
Abstract
Background
Elevated lipoprotein(a) [Lp(a)] and fibrinogen (Fib) are both associated with coronary artery disease (CAD). The atherogenicity of Lp(a) can be partly due to the potentially antifibrinolytic categories. We hypothesize that patients with higher Lp(a) and Fib may have worse outcomes.
Methods
In this prospective study, we consecutively enrolled 8,417 Chinese patients with stable CAD from March 2011 to March 2017. All subjects were divided into 9 groups according to Lp(a) (Lp(a)-Low, Lp(a)-Medium, Lp(a)-High) and Fib levels (Fib-Low, Fib-Medium, Fib-High) and followed up for CVEs, including nonfatal acute myocardial infarction, stroke, and cardiovascular mortality. Kaplan–Meier, Cox regression and C-statistic analyses were performed.
Results
During a median of 37.1 months’ follow-up, 395 (4.7%) CVEs occurred. The occurrence of CVEs increased by Lp(a) (3.5 vs. 5.3 vs. 5.6%, p = 0.001) and Fib (4.0 vs. 4.4 vs. 6.1%, p < 0.001) categories. When further classified into 9 groups by Lp(a) and Fib levels, the CVEs were highest in the 9th (Lp(a)-High and Fib-High) compared with the 1st (Lp(a)-Low and Fib-Low) group (7.2 vs. 3.3%, p < 0.001). The highest risk of subsequent CVEs was found in the 9th group (HRadjusted 2.656, 95% CI 1.628–4.333, p < 0.001), which was more significant than Lp(a)-High (HRadjusted 1.786, 95% CI 1.315–2.426, p < 0.001) or Fib-High (HRadjusted 1.558, 95% CI 1.162–2.089, p = 0.003) group. Moreover, adding the combined Lp(a) and Fib increased the C-statistic by 0.013.
Conclusion
Combining Fib and Lp(a) enhance the prognostic value for incident CVEs beyond Lp(a) or Fib alone.
Similar content being viewed by others


Background
Despite significant advances in the diagnosis and therapy of cardiovascular disease (CVD), patients with established coronary artery disease (CAD) are generally at higher risk of developing recurrent cardiovascular events (CVEs) than the primary prevention individuals [1]. Clinical trials revealed that in the short time window only 20–30% of patients benefit even if traditional risk factors were well managed [2, 3]. As a result, identifying additional modifiable risk factors is necessary to further improve CVEs prediction in the management of patients with established CAD.
Evidence have established high lipoprotein(a) (Lp[a]) levels are associated with high risk of CVD, observationally and causally from human genetics [4,5,6]. Multiple studies have indicated that high Lp(a) cause CVD in a primary prevention setting, moreover, Lp(a)-lowering by 50 mg/dL may reduce CVD by 20% in a secondary prevention setting [7]. AS well known, Lp(a) is composed of an LDL-like particle in which apoB is covalently bound by a single disulfide bond to apolipoprotein(a) (apo[a]). Therefore, the pathogenic role of Lp(a) was supposed to be involved in atherosclerosis and thrombosis formation [8]. In fact, the recent study implied that the mortality effect of high lipoprotein(a) is above that explained by its cholesterol content but the number of KIV-2 repeats in the apo(a) [9]. Originally, apo(a) has evolved from the plasminogen gene through duplication and remodeling. Unlike apolipoprotein B, apo(a) does not contain lipid domains or transport lipid, but instead, it potentiates atherothrombosis through additional pathways including proinflammatory, and potentially antifibrinolytic effects by inhibiting plasminogen activation [10]. As one of the important components of fibrinolytic system, plasma fibrinogen (Fib) has been proved to be a pivotal CVD risk factor [11,12,13]. However, little is known about the inter-relationship of Lp(a) and Fib in the CVEs risk prediction in the secondary prevention setting.
As a consequence, we hypothesize that there is a risk interaction between Lp(a) and Fib, and patients with high Lp(a) and Fib may have worse outcomes. We thereby sought to investigate the association of Lp(a) and Fib in predicting CVEs in patients with stable CAD (SCAD) in the current study.
Methods
Study population
Our study complied with the Declaration of Helsinki and was approved by the hospital’s ethics review board (Fu Wai Hospital, National Center for Cardiovascular Diseases). Informed written consents were collected from all patients obtained in this study.
