
Abstract
Background
Hereditary hearing loss is genetically heterogeneous, and hundreds of mutations in than 60 genes are involved in this disease. Therefore, it is difficult to identify the causative gene mutations involved. In this study, we combined targeted genomic capture and massively parallel sequencing (MPS) to address this issue.
Methods
Using targeted genomic capture and MPS, 104 genes and three microRNA regions were selected and simultaneously sequenced in 23 unrelated probands of Chinese families with nonsyndromic hearing loss. The results were validated by Sanger sequencing for all available members of the probands' families. To analyze the possible pathogenic functional effects of the variants, three types of prediction programs (Mutation Taster, PROVEAN and SIFT) were used. A total of 195 healthy Chinese Han individuals were compared as controls to verify the novel causative mutations.
Results
Of the 23 probands, six had mutations in DFNA genes [WFS1 (n = 2), COCH, ACTG1, TMC1, and POU4F3] known to cause autosomal dominant nonsyndromic hearing loss. These included one novel in-frame indel mutation, three novel missense mutations and two reported missense mutations. Furthermore, one proband from a family with recessive DFNB carried two monoallelic mutations in the GJB2 and USH2A genes. All of these mutations co-segregated with the hearing loss phenotype in 36 affected individuals from 7 families and were predicted to be pathogenic.
Conclusions
Mutations in uncommon deafness genes contribute to a portion of nonsyndromic deafness cases. In the future, critical gene mutations may be accurately and quickly identified in families with hereditary hearing loss by targeted genomic capture and MPS.
Similar content being viewed by others


Introduction
Hearing loss is an extremely common problem worldwide, and it is one of the most genetically heterogeneous disorders occurring in humans. Approximately half of the cases have a genetic etiology, including nonsyndromic hearing loss (NSHL) and syndromic hearing loss (SHL). NSHL as the sole defect accounts for seventy percent or more of deafness cases, and different modes of inheritance have been observed. To date, 65 genes with more than 1000 discrete deafness-causing mutations have been identified [1]. SHL comprises the remaining 30% of hearing loss cases, and it appears accompanied by other medical or physical findings. Similar to NSHL, SHL is associated with a growing list of causative genes and hearing loss syndromes [2]. Thus, the discovery of a causative gene (mutation) of hereditary hearing loss is necessary to resolve the clinical and genetic heterogeneity of deafness.
Previously, most deafness genes (mutations) have been identified through traditional positional cloning (Sanger sequencing), which is an expensive and time-consuming process. More recently, next-generation sequencing, which is also known as massively parallel sequencing (MPS), has been introduced as an alternative approach to more traditional methods [3]-[5]. Whole-exome sequencing (WES) allows for the targeted enrichment and resequencing of nearly all exons of protein-coding genes and identifies genetic variation at a single base-pair resolution. Accordingly, WES uses next-generation technologies to provide a transformational approach for identifying causative mutations of Mendelian disorders. Different targeted genomic capture methods and MPS have been successfully applied to detect gene mutations in relatively small sets of deafness families [6]-[9]. In the present study, we performed targeted genomic capture and MPS to screen 104 genes and three microRNA regions that are known to be responsible for hereditary hearing loss in 23 unrelated probands of Chinese families with NSHL. We identified six causative variants in DFNA genes, including four novel mutations, and a novel combination of two monoallelic mutations in GJB2 and USH2A. This study provides a reliable strategy for the routine genetic diagnosis of hearing loss.
Materials and methods
Subjects and pre-exclusion of frequently reported deafness genes
A total of 143 available members of twenty-three Chinese families were recruited from the Otology Clinic of the First Affiliated Hospital at the Nanjing Medical University, Nanjing, China. Each family had at least 2 hearing-impaired individuals. All affected members in these families were diagnosed as having hereditary NSHL by a complete hearing evaluation, general examination and medical history collection. Peripheral blood samples were obtained from available members of these families, and genomic DNA was extracted using a blood genomic DNA extraction kit (TianGen, Beijing, China) according to the manufacturer's protocol.
