
Abstract
Background
The incidence of multiple myeloma (MM), a type of blood cancer affecting monoclonal plasma cells, is rising. Although new drugs and therapies have improved patient outcomes, MM remains incurable. Recent studies have highlighted the crucial role of the chemokine network in MM’s pathological mechanism. Gaining a better understanding of this network and creating an overview of chemokines in MM could aid in identifying potential biomarkers and developing new therapeutic strategies and targets.
Purpose
To summarize the complicated role of chemokines in MM, discuss their potential as biomarkers, and introduce several treatments based on chemokines.
Methods
Pubmed, Web of Science, ICTRP, and Clinical Trials were searched for articles and research related to chemokines. Publications published within the last 5 years are selected.
Results
Malignant cells can utilize chemokines, including CCL2, CCL3, CCL5, CXCL7, CXCL8, CXCL12, and CXCL13 to evade apoptosis triggered by immune cells or medication, escape from bone marrow and escalate bone lesions. Other chemokines, including CXCL4, CCL19, and CXCL10, may aid in recruiting immune cells, increasing their cytotoxicity against cancer cells, and inducing apoptosis of malignant cells.
Conclusion
Utilizing anti-tumor chemokines or blocking pro-tumor chemokines may provide new therapeutic strategies for managing MM. Inspired by developed CXCR4 antagonists, including plerixafor, ulocuplumab, and motixafortide, more small molecular antagonists or antibodies for pro-tumor chemokine ligands and their receptors can be developed and used in clinical practice. Along with inhibiting pro-tumor chemokines, studies suggest combining chemokines with chimeric antigen receptor (CAR)-T therapy is promising and efficient.
Similar content being viewed by others

Introduction
Multiple myeloma (MM) is a blood cancer of monoclonal plasma cells (PCs) characterized by hypercalcemia, renal insufficiency, anemia, or osteolytic lesions [1]. According to the epidemiological landscape of MM in 2022, though the global burden of MM varied from the country, the overall incidence of MM was increasing [2]. Thanks to new drugs such as immunomodulatory agents, proteasome inhibitors, monoclonal antibodies, and so on, the survival of MM patients has been significantly improved [3, 4]. However, MM is still an incurable disease. To develop effective treatments and manage MM patients, researchers should foster an overview of the pathological mechanism of MM cells. In addition to genic mutations in malignant cells, interactions between malignant cells and normal cells also contribute to the progression of MM.
Chemokines are a member of the cytokine superfamily with chemoattractant properties. According to the arrangement of amino-terminal cysteine (C) residues, chemokines are divided into four subfamilies: CXC, CC, XC, and CX3C subfamily. In the tumor microenvironment (TME), chemokines are secreted by different kinds of cells, including immune cells, tumor cells, and tumor-associated cells. Chemokines engage in immune cells’ activation, differentiation, proliferation, migration, and apoptosis and form a complex network in the immune system [5]. Malignant cells can evade apoptosis via secreting chemokines to recruit immunosuppressive cells, while immune cells also migrate to TME to attack tumors via chemokines.
Researchers have developed inhibitors, antibodies, or antagonists to block interactions between chemokine ligands and receptors as adjuvant therapy. CXCR4 antagonists such as plerixafor [6] and ulocuplumab [7] are safe and effective in combination with bortezomib. Plerixafor [8] and motixafortide [9] can improve the mobilization of stem cells. The success of CXCR4 antagonists indicates that targeting chemokines and their receptors is a potential strategy for managing MM. Therefore, insights into the network of chemokine ligands and receptors can contribute to the oncobiology of MM, which further improves treatment strategy and prognosis.
Thus, this review aims to summarize research on the chemokine network in MM in the past 5 years and discuss chemokines in the pathological progression of MM and potential therapy targeting related chemokines.