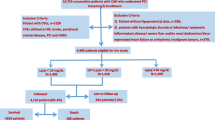
From March 2011 to March 2017, a total of 10,042 Chinese patients with clinical symptoms such as angina pectoris, or chest distress were recruited in our study. The inclusion criteria were patients with stable and angiography-proven CAD (coronary stenosis ≥ 50% of at least one coronary artery). The exclusion criteria were as follows: (1) acute coronary syndrome (ACS); (2) previous myocardial infarction (MI), previous percutaneous coronary artery intervention or bypass grafting; (3) heart failure; (4) other disease status such as severe liver and/or renal insufficiency, thyroid dysfunction, systematic inflammatory disease, and malignant disease. Therefore, 8417 patients were finally enrolled in the current analysis.
Patients were followed up at 6 months intervals by means of direct interview or telephone. The follow-up was performed by trained nurses or physicians who were blinded to the clinical data. The primary end points were cardiovascular mortality, nonfatal MI, and stroke. Nonfatal MI including ST-segment–elevation MI and non–ST-segment–elevation MI was diagnosed as positive cardiac troponins along with typical chest pain or typical electrocardiogram serial changes. Stroke was confirmed by specialist physicians according to the presence of typical symptoms and imaging.
Diabetes mellitus (DM) was diagnosed by fasting plasma glucose ≥ 7.0 mmol/L, the 2 h plasma glucose of the oral glucose tolerance test ≥ 11.1 mmol/L, or current use of hypoglycemic drugs or insulin. Hypertension was defined as self-reported, currently taking antihypertensive drugs, or recorded systolic blood pressure ≥ 140 mmHg or diastolic blood pressure ≥ 90 mmHg three or more consecutive times. Information regarding other disease, family history, and prior therapy of every patient was collected from self-reported medical history.
Laboratory analysis
Blood samples were obtained from each patient from the cubital vein after at least 12 h of fasting. Concentrations of
total cholesterol (TC), triglyceride (TG), low-density lipoprotein-cholesterol (LDL-C), and high-density lipoprotein-cholesterol (HDL-C) were measured using an automatic biochemistry analyzer (7150; Hitachi, Tokyo, Japan) in an enzymatic assay. Lp(a) was determined by immunoturbidimetry method [LASAY Lp(a) auto; SHIMA Laboratories Co., Ltd] with a normal value of < 30 mg/dL. An Lp(a) protein validated standard was used to calibrate the examination, and the coefficient of variation value of repetitive measurements was < 10%. Concentrations of Fib were measured using a Stago auto analyzer by the Clauss method with an STA Fibrinogen kit (Diagnostica Stago, Taverny, France).
Statistical analysis
The values were expressed as the mean ± SD or median (25–75th percentile) for the continuous variables and the number (percentage) for the categorical variables. The Kolmogorov–Smirnov
test was used to test the distribution pattern. The differences in clinical characteristics between groups were analyzed using Student t test, Mann–Whitney U test, χ2 tests, or Fisher exact test when appropriate. The event-free survival rates among groups were estimated by the Kaplan–Meier method and compared by the log-rank test. Univariate and multivariate Cox regression analyses were performed to calculate the hazard ratios (HRs). A P value of less than 0.05 was considered statistically significant. The statistical analyses were performed with SPSS, version 22.0, software (SPSS, Chicago, IL) and R language, version 3.5.2 (Feather Spray).
Results
Baseline characteristics
The baseline characteristics of the study participants were shown in Table 1. Over a median of 37.1 months (25–75th percentile 22.5–55.4 months) follow-up period, 395 CVEs occurred (160 died, 78 suffered non-fatal MI, and 157 had strokes). Patients suffered CVEs tended to be older (p < 0.001), with higher prevalence of hypertension (p = 0.005), DM (p < 0.001), and lower BMI (p = 0.016). There was no significant difference regarding the baseline lipid profiles (TG, TC, LDL-C, HDL-C, apoA1, apoB, all p > 0.05) except Lp(a) levels (p = 0.001). Significantly, the concentration of Fib and D-dimer were higher in patients with CVEs (all p < 0.05). Meanwhile, the rate of statin usage was lower (p = 0.005) at admission while balanced (p > 0.05) at discharge in CVEs compared with patients without events. However, The HR of baseline characteristics with future CVEs were presented in Additional file 1. Table S1.