Each proband of the 23 families was pre-tested for nine hotspot mutations of deafness genes that have been reported in Chinese individuals with a Deafness Gene Mutation Detection Array Kit (Capital Bio Corporation, Beijing, China), as previously described [10]. The mutations included c.35delG, c.176del16bp, c.235delC and c.299delAT in the GJB2 gene, c.538C > T in the GJB3 gene, c.IVS7-2A > G and c.2168A > G in the SLC26A4 gene, and m.1555A > G and m.1494C > T in the MT-RNR1 gene. The analysis revealed that 22 probands were negative for these mutations, and one carried the monoallelic GJB2 c.235delC mutation. To further search for the causative genes in these families, we performed targeted genomic capture and MPS. For cases in which novel variants were detected, segregation analysis was performed to assess the family. A total of 195 ethnicity-matched individuals were selected as controls to confirm the candidate mutations. This study was approved by the Ethical Committee of Nanjing Medical University for Human Studies. All participants provided written informed consent that complied with all Declaration of Helsinki tenets.
Targeted genomic capture and next-generation sequencing
Whole-exon regions of 104 deafness genes and three microRNA regions (Additional file 1: Table S1) were target-enriched using a Target Enrichment Kit (MyGenostics Inc., Beijing, China) as previously described [11]. A minimum of 3 μg of DNA was used to generate indexed Illumina libraries according to the manufacturer's protocol. The final library size was 300 to 400 bp, including the adapter sequences. The enrichment libraries were sequenced on an Illumina HiSeq 2000 sequencer to generate 100 bp paired-end reads. Next, high-quality reads were identified by filtering out low-quality reads and adaptor sequences using Solexa QA package [12] and Cutadapt program, respectively. Variants were first selected if they appeared in the 1000 Genomes Project database with an MAF of >0.05, and then they were selected if they appeared in the 300 local Asian Genome database. The remaining variants were further processed according to the dbSNP database. SNPs and indels were identified using SOAPsnp and GATK programs. Subsequently, the reads were realigned to the reference genome (NCBI37/hg19) using BWA software. Non-synonymous variants were evaluated by four algorithms, including PolyPhen, SIFT, PANTHER and Pmut, to determine pathogenicity [13].
Mutation validation and analysis
Sanger sequencing was performed on 51 available members of seven families. Seven pairs of primers surrounding the suspected variants were designed with Primer Premier 5.0 (Premier Biosoft) (Additional file 2: Table S2). PCR products were purified and sequenced with a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) and ABI 3730 DNA Sequencer (Applied Biosystems, Foster City, CA, USA) with Chromas Lite v2.01 (Technelysium Pty Ltd., Tewantin, QLD, Australia) software. DNA sequence variation was identified through a comparison of subjects' DNA sequences to those of WFS1 (NM_001145853), COCH (NM_001135058), ACTG1 (NM_001199954), TMC1 (NM_138691), POU4F3 (NM_002700), and USH2A (NM_206933).
Possible pathogenic effects caused by the rare or novel non-synonymous SNPs were evaluated by Mutation Taster (http://www.mutationtaster.org), PROVEAN (http://provean.jcvi.org) and SIFT (with cut-off scores of −1.3 and 0.05, respectively; http://sift.jcvi.org). Multiple sequence alignments were performed using ClustalW, with the default setting of nine species.
Results
Deaf panel enrichment and targeted sequencing
The enriched libraries were labeled with a unique barcoding sequence, pooled, and sequenced on an Illumina HiSeq 2000 to generate paired-end reads of 100 bp in length. As shown in Table 1, the average sequencing depths of the targeted regions were 188 to 264. Over 98% coverage of the targeted regions was achieved for each proband. The coverage of the targeted exons for the >10× reads ranged from 94.10% to 96.20% and from 92.60% to 94.80% for the >20× reads. All mitochondrial DNA and microRNA regions were sequenced at a depth of over 100×. Using SOAPsnp, we identified an average of 485 to 656 variants for each sample. To give priority to identification of the deleterious mutations, we adopted a series of filtering strategies to focus on non-synonymous variants (missense, nonsense and splice variants) using a combination of filtering against HapMap 28, SNP databases and mutiple algorithms (PolyPhen, SIFT, PANTHER and Pmut). We found six deleterious non-synonymous mutations: COCH, p.G38D; WFS1, p.R653C; ACTG1, p.E316K; TMC1, p.D572N; POU4F3, p.P164R and USH2A, p.P1684L. Of the indels, a total of 5 ~ 26 coding indels initially discovered in the probands using the GATK program, only one deletion (WFS1, p.E680del) to be identified based on the sample filtering strategy. Through the above analysis, we successfully validated the deleterious candidate variants in the seven families.
