's disease. J Alzheimer's Dis. 2018;65(4):1353–64.
Zou R, Zhang D, Lv L, Shi W, Song Z, Yi B, Lai B, Chen Q, Yang S, Hua P. Bioinformatic gene analysis for potential biomarkers and therapeutic targets of atrial fibrillation-related stroke. J Translat Med. 2019;17(1):45.
Wei CY, Zhu MX, Lu NH, Peng R, Yang X, Zhang PF, Wang L, Gu JY. Bioinformatics-based analysis reveals elevated MFSD12 as a key promoter of cell proliferation and a potential therapeutic target in melanoma. Oncogene. 2019;38(11):1876–91.
Lee RC, Feinbaum RL, Ambros V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell. 1993;75(5):843–54.
Treiber T, Treiber N, Meister G. Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat Rev Mol Cell Biol. 2019;20(1):5–20.
Reed ER, Latourelle JC, Bockholt JH, Bregu J, Smock J, Paulsen JS, Myers RH. MicroRNAs in CSF as prodromal biomarkers for Huntington disease in the PREDICT-HD study. Neurology. 2018;90(4):e264–e272272.
Bagnoli M, Canevari S, Califano D, Losito S, Maio MD, Raspagliesi F, Carcangiu ML, Toffoli G, Cecchin E, Sorio R, et al. Development and validation of a microRNA-based signature (MiROvaR) to predict early relapse or progression of epithelial ovarian cancer: a cohort study. Lancet Oncol. 2016;17(8):1137–46.
Borovecki F, Lovrecic L, Zhou J, Jeong H, Then F, Rosas HD, Hersch SM, Hogarth P, Bouzou B, Jensen RV, et al. Genome-wide expression profiling of human blood reveals biomarkers for Huntington’s disease. Proc Natl Acad Sci USA. 2005;102(31):11023–8.
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, et al. NCBI GEO: archive for functional genomics data sets-update. Nucleic Acids Res. 2013;41(Database issue):D991–D99595.
Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, et al. STRING v9.1: protein–protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41(1):D808–D815815.
Spinelli L, Gambette P, Chapple CE, Robisson B, Baudot A, Garreta H, Tichit L, Guenoche A, Brun C. Clust&See: a Cytoscape plugin for the identification, visualization and manipulation of network clusters. Bio Syst. 2013;113(2):91–5.
da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57.
Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20.
Wang X. Computational prediction of microRNA targets. Methods Mol Biol. 2010;667:283–95.
Camp RL, Dolled-Filhart M, Rimm DL. X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clinical Cancer Res. 2004;10(21):7252–9.
Lee ST, Chu K, Im WS, Yoon HJ, Im JY, Park JE, Park KH, Jung KH, Lee SK, Kim M, et al. Altered microRNA regulation in Huntington’s disease models. Exp Neurol. 2011;227(1):172–9.
Sinha M, Ghose J, Das E, Bhattarcharyya NP. Altered microRNAs in STHdh(Q111)/Hdh(Q111) cells: miR-146a targets TBP. Biochem Biophys Res Commun. 2010;396(3):742–7.
Marti E, Pantano L, Banez-Coronel M, Llorens F, Minones-Moyano E, Porta S, Sumoy L, Ferrer I, Estivill X. A myriad of miRNA variants in control and Huntington's disease brain regions detected by massively parallel sequencing. Nucleic Acids Res. 2010;38(20):7219–35.
Kerschbamer E, Biagioli M. Huntington's disease as neurodevelopmental disorder: altered chromatin regulation, coding, and non-coding RNA transcription. Front Neurosci. 2015;9:509.
Arteaga-Bracho EE, Gulinello M, Winchester ML, Pichamoorthy N, Petronglo JR, Zambrano AD, Inocencio J, De Jesus CD, Louie JO, Gokhan S, et al. Postnatal and adult consequences of loss of huntingtin during development: Implications for Huntington's disease. Neurobiol Dis. 2016;96:144–55.
Wiatr K, Szlachcic WJ, Trzeciak M, Figlerowicz M, Figiel M. Huntington disease as a neurodevelopmental disorder and early signs of the disease in stem cells. Mol Neurobiol. 2018;55(4):3351–71.