Association of plasma Lp(a) Levels and CVEs
In the current analysis, the subjects were assigned to 3 groups according to Lp(a) levels (Lp(a)-L: < 10 mg/dL, Lp(a)-M:10–29.9 mg/dL, Lp(a)-H ≥ 30 mg/dL). As shown in Fig. 1, the prevalence of CVEs in the Lp(a)-L, Lp(a)-M, and Lp(a)-H groups was 3.5%, 5.3%, and 5.6%, respectively (p < 0.001). Kaplan–Meier analysis (Fig. 2a) showed that Lp(a)-H subjects had the lowest event-free survival rate among the three groups (p = 0.001). As presented in Table 2, univariate Cox regression models showed that Lp(a)-M, and Lp(a)-H group had 1.468-fold and 1.580-fold higher risk of CVEs compared with Lp(a)-L group [Lp(a)-M: HR (95% CI) 1.468 (1.142–1.886), p = 0.003; Lp(a)-H: HR (95% CI) 1.580 (1.227–2.033), p < 0.001]. Additional adjustment for other variables in the multivariate Cox regression models did not change the significance of the association [Lp(a)-M: HR (95% CI) 1.531 (1.128–2.079), p = 0.006; Lp(a)-H: HR (95% CI) 1.786 (1.315–2.426), p < 0.001; Table 3].

The KM rates of CVEs in Fib, Lp(a), and combined groups

The event-free survival rate in Fib, Lp(a), and combined groups
Association of plasma Fib Levels and CVEs
Similarly, patients were divided into 3 groups according to Fib levels (Fib-L: < 2.84 g/L, Fib-M:2.85–3.42 g/L, Fib-H: ≥ 3.43 g/L). The prevalence of CVEs in the Fib-L, Fib-M, and Fib-H groups was 4.0%, 4.4%, and 6.1%, respectively (p < 0.001). The event-free survival rate was lowest in the Fib-H group (p < 0.001, Fig. 2b). Compared to Fib-L group, the Fib-H group had 1.631-fold higher risk of CVEs [HR (95% CI) 1.631 (1.282–2.074), p < 0.001] even after adjusting for potential confounders [HR (95% CI) 1.558 (1.162–2.089), p = 0.003].
Inter-relationship of Lp(a), Fib Levels and CVEs
To evaluate an interaction between plasma Lp(a) and Fib levels on the risk of CVEs, the subjects were assigned to 9 groups according to Lp(a) and Fib levels (G1(Lp(a)-L + Fib-L, G2(Lp(a)-L + Fib-M, G3(Lp(a)-L + Fib-H, G4(Lp(a)-M + Fib-L, G5(Lp(a)-M + Fib-M, G6(Lp(a)-M + Fib-H, G7(Lp(a)-H + Fib-L, G8(Lp(a)-H + Fib-M, G9(Lp(a)-H + Fib-H).
The occurrence of CVEs in the 9 groups was 3.3%, 3.5%, 4.0%, 4.1%, 4.9%, 7.0%, 4.8%, 5.4%, and 7.2%, respectively (p < 0.001, Fig. 1). As shown in Fig. 2c, the event-free survival rate was lowest in the 6th and 9th group (p < 0.001). Hazard ratios were calculated for each group using the G1 (group 1, Lp(a)-L and Fib-L) as a reference (Table 2). After adjusting for potential confoundings, the 6th group (Lp(a)-M and Fib-H) and 9th group (Lp(a)-H and Fib-H) had 2.307-fold and 2.656-fold higher risk of CVEs [HR (95% CI) 2.307 (1.409–3.777), p = 0.001; 2.656 (1.628–4.333), p < 0.001, respectively, Table 3).
In the original model, the C-statistic values were 0.633 (95% CI 0.603–0.664) with traditional risk factors, (Table 4). Addition of Lp(a) categories to the original model induced slightly improvement in C-statistic [ΔC-statistic 0.010 (− 0.001–0.023), p = 0.088] but did not reach statistical significance. When added Fib categories to the original model did not improve the C-statistic [ΔC-statistic 0.003 (− 0.005–0.012), p = 0.443]. Nonetheless, the combined Lp(a) and Fib categories resulted in a slightly improvement in C-statistic [ΔC-statistic 0.013 (0.002–0.027), p = 0.033].