Verification of candidate mutations in seven families
Six families with candidate variants (JSNY-021, JSNY-027, JSNY-033, JSNY-043, JSNY-053 and JSNY-056) showed autosomal dominant inheritance (Figure 1a). For all affected members of each family, late-onset, progressive hearing loss was observed. Pure-tone audiograms showed bilaterally symmetric, sensorineural, mild-to-profound deafness (Figure 1b). With regard to audiometric configurations, two pedigrees were up-sloping (JSNY-021 and JSNY-033), and the others exhibited down-sloping or flat patterns. Aside from their hearing loss, the patients were phenotypically normal. None of them complained of vestibular symptoms. In each of the probands, multiple potentially functional variants with predicted damaging effects were identified by our approach and validated by Sanger sequencing. Each validated variant was tested for complete co-segregation with hearing loss in the proband's family, and this assessment included an additional 28 affected individuals and 13 unaffected family members. Finally, six variants were successfully confirmed in the WFS1 (n = 2), COCH, ACTG1, TMC1, and POU4F3 genes (Table 2). Of them, three were novel missense mutations (p.G38D in COCH, p.E316K in ACTG1, and p.P164R in POU4F3), one was a novel in-frame indel mutation (c.2036-2038delAGG in WFS1), and two were known deafness-causing mutations that have been previously reported (p.R653C in WFS1 and p.D572N in TMC1).

Pedigrees and audiograms of 6 DFNA families. (a) Family pedigrees showing autosomal dominant inheritance. Open symbols, unaffected; solid black symbols, affected; squares, men; circles, female; arrows at lower left, probands. (b) Pure-tone audiograms of affected members in each family. All hearing thresholds shown here are from the better ear. The red line indicates the proband.
Another family possessed a candidate variant (JSNY-045) exhibiting autosomal recessive inheritance. Both siblings II-2 and II-3 suffered from severe sensorineural hearing loss that was congenital and non-progressive (Figure 2a). None of them reported eye problems. Complete ophthalmic examination showed negative findings (Table 3). During routine mutation detection, the proband (II-3) was found to be a carrier of the monoallelic GJB2 c.235delC mutation. Subsequent targeted capture sequencing of known deafness genes confirmed the presence of this mutation as well as of a newly detected heterozygous USH2A gene mutation (c.5051C > T, p.P1684L) located in exon 25 (Figure 2c). We then used Sanger sequencing to screen the proband's sibling and parents. The results showed that the sibling possessed the same genotype as the proband (Figure 2b), while the parents were each unaffected heterozygotes carrying a GJB2 c.235delC monoallelic mutation (the father) or a USH2A c.5051C > T monoallelic mutation (the mother).

DFNB in the affected family segregated with the GJB2 c.235delC and USH2A c.5051C > T mutations. (a) Family pedigree of JSNY-045 and audiograms of two deaf siblings and their parents. (b) The average coverage of each exon of the GJB2 and USH2A genes in the proband (left) and sequencing electropherograms of the heterozygous GJB2 c.235delC and USH2A c.5051C > T mutations (right). (c) A novel c.5051C > T variant and six previously reported mutations in USH2A exon 25. (d) Conservation analysis of the novel missense mutation. The USH2A p.P1684L mutation occurs at an evolutionarily conserved amino acid (in red box).
To predict the probable pathogenic effects of the above variants, we analyzed the evolutionary conservation and damaging effects of the amino acid substitutions. The results showed that all 7 of these variants were highly conserved among multiple vertebrate species (Figure 2d, Additional file 3: Figure S1). The results from the prediction analysis are shown in Table 2.
None of the candidate mutations was found in the 195 Chinese Han healthy controls.