Main text
Chemokines as biomarkers
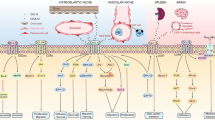
According to previous studies, chemokines can serve as biomarkers and targets for various tumors, including hepatocellular carcinoma [10], endometrial cancer [11], and colorectal cancer [12]. Recent studies highlighted the role of chemokines in the pathological progression of MM (Fig. 1). On the one hand, chemokines participate in the body’s metabolism. Chemokines directly secreted or indirectly induced by malignant cells may disturb the normal function of chemokines and lead to several complications in MM. The breakup of chemokine balances can cause common complications, including bone destruction and anemia, in MM. It has been suggested that a higher level of activated osteoclastic chemokines, such as CXCL7, aggravated bone destruction in MM [13]. Besides, MM cells can secret CCL3 to disrupt erythrocyte differentiation and cause anemia [14]. These complications caused by the abnormal level of chemokines exacerbate the disease and the pain of patients. On the other hand, chemokines take part in the formation of the immune environment, which impacts the clearance of malignant cells. Malignant cells can utilize chemokines to recruit immune cells to protect themselves from apoptosis [15]. The level of chemokines in MM patients also influences the proliferation, migration, and recruitment of immune cells, which is associated with the effectiveness of chimeric antigen receptor (CAR)-T therapy [16, 17]. MM cells can also utilize chemokines to migrate, which causes extramedullary infiltration and exacerbates patients’ burden [18]. Experts proposed chemokines could be potential biomarkers for MM to predict progression and prognosis. Here, we will delve into chemokines’ function and significance in MM (Fig. 2).

The chemokine network in multiple myeloma

The function and mechanism of chemokines in multiple myeloma
Chemokines are mainly comprised of the CXC subfamily and CC subfamily chemokines. According to their function in MM, they can be divided into pro-tumor, anti-tumor, and dual-effect chemokines. Pro-tumor chemokines can promote extramedullary infiltration, bone destruction, and tumor proliferation by interaction with MΦs, monocytes, neutrophils, Tregs, or PBMCs. Contrastingly, anti-tumor chemokines can bind to their receptors to induce NK cells, CD8 + T cells, DCs, or endothelial cells to enhance their anti-tumor efficacy and tumor apoptosis. Or some chemokines, including CCL5 and CXCL10, are double-edge swords in MM. MΦs, macrophages; Tregs, regulatory T cells; PBMCs, peripheral blood mononuclear cells; NK cells, natural killer cells; DCs, dendritic cells; MM, multiple myeloma.
CC chemokine/receptor subfamily
CC chemokines are a group of cytokines with an N-terminal CC domain. These chemokines reveal both anti-tumor and pro-tumor functions in MM. While some may lead to deteriorated anemia, chemoresistance, and tumor dissemination, others may aid in the recruitment of dendritic cells (DCs) essential for the immune system to attack malignant cells.
To resist apoptosis induced by chemical drugs, MM cells may attempt to recruit tumor-associated macrophages (TAMs) or myeloid-derived suppressor cells (MDSCs) to form TME. Newly published studies suggest that chemokines CCL2, CCL3, and CCL5, along with their receptors, participate in the formation of chemoresistance in MM.
CCL2, known as monocyte chemoattractant protein-1 (MCP-1), is mainly secreted by monocytes, macrophages, and DCs [19]. It interacts with CCR2 to promote tumor growth and progression through various mechanisms. Previous studies have shown that the CCL2-CCR2 axis is related to increased angiogenesis, recruitment of immunosuppressive cells, and the proliferation and survival of malignant cells [20]. Recent research suggests that CCL2 is linked to M2 polarization and contributes to MM chemoresistance [15]. MM cells upregulate CCL2 expression in macrophages, further upregulating MCP-1-induced protein (MCPIP1) via the JAK2-STAT3 signaling pathway. Although CCL2 does not directly affect MM cells’ proliferation and chemoresistance, MCPIP1 plays a role in M2 polarization and enhances the protective effect of macrophages in MM. The research indicates that CCL2 expression is linked to therapeutic status and can be a good prognostic factor.