Discussion
In this prospective, large-cohort study, we investigated the association of plasma Lp(a) and Fib on the prediction of CVEs in angiography-proven stable CAD patients. Our data clearly found that both Lp(a) and Fib were independent predictors of CVEs in patients with stable CAD. More interestingly, the study firstly indicated that the combined Lp(a) and Fib categories enhanced the predicting values by incrementally increasing risk of CVEs in this population. The adjusted HR for CVEs was 2.656-fold and 2.307-fold higher among stable CAD patients in the Fib-H with Lp(a)-H or Lp(a)-M group, respectively. Finally, adding Lp(a) and Fib to the Cox model increased the C-statistic by 0.013 beyond that achieved with any single biomarker. These findings suggested that the combination of Lp(a), a complex marker of cholesterol and anti-fibrinolysis, and Fib, a marker of coagulation state, could enhance the predictive value, which would help the future risk stratification of stable CAD patients.
It is uncertain whether plasma Lp(a) levels are associated with CVEs in patients with stable CAD although several studies have suggested an association of elevated Lp(a) concentrations with the risk of CVD including the primary prevention population, familial hypercholesterolemia, statin-treated patients, and so forth. Concerning the secondary prevention setting, especially in patients with stable CAD, the results were controversial due to unknown causes. The Copenhagen City Heart Study showed that for patients with Lp(a) concentrations between 30 and 76 mg/dL, 77 and 117 mg/dL, and above 117 mg/dL, the risk of MI increased by a 1.6-fold, 1.9-fold, and 2.6-fold compared with those below 5 mg/dL in the primary prevention setting [4]. Data in patients with FH showed that the high cardiovascular risk in these patients is further increased by their unusual Lp(a) concentrations, which tend to be 2–3 fold higher than in the general population [14]. Of note, in the secondary prevention setting for patients with established CAD, inconsistent data were observed [15, 16]. Among 569 patients having undergone PCI and LDL-C levels were well-controlled (< 100 mg/dL), those with higher Lp(a) levels had significantly higher risk of MACEs compared to patients with lower Lp(a) levels, while elevated Lp(a) values were an independent predictor of mortality and recurrence of ACS [17]. Recently, our data proved that elevated Lp(a) levels were significantly associated with the risk of MACEs in patients with CAD combined with DM or pre-DM [18]. However, for patients with recent ACS who are treated with statins, Lp(a) concentration was not associated with MACEs [19]. Based on this situation, we consecutively enrolled 8,417 patients who had angiography-proven stable CAD and followed up for a median of 37.1 months. The data clearly showed that high Lp(a) (≥ 30 mg/dL) was resulted in 1.786-fold CVEs risk compared with low Lp(a) levels (< 10 mg/dL).
Next, previous including our studies supported the notion that Fib, a coagulation factor, is also a marker for risk of CVD [20, 21]. In this study, we re-examined the role of Fib in prediction of CVEs, and finally proved that high Fib was related to 1.631-fold higher risk of CVEs compared with the low Fib level. Till now, Lp(a) is thought to mediate clinical events by 3 main mechanisms, pro-atherogenic effects via its LDL-C moiety [3, 22], pro-inflammatory effects via its content of oxidized phospholipids [23] and anti-fibrinolytic effects via its apolipoprotein(a) component [24]. Lp(a) has high homology (75–99%) to plasminogen but lacks protease activity, and therefore has been hypothesized to inhibit fibrinolysis and mediate prothrombotic potential. Therefore, we hypothesize that there might be an enhanced impact of Lp(a) and Fib due to their pathophysiological action and previous evidence. A previous study indicated that high Fib associated with high Lp(a) levels significantly increased the risk of CAD [25], the study was designed for the primary prevention and was restricted by the male population. Hence, in this secondary prevention population, we divided our patients into 9 subgroups and found that the Fib-H with Lp(a)-H or Lp(a)-M group had 2.656-fold and 2.307-fold higher risk of CVEs, and the combination of Lp(a) and Fib categories improved the predictive value for CVEs beyond any biomarker alone.