Discussion
In the present study, we confirmed the presence of seven non-synonymous variants in 7 of the 23 deafness families. Six families likely had causative mutations in DFNA genes [WFS1 (n = 2), ACTG1, POU4F3, COCH and TMC1], and one showed two monoallelic and most likely causative mutations in the GJB2 and USH2A genes, respectively. With the exception of the p.R653C mutation in WFS1 and the p.D572N mutation in TMC1, the other five variants have not been previously reported to be associated with hereditary hearing loss. The causative genes detected in the remaining 16 families require further examination, presumably by linkage analysis and/or WES. It is probable that hearing loss in these families is due to mutations in unidentified deafness-related genes. Alternatively, pathogenic mutations might exist in those regions not covered in our sequencing analysis, including intronic regulatory sequences.
In the JSNY-021 family, a novel in-frame indel mutation caused by c.2036_2038delAGG (p.E680del) was detected in WFS1, which is responsible for DFNA6/14/38 hearing loss. This gene encodes wolframin, which is a membrane glycoprotein predominantly located in the endoplasmic reticulum (ER). This protein is essential for maintaining correct levels of Ca2+ and other charged particles necessary for hearing, and its lack of function induces apoptotic input signaling in the ER [16],[17]. Although it remains unknown whether wolframin is expressed in the human cochlea, mutations in the WFS1 gene, such as c.511G > A (p.D171N), c.2005 T > C (p.Y669H) and c.2590G > A (p.E864K), have been identified as frequent causes of autosomal dominant low-frequency hearing loss in different ethnicities [18]-[20]. The heterozygous 3-bp deletion (c.2036_2038delAGG) identified in the present study is expected to cause the loss of the E680 codon, which might affect the three-dimensional shape or properties of the wolframin protein and consequently interfere the normal function of the wolframin tetramer. Considering previous studies and the low-frequency NSHL phenotype found in the JSNY-021 family in this study, we hypothesize that this mutation likely has a pathogenic effect.
ACTG1 encodes cytoskeletal actin gamma 1, which is known to be the building block of hair cell stereocilia. These stereocilia are constantly undergoing actin polymerization at their tips and depolymerization at their bases [21]. In auditory hair cells of the cochlea and intestinal epithelial cells, actin gamma 1 has a predominant and unique expression pattern. Mutations in the ACTG1 gene have been mainly associated with autosomal dominant progressive sensorineural deafness 20/26 (DFNA20/26), and some have been linked to Baraitser-Winter syndrome, which is a rare autosomal recessive disorder characterized by developmental delay, facial dysmorphology, brain malformations, coloboma, and variable hearing loss [22]-[24]. To date, a total of 19 ACTG1 mutations have been reported in patients with DFNA 20/26 and Baraitser-Winter syndrome, of which 12 have been identified in NSHL families (http://www.hgmd.org/, designed by P.D.Stenson HGMD®). In this study, we found the novel missense mutation p.E316K, which was caused by a c.946 G > A transition in ACTG1, in the JSNY-043 family. p.E316K is located in subdomain 3 of actin gamma 1, which is a highly conserved actin domain of ACTG1. Until now, all reported ACTG1 missense mutations have been located in this domain.
The POU4F3 protein is a well-known transcription factor encoded by POU4F3. This protein belongs to the POU-domain class IV transcription factor family and plays an important role in the maturation, differentiation and survival of hair cells [25]. Mutations in the POU4F3 gene have been described in patients with nonsyndromic sensorineural deafness autosomal dominant type 15 (DFNA15). To date, more than five different mutation types, including the deletion of the entire POU4F3 gene sequence, have been reported worldwide in different ethnic groups [26],[27], but none have been reported in the Asian population. In the present study, the POU4F3 p.P164R missense mutation caused by c.491C > G was identified as a novel mutation in the JSNY-056 Chinese family.