Aside from CCL2, CCL3, known as macrophage inflammatory protein-1α (MIP-1α), can bind to CCR1 to protect MM cells from apoptosis induced by melphalan and bortezomib. Increased CCR1 is associated with upregulated Bcl-2, Bcl- xl, survivin, and downregulated Bim, leading to chemoresistance [21]. MM cells can also induce the differentiation of peripheral blood mononuclear cells (PBMCs) towards MDSCs by secreting CCL5 [22]. In vitro experiments show that the secretion of CCL5 and CCL3 is significantly higher in MDSCs-inducible groups than in MDSCs-non-inducible groups. Moreover, MDSCs-non-inducible groups gained the ability to induce MDSCs by adding CCL5, while supplementation of CCL3 did not promote MDSCs differentiation. The serum level of CCL5 in patients with better response significantly decreased after receiving combination therapy of carfilzomib, lenalidomide (LEN), and dexamethasone, suggesting that CCL5 may promote disease progression. It may be practical to monitor the CCL5 level to manage the disease.
In addition to CCL2 and CCL5, CCL3 shows multifunctional properties that can aggravate the disease. The CCL3-CCR1 axis can attract M2 macrophages into TME, resulting in anemia and metastasis in MM. Research shows that elevated CCL3 levels in the myeloma microenvironment impair erythrocyte differentiation of hematopoietic stem and progenitor cells (HSPCs), leading to anemia [14]. CCL3 suppresses the expression of GATA1 via p38 signaling, which hinders erythroid differentiation in CD34 + HSPCs. The suppressive effect of CCL3 on erythropoiesis can be blocked by CCR1 antagonists, suggesting that CCL3/CCR1/phos-p38 is critical to CCL3-induced anemia in MM. A high level of CCR1 is observed in MM PCs and indicates a poor prognosis [23]. Moreover, CCR1 is an independent prognosis factor of MM patients and induces splenic and bone dissemination of MM cells [18]. The CCL3-CCR1 axis plays a vital role in overcoming retention caused by CXCL12 and the migration of MM cells from the bone environment to peripheral blood [18]. Based on the current comprehension of CCL3, its prognostic and therapeutic value may be worth further exploration.
Some chemokines utilized by MM cells can promote migration and chemoresistance, while others may participate in antigen uptake, cytotoxicity, and proliferation of lymphocytes. For instance, the CCL19-CCR7 axis induces migration of DCs towards sites with a higher concentration of CCL19 and mediates DC homing [24, 25]. DC vaccines utilizing DCs from patients to activate immune responses have been used in clinical practice, and immunogenicity determines DC vaccines’ efficacy. Inspired by the function of CCL19-CCR7, research finds that monocyte-derived DCs have a lower transcript and protein level of CCR7, leading to migration dysfunction [26]. By contrast, hematopoietic stem cell-derived DCs are preferable sources of DC vaccine, as stem cell-derived DCs from MM patients show similarities in cell yield, morphology, and phenotype compared to healthy donors. It indicates that chemokines can also be a possible reference standard to select cells used in autologous cell transplantation.
CXC chemokine/receptor subfamily
The CC chemokine subfamily and the CXC chemokine subfamily comprise most of the chemokine family. Like CC chemokines, CXC chemokines are a double-edged sword in MM. While some induce bone destruction [13, 27, 28], chemoresistance [29], and tumor metastasis [30], others can enhance the cytotoxicity of CD8 + T cells or induce apoptosis of MM cells to protect the body against tumors [17].
Chemokines such as CXCL7, CXCL8, CXCL12, and CXCL13 by MM cells and surrounding cells contribute to disease progress and decrease the overall survival in patients [13, 28, 31]. CXCL7, for example, is processed by neutrophils and interacts with CXCR2 to recruit neutrophils [32]. It also plays a role in osteoclastogenesis and the formation of osteoclasts that break down bone tissue, a common characteristic of MM [33].