Nevertheless, our study had several limitations. First of all, this is a study among Chinese population with stable CAD in the statin era, and whether the data applied to other populations need to be testified. Secondly, the Lp(a) and Fib concentrations were only measured at baseline, and the alterations of these biomarkers may also be clinically significant during the follow-up period. Finally, as this was an observational study, further investigations are needed to clarify the underlying mechanism of the associations.
Conclusions
In conclusion, according to the functional similarity of Lp(a) and Fib in pro-atherogenic and anti-fibrinolytic effects, we examined the potential role of combining Lp(a) with Fib for predicting CVE in 8,417 patients with stable CAD and followed up an average of 37.1 months. Data firstly suggested that Lp(a) plus Fib could significantly enhanced predicting value for cardiovascular outcome in patients with stable CAD compared to that of Lp(a) or Fib alone.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- CVD:
-
Cardiovascular disease
- CAD:
-
Coronary artery disease
- CVEs:
-
Cardiovascular events
- Lp[a]:
-
Lipoprotein(a)
- Apo(a):
-
Apolipoprotein(a)
- Fib:
-
Fibrinogen
- ACS:
-
Acute coronary syndrome
- MI:
-
Myocardial infarction
- TC:
-
Total cholesterol
- TG:
-
Triglyceride
- LDL-C:
-
Low-density lipoprotein-cholesterol
- HDL-C:
-
High-density lipoprotein-cholesterol
References
Omland T, White HD. State of the art: blood biomarkers for risk stratification in patients with stable ischemic heart disease. Clin Chem. 2017;63:165–76.
Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Das SR, Delling FN, Djousse L, Elkind MSV, Ferguson JF, Fornage M, Jordan LC, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, O'Flaherty M, Pandey A, Perak AM, Rosamond WD, Roth GA, Sampson UKA, Satou GM, Schroeder EB, Shah SH, Spartano NL, Stokes A, Tirschwell DL, Tsao CW, Turakhia MP, VanWagner LB, Wilkins JT, Wong SS, Virani SS, American Heart Association Council on Epidemiology, and Prevention Statistics Committee, and Stroke Statistics Subcommittee. Heart disease and stroke statistics-2019 update: a report from the American Heart Association. Circulation. 2019;139:e56–e528.
Tsimikas S. A test in context: lipoprotein(a): diagnosis, prognosis, controversies, and emerging therapies. J Am Coll Cardiol. 2017;69(6):692–711.
Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301(2331):9.
Clarke R, Peden JF, Hopewell JC, Kyriakou T, Goel A, Heath SC, Parish S, Barlera S, Franzosi MG, Rust S, Bennett D, Silveira A, Malarstig A, Green FR, Lathrop M, Gigante B, Leander K, de Faire U, Seedorf U, Hamsten A, Collins R, Watkins H, Farrall M. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361:2518–28.
Nordestgaard BG, Langsted A. Lipoprotein(a) as a cause of cardiovascular disease: insights from epidemiology, genetics, and biology. J Lipid Res. 2016;57:1953–75.
Madsen CM, Kamstrup PR, Langsted A, Varbo A, Nordestgaard BG. Lipoprotein(a)-Lowering by 50 mg/dL (105 nmol/L) May Be Needed to Reduce Cardiovascular Disease 20% in Secondary Prevention: A Population-Based Study. Arterioscler Thromb Vasc Biol. 2020;40:255–66.
Nordestgaard BG, Chapman MJ, Ray K, Boren J, Andreotti F, Watts GF, Ginsberg H, Amarenco P, Catapano A, Descamps OS, Fisher E, Kovanen PT, Kuivenhoven JA, Lesnik P, Masana L, Reiner Z, Taskinen MR, Tokgozoglu L, Tybjaerg-Hansen A. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31:2844–53.
Langsted A, Kamstrup PR, Nordestgaard BG. High lipoprotein(a) and high risk of mortality. Eur Heart J. 2019;40(33):2760–70.
Schmidt K, Noureen A, Kronenberg F, Utermann G. Structure, function, and genetics of lipoprotein(a). J Lipid Res. 2016;57:1339–59.