The COCH gene encodes a 550-amino acid protein with multiple domains, including a signal peptide (SP), an LCCL module and two von Willebrand factor A (vWFA) domains [28]. The COCH protein, cochlin, is abundantly expressed in the cochlea and vestibular system of the inner ear. Mutations in the COCH gene lead to autosomal dominant nonsyndromic sensorineural deafness 9 (DFNA9), which has been clinically characterized by progressive late-onset hearing loss with or without vestibular dysfunction [29]. Presently, 18 COCH mutations have been identified in DFNA9 families (http://www.hgmd.org/, designed by P.D.Stenson HGMD®), most of which are located in the LCCL region. Here, we identified a novel missense mutation (c.113G > A, p.G38D) in exon 3 of the COCH gene, which is the LCCL domain of cochlin. Mutations in this domain are expected to cause misfolding and protein aggregation in a dominant-negative manner, leading to cytotoxicity of the inner ear fibrocytes [30]. The clinical features associated with this novel mutation in the JSNY-027 family lends support to the pathogenic nature of the p.G38D variant.
In the present study, we also identified one novel mutation of USH2A c.5051C > A (p.P1684L) in the JSNY-045 family. USH2 mutations result in an autosomal recessive disorder characterized by retinitis pigmentosa and mild-to-moderate sensorineural hearing loss, and the USH2A gene is most commonly mutated. This gene is located at 1q41 and encodes a protein with a predicted size of 171.5 kDa [31],[32]. To date, more than 120 different disease-causing mutations have been reported in the USH2A gene, which are widely distributed over the coding regions of all 72 exons. Interestingly, the novel p.P1684L mutation, which is located in exon 25 of the USH2A gene, was identified together with the GJB2 c.235delC mutation in two deaf siblings of the JSNY-045 family, while their hearing parents each carried only one monogenic recessive mutation. All subjects exhibited normal vestibular and visual functions. We therefore deduced that the USH2A monogenic recessive mutation failed to cause the Usher phenotype but may have contributed to the pathogenesis of the GJB2 c.235delC mutation, resulting in phenotypic hearing loss in the two patients. In fact, digenic inheritance of nonsyndromic deafness caused by mutations in the GJB2 gene and other connexin genes, such as GJB3, GJB6, GJB4, or GJA1, have been previously reported in several deaf patients [33]-[35]. In addition, a Japanese family with comorbid GJB2 and WFS1 mutations was also described in 2012. In this family, one individual with mutations in both GJB2 and WFS1 presented with a GJB2 phenotype [36]. Our results together with previous findings suggest that monoallelic GJB2 mutations may contribute to NSHL as a result of their co-inheritance with other deafness-causing genes. However, the pathogenic mechanisms underlying the cooperation of these genes with GJB2 require further research.
Conclusions
Using targeted genomic capture and MPS, we have successfully identified causative gene mutations in six families with autosomal dominant NSHL, and in patients from a family with recessive DFNB, we detected two monoallelic mutations in the GJB2 and USH2A genes, respectively. The USH2A gene may have cooperated with GJB2 to cause the NSHL phenotype in this Chinese family. Our results suggest that targeted genomic capture combined with MPS can serve as a useful technique in the etiological diagnosis of sensorineural hearing loss.
Additional files
References
Deafness Variation Database [], [http://deafnessvariationdatabase.org]
Friedman TB, Griffith AJ: Human nonsyndromic sensorineural deafness. Annu Rev Genomics Hum Genet. 2003, 4: 341-402. 10.1146/annurev.genom.4.070802.110347.
Mestan KK, Ilkhanoff L, Mouli S, Lin S: Genomic sequencing in clinical trials. J Transl Med. 2011, 9: 222-10.1186/1479-5876-9-222.
Rabbani B, Tekin M, Mahdieh N: The promise of whole-exome sequencing in medical genetics. J Hum Genet. 2014, 59: 5-15. 10.1038/jhg.2013.114.
Boycott KM, Dyment DA, Sawyer SL, Vanstone MR, Beaulieu CL: Identification of genes for childhood heritable diseases. Annu Rev Med. 2014, 65: 19-31. 10.1146/annurev-med-101712-122108.
Choi BY, Park G, Gim J, Kim AR, Kim BJ, Kim HS, Park JH, Park T, Oh SH, Han KH, Park WY: Diagnostic application of targeted resequencing for familial nonsyndromic hearing loss. PLoS One. 2013, 8: e68692-10.1371/journal.pone.0068692.