Matrix metalloproteinase-13 (MMP13) secreted by mesenchymal stromal cells (MSCs) can enhance the activation and formation of osteoclasts by promoting the bioavailability of CXCL7 [13]. However, an in vivo experiment demonstrated that mice with MM cells had elevated MMP-13 in mRNA level, resulting in bone lesions and lower overall survival. Though MMP-13 expression is not observed in all MM patients, analysis shows that it is associated with overall survival. Understanding the role of MMP-13 in regulating CXCL7 bioavailability may provide insight into bone lesions in MM.
CXCL8, also known as interleukin-8 (IL-8), can activate CXCR1 and CXCR2 to recruit neutrophils [27], similar to CXCL7. In patients with MM, CXCL8 levels are higher than in healthy individuals, indicating an association between CXCL8 and MM [34]. Interestingly, there is a difference in IL-8 concentration between males and females worth exploring. Additionally, it has been reported that IL-8 is linked to osteolysis in breast cancer, underscoring its role in complications in MM [35]. In MM, MM cells can secrete exosomes that MSCs can internalize, leading to upregulation of the expression of IL-8 [31]. This process relies on amphiregulin (AREG) packed into exosomes and its interaction with epidermal growth factor receptor (EGFR). According to the study, targeting EGFR may offer a practical and innovative strategy to inhibit the CXCL8-CXCR1/2 axis in MM.
Though studies have proved the role of CXCL13 in several hematologic diseases [36,37,38], it has been suggested recently that the CXCL13-CXCR5 axis contributes to tumor progress. Higher levels of CXCL13 are not only due to the direct secretion of malignant cells but also bone marrow stromal cells (BMSCs) and macrophages stimulated by malignant cells [28]. MM cells can rely on Bruton’s tyrosine kinase (BTK) signaling to induce M2 polarization, while M2 polarized macrophages mutually upregulate CXCL13 expression in MM cells via TGFβ. With a higher level of CXCL13, enhanced formation of osteoclasts and elevated receptor activator of nuclear kappa B ligand (RANKL) is observed, indicating the role of CXCL13 in osteolytic disease. In addition to lytic lesions, biopsies suggest the relationship between CXCL13 and extramedullary disease. And MSCs have been found to mediate chemoresistance to bortezomib depending on CXCL13 [39]. However, there are currently no small molecule inhibitors directly targeting CXCL13. Future research may attempt to complement the lack of current study due to its function on bone destruction, chemoresistance, and extramedullary disease.
Besides pro-tumor CXC chemokines mentioned above, CXCL12, also known as stromal cell-derived factor-1 (SDF-1), attracts great interest from researchers due to its complex function in MM. CXCL12 can bind to CXCR4 to participate in various dysfunctions in hematological diseases [40, 41]. Emerging studies suggest the role of CXCL12 in multifaceted function in MM, including tumor migration [30] and chemoresistance [29, 42]. CXCL12 has two isoforms, CXCL12alpha and CXCL12gamma, each with distinct stability and immobilization properties [43]. CXCL12alpha is known to promote the phosphorylation of phosphoinositide 3-kinase (PI3K) and protein kinase B (PKB), which further leads to the overexpression of interleukin-6 (IL-6) [42]. Furthermore, the CXCL12alpha-CXCR4 axis contributes to a higher adhesion rate between MM cells and BMSCs, contributing to lower apoptosis in the coculture state. CXCL12gamma, expressed in BMSCs, mediates chemoresistance and requires heparan sulfate proteoglycans (HSPGs) to be immobilized to the membrane [29]. Notably, protection provided by BMSCs functions via adhesion instead of solvable molecules and can be abolished by CXCL12gamma-CXCR4 inhibition. Regardless of chemoresistance, the CXCL12-CXCR4 axis may also induce extramedullary migration [30]. It was known that MM cells could emigrate from bone marrow to form circulating plasma cells (cPCs) and invade other tissues in the past [44]. However, the mechanism is still unclear and remains to be discussed. Recent single-cell sequencing results have revealed increased secretion of CXCL12 in cPCs, indicating its potential role in extramedullary plasmacytoma.