Liu M, Xu Z, Du Z, Wu B, Jin T, Xu K, Xu L, Li E, Xu H. The Identification of key genes and pathways in glioma by bioinformatics analysis. J Immunol Res. 2017;2017:1278081.
Mukherjee S, Klaus C, Pricop-Jeckstadt M, Miller JA, Struebing FL. A microglial signature directing human aging and neurodegeneration-related gene networks. Front Neurosci. 2019;13:2.
Bauer VW, Swaffield JC, Johnston SA, Andrews MT. CADp44: a novel regulatory subunit of the 26S proteasome and the mammalian homolog of yeast Sug2p. Gene. 1996;181(1–2):63–9.
Tanahashi N, Suzuki M, Fujiwara T, Takahashi E, Shimbara N, Chung CH, Tanaka K. Chromosomal localization and immunological analysis of a family of human 26S proteasomal ATPases. Biochem Biophys Res Commun. 1998;243(1):229–32.
Labbadia J, Morimoto RI. Huntington's disease: underlying molecular mechanisms and emerging concepts. Trends Biochem Sci. 2013;38(8):378–85.
Rousseau E, Kojima R, Hoffner G, Djian P, Bertolotti A. Misfolding of proteins with a polyglutamine expansion is facilitated by proteasomal chaperones. J Biol Chem. 2009;284(3):1917–29.
Moslehi R, Mills JL, Signore C, Kumar A, Ambroggio X, Dzutsev A. Integrative transcriptome analysis reveals dysregulation of canonical cancer molecular pathways in placenta leading to preeclampsia. Sci Rep. 2013;3:2407.
van Roon-Mom WM, Reid SJ, Jones AL, MacDonald ME, Faull RL, Snell RG. Insoluble TATA-binding protein accumulation in Huntington's disease cortex. Brain Res Mol Brain Res. 2002;109(1–2):1–10.
Chan DC. Dissecting mitochondrial fusion. Dev Cell. 2006;11(5):592–4.
Zhao Y, Sun X, Qi X. Inhibition of Drp1 hyperactivation reduces neuropathology and behavioral deficits in zQ175 knock-in mouse model of Huntington's disease. Biochem Biophys Res Commun. 2018;507(1–4):319–23.
Jiang M, Zheng J, Peng Q, Hou Z, Zhang J, Mori S, Ellis JL, Vlasuk GP, Fries H, Suri V, et al. Sirtuin 1 activator SRT2104 protects Huntington’s disease mice. Ann Clin Transl Neurol. 2014;1(12):1047–52.
Smith MR, Syed A, Lukacsovich T, Purcell J, Barbaro BA, Worthge SA, Wei SR, Pollio G, Magnoni L, Scali C, et al. A potent and selective Sirtuin 1 inhibitor alleviates pathology in multiple animal and cell models of Huntington’s disease. Hum Mol Genet. 2014;23(11):2995–3007.
Tang Q, Len Q, Liu Z, Wang W. Overexpression of miR-22 attenuates oxidative stress injury in diabetic cardiomyopathy via Sirt 1. Cardiovasc Therap. 2018;36(2):e12318.
Naia L, Rosenstock TR, Oliveira AM, Oliveira-Sousa SI, Caldeira GL, Carmo C, Laco MN, Hayden MR, Oliveira CR, Rego AC. Comparative mitochondrial-based protective effects of resveratrol and nicotinamide in Huntington's disease models. Mol Neurobiol. 2017;54(7):5385–99.
Ho DJ, Calingasan NY, Wille E, Dumont M, Beal MF. Resveratrol protects against peripheral deficits in a mouse model of Huntington's disease. Exp Neurol. 2010;225(1):74–84.
Rai AN, Vargas ML, Wang L, Andersen EF, Miller EL, Simon JA. Elements of the polycomb repressor SU(Z)12 needed for histone H3–K27 methylation, the interface with E(Z), and in vivo function. Mol Cell Biol. 2013;33(24):4844–56.
Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469(7330):343–9.