Brownstein Z, Abu-Rayyan A, Karfunkel-Doron D, Sirigu S, Davidov B, Shohat M, Frydman M, Houdusse A, Kanaan M, Avraham KB: Novel myosin mutations for hereditary hearing loss revealed by targeted genomic capture and massively parallel sequencing. Eur J Hum Genet. 2014, 22: 768-775. 10.1038/ejhg.2013.232.
Vona B, Muller T, Nanda I, Neuner C, Hofrichter MA, Schroder J, Bartsch O, Lassig A, Keilmann A, Schraven S, Kraus F, Shehata-Dieler W, Haaf T: Targeted next-generation sequencing of deafness genes in hearing-impaired individuals uncovers informative mutations.Genet Med 2014. doi:10.1038/gim.2014.65. [Epub ahead of print].,
Vozzi D, Morgan A, Vuckovic D, D'Eustacchio A, Abdulhadi K, Rubinato E, Badii R, Gasparini P, Girotto G: Hereditary hearing loss: a 96 gene targeted sequencing protocol reveals novel alleles in a series of Italian and Qatari patients. Gene. 2014, 542: 209-216. 10.1016/j.gene.2014.03.033.
Li CX, Pan Q, Guo YG, Li Y, Gao HF, Zhang D, Hu H, Xing WL, Mitchelson K, Xia K, Dai P, Cheng J: Construction of a multiplex allele-specific PCR-based universal array (ASPUA) and its application to hearing loss screening. Hum Mutat. 2008, 29: 306-314. 10.1002/humu.20622.
Wu J, Matthaei H, Maitra A, Dal Molin M, Wood LD, Eshleman JR, Goggins M, Canto MI, Schulick RD, Edil BH, Wolfgang CL, Klein AP, Diaz LA, Allen PJ, Schmidt CM, Kinzler KW, Papadopoulos N, Hruban RH, Vogelstein B: Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med. 2011, 3: 92ra66-10.1126/scitranslmed.3002543.
Cox MP, Peterson DA, Biggs PJ: SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics. 2010, 11: 485-10.1186/1471-2105-11-485.
Yang Y, Wu J, Liu H, Chen X, Wang Y, Zhao M, He X: Two homozygous nonsense mutations of GNPTAB gene in two Chinese families with mucolipidosis II alpha/beta using targeted next-generation sequencing. Genomics. 2013, 102: 169-173. 10.1016/j.ygeno.2013.06.001.
Awata T, Inoue K, Kurihara S, Ohkubo T, Inoue I, Abe T, Takino H, Kanazawa Y, Katayama S: Missense variations of the gene responsible for Wolfram syndrome (WFS1/wolframin) in Japanese: possible contribution of the Arg456His mutation to type 1 diabetes as a nonautoimmune genetic basis. Biochem Biophys Res Commun. 2000, 268: 612-616. 10.1006/bbrc.2000.2169.
Kurima K, Peters LM, Yang Y, Riazuddin S, Ahmed ZM, Naz S, Arnaud D, Drury S, Mo J, Makishima T, Ghosh M, Menon PS, Deshmukh D, Oddoux C, Ostrer H, Khan S, Riazuddin S, Deininger PL, Hampton LL, Sullivan SL, Battey JF, Keats BJ, Wilcox ER, Friedman TB, Griffith AJ: Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair-cell function. Nat Genet. 2002, 30: 277-284. 10.1038/ng842.
Cryns K, Sivakumaran TA, Van den Ouweland JM, Pennings RJ, Cremers CW, Flothmann K, Young TL, Smith RJ, Lesperance MM, Van Camp G: Mutational spectrum of the WFS1 gene in Wolfram syndrome, nonsyndromic hearing impairment, diabetes mellitus, and psychiatric disease. Hum Mutat. 2003, 22: 275-287. 10.1002/humu.10258.
Philbrook C, Fritz E, Weiher H: Expressional and functional studies of Wolframin, the gene function deficient in Wolfram syndrome, in mice and patient cells. Exp Gerontol. 2005, 40: 671-678. 10.1016/j.exger.2005.06.008.