While some CXC chemokines, as mentioned earlier, act as tumor promoters in MM, others, like CXCL4 and CXCL10, impede malignant cell growth. CXCL4, called platelet factor 4 (PF-4), can induce MM cell apoptosis by enhancing SOCS3 to regress STAT3, validating its potential predictive value [45]. A recent study shows that higher PF-4 levels correlate with better outcomes, and higher PF-4 is also observed in healthy individuals [46]. Differently, CXCL10 can recruit natural killer (NK) cells, cytotoxic T lymphocytes, and macrophages, playing a crucial role in anti-tumor immunity through CXCL9/10/11-CXCR3 axes [47]. As CXCL10 is induced by interferon, including IFN-α/β and IFN-γ, it is also named IFN-γ-induced protein-10, and its association with chemotaxis, cytotoxicity, and proliferation is well-documented [48]. CXCL10 has been shown to enhance CAR-T cells’ proliferation and anti-tumor ability in vitro [17]. And patients undergoing CAR-T therapy with higher CXCL10 at baseline reveal better outcomes than those with lower CXCL10. However, MM cells may utilize the CXCRL9/10-CXCR3 axes to interfere with the bone marrow localization of NK cells to evade immune surveillance [49]. Blocking CXCR3 has been shown to improve NK cells’ infiltration in the bone marrow and reinforce IL-15-activated NK cells’ anti-tumor activity [50]. Therefore, due to their complicated functions, the decision to boost or inhibit CXCL9/10-CXCR3 axes should be based on specific clinical backgrounds.
Therapy based on chemokines
There are two mainstream strategies for utilizing chemokines to treat tumors: targeting pro-tumor chemokines and increasing the concentration of anti-tumor chemokines [51]. Additionally, some studies are exploring anti-tumor chemokines with adoptive cellular therapy (ACT) to enhance its effectiveness. Here, aimed at pro-tumor and anti-tumor chemokines, we will discuss the use of chemokines in treating MM, respectively (Table 1).
Targeting pro-tumor chemokines
Malignant cells can trigger other cells or autonomously secrete chemokines to evade immune supervision. This creates an environment known as TME that protects MM cells from immune cells, making them less susceptible to treatment and leading to refractoriness. Thus, clinical practices focus on blocking signaling between MM and normal cells with inhibitors or small molecules to break down these barriers.
Chemokines such as CCL2, CCL3, CCL5, and CXCL12 contribute to immune suppression in MM, leading to chemoresistance. The CCL2-CCR2 axis is responsible for M2 polarization and prevents MM cells from apoptosis caused by chemotherapy drugs [15]. Studies are currently being conducted to address this issue by targeting the CCL2-CCR2 axis associated with the recruitment of TAMs. For instance, carlumab has shown anti-tumor activity in preclinical and clinical trials. However, subsequent tests have shown no long-term suppression of serum CCL2 or significant anti-tumor effects [56].
Considering the protective effect of CCL3 on malignant cells, researchers have combined CCL3-neutralizing antibodies with melphalan and bortezomib to enhance cytotoxicity in MM cells [21]. Inspired by the complicated impact of the CCL3-CCR1 axis, various CCR1 antagonists have been developed to block the CCL3-CCR1 axis, although their effectiveness requires further evaluation [50]. It is revealed that BX471, an antagonist of CCR1, can reverse the reduced erythropoiesis induced by the CCL3-CCR1 axis in ex vivo [14]. Moreover, another CCR1 antagonist, CCX9588, has been proven to prevent malignant cells from migrating to CCL3 in vitro and disseminating to the bone in vivo [18]. Though most modulators are preclinical, CCL3 and CCR1 are potential therapeutic targets in MM and are worth exploring.