Cooper S, Grijzenhout A, Underwood E, Ancelin K, Zhang T, Nesterova TB, Anil-Kirmizitas B, Bassett A, Kooistra SM, Agger K, et al. Jarid2 binds mono-ubiquitylated H2A lysine 119 to mediate crosstalk between Polycomb complexes PRC1 and PRC2. Nat Commun. 2016;7:13661.
Dong X, Tsuji J, Labadorf A, Roussos P, Chen JF, Myers RH, Akbarian S, Weng Z. The role of H3K4me3 in transcriptional regulation is altered in Huntington's disease. PLoS ONE. 2015;10(12):e0144398.
Schmitges FW, Prusty AB, Faty M, Stutzer A, Lingaraju GM, Aiwazian J, Sack R, Hess D, Li L, Zhou S, et al. Histone methylation by PRC2 is inhibited by active chromatin marks. Mol Cell. 2011;42(3):330–41.
Seong IS, Woda JM, Song JJ, Lloret A, Abeyrathne PD, Woo CJ, Gregory G, Lee JM, Wheeler VC, Walz T, et al. Huntingtin facilitates polycomb repressive complex 2. Hum Mol Genet. 2010;19(4):573–83.
Han L, Wang P, Zhao G, Wang H, Wang M, Chen J, Tong T. Upregulation of SIRT1 by 17 beta-estradiol depends on ubiquitin-proteasome degradation of PPAR-gamma mediated by NEDD4-1. Protein Cell. 2013;4(4):310–21.
Ling S, Li J, Shan Q, Dai H, Lu D, Wen X, Song P, Xie H, Zhou L, Liu J, et al. USP22 mediates the multidrug resistance of hepatocellular carcinoma via the SIRT1/AKT/MRP1 signaling pathway. Mol Oncol. 2017;11(6):682–95.
Liu H, Liu N, Zhao Y, Zhu X, Wang C, Liu Q, Gao C, Zhao X, Li J. Oncogenic USP22 supports gastric cancer growth and metastasis by activating c-Myc/NAMPT/SIRT1-dependent FOXO1 and YAP signaling. Aging. 2019;11:9643.
Zou Q, Tang Q, Pan Y, Wang X, Dong X, Liang Z, Huang D. MicroRNA-22 inhibits cell growth and metastasis in breast cancer via targeting of SIRT1. Exp Ther Med. 2017;14(2):1009–166.
Hu Y, Chopra V, Chopra R, Locascio JJ, Liao Z, Ding H, Zheng B, Matson WR, Ferrante RJ, Rosas HD, et al. Transcriptional modulator H2A histone family, member Y (H2AFY) marks Huntington disease activity in man and mouse. Proc Natl Acad Sci USA. 2011;108(41):17141–6.
Acknowledgements
We thank Dr. Dimitri Krainc, Harvard University, Boston, MA, USA for the excellent work of the original dataset published in the journal Proc Natl Acad Sci USA (https://doi.org/10.1073/pnas.0504921102)
Funding
This work was supported by the National Natural Science Foundation of China (No. 81371271), the “Liaoning BaiQianWan Talents Program” and was also sponsored by 345 Talent Project of Shengjing Hospital of China Medical University.
Author information
Authors and Affiliations
Contributions
CX and SC: takes responsibility for all aspects of the reliability and freedom from bias of the data presented and their discussed interpretation, drafting the article. CX and SC contributed equally. BL: takes responsibility for statistical analyses, and interpretation of data. SC: takes responsibility for the design of the study, full text evaluation and guidance. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Differentially expressed genes involved in HD and preHD samples.
Additional file 2: Table S2.
All of the hub genes and their scores in two groups.
Additional file 3: Table S3.
Gene Ontology (GO) terms enrichment analysis of hub genes in Group 1 and Group 2.
Additional file 4: Table S4.
KEGG pathways of the hub genes in two groups.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xiang, C., Cong, S., Liang, B. et al. Bioinformatic gene analysis for potential therapeutic targets of Huntington’s disease in pre-symptomatic and symptomatic stage. J Transl Med 18, 388 (2020). https://doi.org/10.1186/s12967-020-02549-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-020-02549-9