Fibrinogen Studies Collaboration, Danesh J, Lewington S, Thompson SG, Lowe GD, Collins R, Kostis JB, Wilson AC, Folsom AR, Wu K, Benderly M, Goldbourt U, Willeit J, Kiechl S, Yarnell JW, Sweetnam PM, Elwood PC, Cushman M, Psaty BM, Tracy RP, Tybjaerg-Hansen A, Haverkate F, de Maat MP, Fowkes FG, Lee AJ, Smith FB, Salomaa V, Harald K, Rasi R, Vahtera E, Jousilahti P, Pekkanen J, D'Agostino R, Kannel WB, Wilson PW, Tofler G, Arocha-Piñango CL, Rodriguez-Larralde A, Nagy E, Mijares M, Espinosa R, Rodriquez-Roa E, Ryder E, Diez-Ewald MP, Campos G, Fernandez V, Torres E, Marchioli R, Valagussa F, Rosengren A, Wilhelmsen L, Lappas G, Eriksson H, Cremer P, Nagel D, Curb JD, Rodriguez B, Yano K, Salonen JT, Nyyssönen K, Tuomainen TP, Hedblad B, Lind P, Loewel H, Koenig W, Meade TW, Cooper JA, De Stavola B, Knottenbelt C, Miller GJ, Cooper JA, Bauer KA, Rosenberg RD, Sato S, Kitamura A, Naito Y, Palosuo T, Ducimetiere P, Amouyel P, Arveiler D, Evans AE, Ferrieres J, Juhan-Vague I, Bingham A, Schulte H, Assmann G, Cantin B, Lamarche B, Després JP, Dagenais GR, Tunstall-Pedoe H, Woodward M, Ben-Shlomo Y, Davey Smith G, Palmieri V, Yeh JL, Rudnicka A, Ridker P, Rodeghiero F, Tosetto A, Shepherd J, Ford I, Robertson M, Brunner E, Shipley M, Feskens EJ, Kromhout D, Dickinson A, Ireland B, Juzwishin K, Kaptoge S, Lewington S, Memon A, Sarwar N, Walker M, Wheeler J, White I, Wood A. Plasma fibrinogen level and the risk of major cardiovascular diseases and nonvascular mortality: an individual participant meta-analysis. JAMA. 2005;294:1799–809.
Mjelva OR, Svingen GFT, Pedersen EKR, Seifert R, Kvaloy JT, Midttun O, Ueland PM, Nordrehaug JE, Nygård O, Nilsen DWT. Fibrinogen and neopterin is associated with future myocardial infarction and total mortality in patients with stable coronary artery disease. Thromb Haemost. 2018;118:778–90.
Ang L, Behnamfar O, Palakodeti S, Lin F, Pourdjabbar A, Patel MP, Reeves RR, Mahmud E. Elevated baseline serum fibrinogen: effect on 2-yearmajor adverse cardiovascular events following percutaneous coronary intervention. J Am Heart Assoc. 2017;6:e006580.
Jansen AC, van Aalst-Cohen ES, Tanck MW, Trip MD, Lansberg PJ, Liem AH, van Lennep HW, Sijbrands EJ, Kastelein JJ. The contribution of classical risk factors to cardiovascular disease in familial hypercholesterolaemia: data in 2400 patients. J Intern Med. 2004;256:482–90.
Schwartz GG, Ballantyne CM, Barter PJ, Kallend D, Leiter LA, Leitersdorf E, McMurray JJV, Nicholls SJ, Olsson AG, Shah PK, Tardif JC, Kittelson J. Association of Lipoprotein(a) With Risk of Recurrent Ischemic Events Following Acute Coronary Syndrome: Analysis of the dal-Outcomes Randomized Clinical Trial. JAMA Cardiol. 2018;3:164–8.
Zhou BY, Sun D, Wang C, Wu NQ, Guo YL, Zhu CG, Gao Y, Liu G, Dong Q, Li JJ. Plasma lipoprotein(a) concentration is associated with the coronary severity but not with events in stable coronary artery disease patients: a Chinese Cohort Study. Heart Lung Circ. 2019;28:1009–177.
Konishi H, Miyauchi K, Kasai T, Tsuboi S, Ogita M, Naito R, Sai E, Fukushima Y, Katoh Y, Okai I, Tamura H, Okazaki S, Daida H. Impact of lipoprotein(a) as residual risk on long-term outcomes in patients after percutaneous coronary intervention. Am J Cardiol. 2015;115:157–60.