Though CCL2 and CCL3 may be potential therapeutic targets in MM, antibodies or small molecules aimed at them have yet to be widely applied in clinical practice. Notably, inhibition of CXCL12 has been confirmed as an efficient approach to managing MM patients and is currently being used. Among CXCR4 antagonists, plerixafor (AMD3100) is the first and the only chemokine modulator approved for treating multiple myeloma patients. Although initially used in stem cell mobilization [8], plerixafor has undergone phase I/II clinical trial with safety and efficacy in combination with bortezomib [6]. Despite investigating the new use of plerixafor, new modulators are being developed and in clinical trials. CXCR4 antagonist motixafortide (BKT140) reveals satisfying outcomes in mobilizing stem cells in phase III trial and is likely to be popularized in the treatment [9]. Besides, the phase Ib/II trial of ulocuplumab also received exhilarating results [7]. Inspired by the success of plerixafor and ulocuplumab and the complicated role of the CXCL12-CXCR4 axis in MM, more efforts are made to explore inhibitors, antagonists, and antibodies aimed at the interaction between CXCL12 and its receptor. Olaptesed pegol (NOX-A12), which can bind to CXCL12, shows benefits in combination with dexamethasone, indicating the strategy to target CXCL12 rather than CXCR4 [57].
Unlike CCL2, CCL3, and CXCL12, CCL5 regulates the formation of MDSCs. To prevent the protective effects of CCL5 on MM cells, immunomodulatory drugs are used to block the CCL5-CCR5 axis and interfere with MDSCs induction [22]. LEN and pomalidomide can downgrade the expression of CCR5 and increase interferon regulatory factor 8 (IRF8) in the mRNA level in peripheral blood mononuclear cells (PBMCs) while also hindering the expression of CCL5 in MM cells. By acting on both normal PBMCs and malignant cells, these drugs can decrease the protection of MDSCs against MM cells and improve disease progression.
In addition to abating chemoresistance in MM, targeting pro-tumor chemokines can relieve bone destruction. CXCL8 participants in osteolytic lesions in MM, so antibodies targeting CXCL8 have been developed to abrogate the CXCL8-CXCR1/2 axis [58,59,60]. However, CXCL8 antibodies have yet to be applied in managing MM patients. Despite small molecules or antibodies directly binding to CXCL8 or CXCR1/2, targeting related signaling to reduce CXCL8 secretion is also feasible. Gefitinib, an EGFR inhibitor, can block AREG-EGFR signaling to relieve bone destruction in MM [31, 61], while JQ1 acts on BMSCs to disturb CXCL8 synthesis and reveal anti-tumor efficacy [52].
Instead of directly focusing on the concentration of chemokines, another perspective considers regulating their bioavailability. As MMP13 regulates the bioavailability of CXCL7, Lo et al. conducted both cell and animal experiments to explore the efficacy of MMP13 inhibitors [13]. The result indicated that MMP13 inhibitors reduced osteoclastogenesis and restrained the growth of malignant cells, leading to improved overall survival. Unlike generally inhibiting CXCL7 or CXCR2, the mechanism of CXCL7 bio-utilization provides a new view to block the CXCL7-CXCR2 axis. Besides using inhibitors or small molecules to interfere with the axis, it is practical to decrease the bioavailability of CXCL7 to improve bone lesions in MM. Some studies on the CXCL12-CXCR4 axis also attempt to regulate CXCL12 concentration via associated signaling pathways. For example, ruxolitinib can block the JAK1/2 pathway and further downgrade the expression of CXCL12 in monocytes and CXCR4 in MM cells when coculturing [53]. Similarly, some studies utilize inhibitors to block responses induced by the CXCL12-CXCR4 axis. Copanlisib, a PI3K inhibitor, can interfere with CXCL12-dependent chemotaxis to reduce fibroblast migration and restrict MM cell chemoresistance [54]. According to the complicated biological function of the CXCL12-CXCR4 axis in MM, further exploration may unveil more signaling pathways that interact with the axis and provide additional potential targets to abate pro-tumor effects.
Studies on CXCL13 have mainly focused on indirect regulation rather than directly targeting CXCL13 or its receptor, CXCR5, alongside CXCL7 and CXCL12. As the interaction between MM cells and macrophages depends on BTK, it is also a promising approach to inhibit pro-inflammatory reactions in macrophages. Ibrutinib, a BTK inhibitor, can reduce abnormal overexpression of CXCL13 [28] in vivo experiments, and the clinical practice achieved satisfying safety and efficacy [62]. Future studies may utilize BTK inhibitors, including ibrutinib, to improve the outcome of patients with refractory MM.