Goncalves AC, Matos TD, Simoes-Teixeira HR, Pimenta Machado M, Simao M, Dias OP, Andrea M, Fialho G, Caria H: WFS1 and non-syndromic low-frequency sensorineural hearing loss: a novel mutation in a Portuguese case. Gene. 2014, 538: 288-291. 10.1016/j.gene.2014.01.040.
Haghighi A, Setoodeh A, Saleh-Gohari N, Astuti D, Barrett TG: Identification of homozygous WFS1 mutations (p.Asp211Asn, p.Gln486*) causing severe Wolfram syndrome and first report of male fertility. Eur J Hum Genet. 2013, 21: 347-351. 10.1038/ejhg.2012.154.
Tsai HT, Wang YP, Chung SF, Lin HC, Ho GM, Shu MT: A novel mutation in the WFS1 gene identified in a Taiwanese family with low-frequency hearing impairment. BMC Med Genet. 2007, 8: 26-10.1186/1471-2350-8-26.
Sonnemann KJ, Fitzsimons DP, Patel JR, Liu Y, Schneider MF, Moss RL, Ervasti JM: Cytoplasmic gamma-actin is not required for skeletal muscle development but its absence leads to a progressive myopathy. Dev Cell. 2006, 11: 387-397. 10.1016/j.devcel.2006.07.001.
Park G, Gim J, Kim AR, Han KH, Kim HS, Oh SH, Park T, Park WY, Choi BY: Multiphasic analysis of whole exome sequencing data identifies a novel mutation of ACTG1 in a nonsyndromic hearing loss family. BMC Genomics. 2013, 14: 191-10.1186/1471-2164-14-191.
Liu P, Li H, Ren X, Mao H, Zhu Q, Zhu Z, Yang R, Yuan W, Liu J, Wang Q, Liu M: Novel ACTG1 mutation causing autosomal dominant non-syndromic hearing impairment in a Chinese family. J Genet Genomics. 2008, 35: 553-558. 10.1016/S1673-8527(08)60075-2.
Perrin BJ, Sonnemann KJ, Ervasti JM: β-actin and γ-actin are each dispensable for auditory hair cell development but required for Stereocilia maintenance. PLoS Genet. 2010, 6: e1001158-10.1371/journal.pgen.1001158.
Collin RW, Chellappa R, Pauw RJ, Vriend G, Oostrik J, van Drunen W, Huygen PL, Admiraal R, Hoefsloot LH, Cremers FP, Xiang M, Cremers CW, Kremer H: Missense mutations in POU4F3 cause autosomal dominant hearing impairment DFNA15 and affect subcellular localization and DNA binding. Hum Mutat. 2008, 29: 545-554. 10.1002/humu.20693.
Lee HK, Park HJ, Lee KY, Park R, Kim UK: A novel frameshift mutation of POU4F3 gene associated with autosomal dominant non-syndromic hearing loss. Biochem Biophys Res Commun. 2010, 396: 626-630. 10.1016/j.bbrc.2010.04.132.
Freitas EL, Oiticica J, Silva AG, Bittar RS, Rosenberg C, Mingroni-Netto RC: Deletion of the entire POU4F3 gene in a familial case of autosomal dominant non-syndromic hearing loss. Eur J Med Genet. 2014, 57: 125-128. 10.1016/j.ejmg.2014.02.006.
Chen DY, Chai YC, Yang T, Wu H: Clinical characterization of a novel COCH mutation G87V in a Chinese DFNA9 family. Int J Pediatr Otorhinolaryngol. 2013, 77: 1711-1715. 10.1016/j.ijporl.2013.07.031.
Gallant E, Francey L, Fetting H, Kaur M, Hakonarson H, Clark D, Devoto M, Krantz ID: Novel COCH mutation in a family with autosomal dominant late onset sensorineural hearing impairment and tinnitus. Am J Otolaryngol. 2013, 34: 230-235. 10.1016/j.amjoto.2012.11.002.
Gao J, Xue J, Chen L, Ke X, Qi Y, Liu Y: Whole exome sequencing identifies a novel DFNA9 mutation, C162Y. Clin Genet. 2013, 83: 477-481. 10.1111/cge.12006.