Jin JL, Cao YX, Zhang HW, Sun D, Hua Q, Li YF, Guo YL, Wu NQ, Zhu CG, Gao Y, Dong QT, Liu HH, Dong Q, Li JJ. Lipoprotein(a) and cardiovascular outcomes in patients with coronary artery disease and prediabetes or diabetes. Diabetes Care. 2019;42:1312–8.
Schwartz GG, Ballantyne CM, Barter PJ, Kallend D, Leiter LA, Leitersdorf E, McMurray JJV, Nicholls SJ, Olsson AG, Shah PK, Tardif JC, Kittelson J. Association of lipoprotein(a)with risk of recurrent ischemic events following acute coronary syndrome: analysis of the dal-Outcomes Randomized Clinical Trial. JAMA Cardiol. 2018;3:164–8.
Lindahl B, FRISC Study Group. Markers of myocardial damage and inflammation in relation to long-term mortality in unstable coronary artery disease. Fragmin during instability in coronary artery disease. N Engl J Med. 2000;343:1139–47.
Zhang Y, Zhu CG, Guo YL, Xu RX, Li S, Dong Q, Li JJ. Higher fibrinogen level is independently linked with the presence and severity of new-onset coronary atherosclerosis among Han Chinese population. PLoS ONE. 2014;9:e113460.
Tsimikas S, Fazio S, Ferdinand KC, Ginsberg HN, Koschinsky ML, Marcovina SM, Moriarty PM, Rader DJ, Remaley AT, Reyes-Soffer G, Santos RD, Thanassoulis G, Witztum JL, Danthi S, Olive M, Liu L. NHLBI Working Group recommendations to reduce lipoprotein(a)-mediated risk of cardiovascular disease and aortic stenosis. J Am Coll Cardiol. 2018;71:177–92.
van der Valk FM, Bekkering S, Kroon J, Yeang C, Van den Bossche J, van Buul JD, Ravandi A, Nederveen AJ, Verberne HJ, Scipione C, Nieuwdorp M, Joosten LA, Netea MG, Koschinsky ML, Witztum JL, Tsimikas S, Riksen NP, Stroes ES. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. 2016;134:611–24.
Boffa MB, Koschinsky ML. Lipoprotein (a): truly a direct prothrombotic factor in cardiovascular disease? J Lipid Res. 2016;57:745–57.
Cantin B, Després JP, Lamarche B, Moorjani S, Lupien PJ, Bogaty P, Bergeron J, Dagenais GR. Association of fibrinogen and lipoprotein(a) as a coronary heart disease risk factor in men (The Quebec Cardiovascular Study). Am J Cardiol. 2002;89:662–6.
Acknowledgements
The authors wish to thank the participants and staff of this prospective population study.
Funding
This work was partially supported by the Capital Health Development Fund (201614035), CAMS Major Collaborative Innovation Project (2016-I2M-1-011), the Fundamental Research Funds for the Central Universities (2018-F05), and Youth Research Fund of Peking Union Medical College (2018-XHQN03).
Author information
Authors and Affiliations
Contributions
YZ completed the project, analyzed data, and wrote the manuscript. JLJ, YXC, and HWZ contributed to data collection. QH. and YFL contributed to the collections of data. YLG, NQW and Y. G. contributed to recruitment of patients, clinical diagnosis of disease, and data collection. HHL, RXX, CJC, GL, QD, and J. S. contributed to the collections of clinical data and procedure of laboratory examination. JJL designed the study, interpreted data, and contributed to critically revising the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Our study was approved by the hospital’s ethics review board (Fu Wai Hospital, National Center for Cardiovascular Diseases). All patients provided informed written consents in this study.
Consent for publication
All the authors and participants have approved the manuscript for publication.
Competing interests
The authors declare that they had no conflicts of interests to disclosure.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
Table S1. Association of baseline clinical variables with CVEs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, Y., Jin, JL., Cao, YX. et al. Prognostic utility of lipoprotein(a) combined with fibrinogen in patients with stable coronary artery disease: a prospective, large cohort study. J Transl Med 18, 373 (2020). https://doi.org/10.1186/s12967-020-02546-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-020-02546-y