Increasing the concentration of anti-tumor chemokines
Despite decreasing the concentration of pro-tumor chemokines, increasing the concentration of anti-tumor chemokines is also a practical strategy in MM treatments. As some chemokines contribute to eliminating tumors, clinical researchers have focused on arming CAR-T cells with anti-tumor chemokines to enhance their potency in removing malignant cells.
One way to utilize anti-tumor chemokines is by increasing the concentration of CCL19, which is associated with antigen-presenting. In addition to using stem-derived DCs instead of monocyte-derived DCs [26], arming CAR-T cells with CCL19 to enhance cytotoxicity is also worth exploring. CAR-T cells secreting CCL19 and IL-7 have been proven to have higher infiltration of DCs and T cells in tumor tissue [63]. While in MM, Duan et al. designed B-cell maturation antigen (BCMA)-7 × 19 CAR-T cells, which overexpressed CCL19 and IL-7 to cure two patients with refractory MM [16]. BCMA-7 × 19 CAR-T cells revealed delayed terminal differentiation, leading to a higher ratio of stem cell-like memory T cells (Tscms) and durability. Although with more potent cytotoxicity towards tumor cells, BCMA-7 × 19 CAR-T cell therapy was safe with self-limiting and revisable adverse effects. Though the clinical trial had a small sample of only two patients, it is valuable to dig out the potential of BCMA-7 × 19 CAR-T cells in MM management.
Similarly, the addition of CXCL10 has been shown to enhance CAR-T cells’ proliferation, cytotoxicity, and chemotaxis, making it a possible solution for the remaining challenges of CAR-T therapy. CXCL10 can reduce PD-1 expression in CAR-T cells and provides a viable solution to improve the exhaustion of CAR-T cells [17]. However, CXCL10 may adversely affect NK cell localization and potential cytotoxicity, attenuating the efficacy of therapies based on NK cells. Limited by its negative effect on NK cell localization and potential cytotoxicity, the application of CXCL10 to CAR-T therapy may have a long way to go. In a word, exploring anti-tumor chemokines can provide CAR-T therapy with novel targets and strategies to solve current challenges in the infiltration and exhaustion of CAR-T cells.
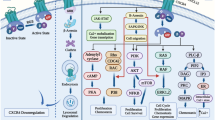
Interactions between chemokines and their receptors interweave a complex network in MM. Blue boxes in the figure indicate the anti-tumor effects of chemokines, while red boxes indicate the pro-tumor effect of chemokines. Malignant cells can utilize the chemokine network to interfere with physiological functions, including erythroid differentiation, osteoclast, and M2 polarization, induce chemoresistance, and downregulate the immune system. MΦs, PBMCs, MDSCs, BMSCs, MSCs, and HSPCs can be influenced and participate in the pathological progression. MM cells can use exosomes, MMPs, HSPGs, or MIF to regulate chemokines and foster TME suitable for themselves. Conversely, chemokines also aid immune cells in migrating into tumor sites, enhancing the immune system and inducing apoptosis of malignant cells. And such function may be applied to develop drugs against MM. MM, multiple myeloma; MΦs, macrophages; PBMCs, peripheral blood mononuclear cells; MDSCs, myeloid-derived suppressor cells; BMSCs, bone marrow stromal cells; MSCs, mesenchymal stromal cells; HSPCs, hematopoietic stem and progenitor cells; MMPs, matrix metalloproteinases; HSPGs, heparan sulfate proteoglycans; MIF, macrophage migratory inhibitory factor; TME, tumor microenvironment.
Future and challenges of chemokines
Interactions between chemokines and their receptors are complex and correlate with pathological progression and prognosis of diseases. Except for MM, chemokine modulators are developed to heal various conditions such as pulmonary fibrosis [64], colon cancer [65], multiple sclerosis (MS) [66], and rheumatoid arthritis (RA) [67,68,69], indicating the potential of chemokines as drug targets.