Dreyer B, Tranebjaerg L, Rosenberg T, Weston MD, Kimberling WJ, Nilssen O: Identification of novel USH2A mutations: implications for the structure of USH2A protein. Eur J Hum Genet. 2000, 8: 500-506. 10.1038/sj.ejhg.5200491.
Baux D, Larrieu L, Blanchet C, Hamel C, Ben Salah S, Vielle A, Gilbert-Dussardier B, Holder M, Calvas P, Philip N, Edery P, Bonneau D, Claustres M, Malcolm S, Roux AF: Molecular and in silico analyses of the full-length isoform of usherin identify new pathogenic alleles in Usher type II patients. Hum Mutat. 2007, 28: 781-789. 10.1002/humu.20513.
Kooshavar D, Tabatabaiefar MA, Farrokhi E, Abolhasani M, Noori-Daloii MR, Hashemzadeh-Chaleshtori M: Digenic inheritance in autosomal recessive non-syndromic hearing loss cases carrying GJB2 heterozygote mutations: assessment of GJB4, GJA1, and GJB3. Int J Pediatr Otorhinolaryngol. 2013, 77: 189-193. 10.1016/j.ijporl.2012.10.015.
Cama E, Melchionda S, Palladino T, Carella M, Santarelli R, Genovese E, Benettazzo F, Zelante L, Arslan E: Hearing loss features in GJB2 biallelic mutations and GJB2/GJB6 digenic inheritance in a large Italian cohort. Int J Audiol. 2009, 48: 12-17. 10.1080/14992020802400654.
Liu XZ, Yuan Y, Yan D, Ding EH, Ouyang XM, Fei Y, Tang W, Yuan H, Chang Q, Du LL, Zhang X, Wang G, Ahmad S, Kang DY, Lin X, Dai P: Digenic inheritance of non-syndromic deafness caused by mutations at the gap junction proteins Cx26 and Cx31. Hum Genet. 2009, 125: 53-62. 10.1007/s00439-008-0602-9.
Minami SB, Masuda S, Usui S, Mutai H, Matsunaga T: Comorbidity of GJB2 and WFS1 mutations in one family. Gene. 2012, 501: 193-197. 10.1016/j.gene.2012.03.060.
Acknowledgments
We sincerely thank all of the family members for their participation in this study. This research was supported by the National Natural Science Foundation of China (No. 31171217), a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institution to XC, and the Grant from Jiangsu Health Administration of China (LJ201120), a Grant from the Research Special Fund for Public Welfare Industry of Health, Ministry of Health of China (No. 201202005) to GX.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
XC and GX conceived and designed the study. QW, HZ, XQ, ZC, JY and YL performed the experiments and analysis. QW and HZ wrote the original manuscript. XC and GX contributed to revisions of the manuscript. All authors read and approved the final version.
Electronic supplementary material
Additional file 2: Table S2.: PCR primers surrounding the suspected variants of 7 genes. (DOC 36 KB)
12967_2014_311_MOESM3_ESM.tiff
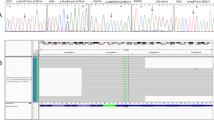
Additional file 3: Figure S1.: Sanger sequencing and conservation analysis of gene mutations identified in 6 autosomal dominant families. (a) WFS1 c.2036_2038delAGG (p.E680del) in-frame indel mutation in the JSNY-021 family. (b) COCH c.113G > A (p.G38D) missense mutation in the JSNY-027 family. (c) WFS1 c.1957C > T (p.R653C) missense mutation in the JSNY-033 family. (d) ACTG1 c.946G > A (p.E316K) missense mutation in the JSNY-043 family. (e) TMC1 c.1714G > A (p.D572N) missense mutation in the JSNY-053 family. (f) POU4F3 c.491C > G (p.P164R) missense mutation in the JSNY-056 family. (TIFF 3 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
Cite this article
Wei, Q., Zhu, H., Qian, X. et al. Targeted genomic capture and massively parallel sequencing to identify novel variants causing Chinese hereditary hearing loss. J Transl Med 12, 311 (2014). https://doi.org/10.1186/s12967-014-0311-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-014-0311-1