Iceberg theory is quite suitable for describing modulators’ developments in MM (Fig. 3). IL-6, matrix metalloproteinases (MMPs), and vascular endothelial growth factor (VEGF) are potential targets in MM, which lay in part close to the water surface. Though inhibition in clinical trials did not receive satisfying outcomes [70,71,72,73,74,75,76], further studies may attempt to find specific patients sensitive to these modulators, explore combinations with other drugs, or develop more efficient modulators. In contrast, G protein-coupled receptor class C group 5 member D (GPRC5D) [77], CD38 [78, 79], signaling lymphocytic activation molecule F7 (SLAMF7) [80,81,82], and BCMA [83,84,85] showed favorable results. Modulators targeting them are in the upper layers; some are already in clinical use (Table 2).

Target inhibitors, antagonists, or antibodies in multiple myeloma
Among drugs aimed at chemokines, only drugs targeting the CXCL12-CXCR4 axis have been used in clinical practice. In contrast, most neutralizing antibodies, antagonists, or inhibitors are still in the stage of experiment or development. Plerixafor is at the tip of the iceberg, which has been uncovered and applied already, while motixafortide has passed the phase III trial and will surface. Early studies mainly utilized plerixafor to mobilize CD34 + hematopoietic cells for ACT [8]. Aimed at reducing the failure of mobilization of autologous hematopoietic cells, motixafortide, known as BKT140, has been developed and has passed phase III study recently [9], which may be a substitution for plerixafor. As scientists have a deeper insight into the CXCL12-CXCR4 axis in MM, a phase I/II clinical research innovatively combined plerixafor with bortezomib to sensitize MM cells and improve outcomes [6]. In addition to plerixafor, ulocuplumab, another CXCR4 antagonist, is also proven efficient in increasing the response rate in refractory MM in phase II clinical study [7]. Regarding the CXCL12-CXCR4 axis, some modulators targeting CXCL12 instead of CXCR4 are also worth further exploring. Olaptesed pegol is a CXCL12 inhibitor that could improve response rates in combination with bortezomib and dexamethasone [57].
Except for CXCL12-CXCR4, CCL3-CCR1 is another hotspot axis, whereas modulators targeting CCR1 still have a long way to go. Given that CCL3-CCR1 participates in inflammation, various antagonists or inhibitors of CCR1 are developed to cure diseases associated with immune disorders. Among them, only CCX354, which is designed for RA, passed phase II [67]; nevertheless, AZD-4818 [88], BMS-817399 [89], and CP-481715 [90] are proved to be either toxic in phase I or limited efficacy in phase II. Besides, most CCR1 antagonists remain uncertain of their efficacy and are in the preclinical phase or phase I [18, 65, 91,92,93,94,95,96,97]. Thus, modulating targeting CCL3-CCR1 resembles the enormous part of the iceberg buried undersea and has great potential to explore. Considering the recent discovery of CXCL13-CXCR5 and the lack of specific inhibitors, the CXCL13-CXCR5 axis is also worth unearthing. Other chemokine-chemokine receptor axes, including CCL5-CCR1, CCL2-CCR2, and CXCL9/10-CXCR3, are also under study, though results revealed the need for improvements of their modulators [55, 56, 64, 66]. Inspired by the newly developed therapy of plerixafor and the appearance of motixafortide, future studies may either invent new modulators targeting chemokines to regulate their biological function or propose innovative uses of drugs approved for market.
Exosomes or proteinases also associates with chemokines [13, 31]. Immunomodulators are proven to participate in regulating the chemokine network [22], while clinical practices that apply anti-tumor chemokines to improve CAR-T therapy demonstrate the feasibility of combining chemokines with ACT [16]. With profound knowledge, more available drugs and treatments would emerge and benefit MM patients. More efforts should be made to probe into the function and pathological mechanism of chemokines in MM to better support the development of new applications of chemokines.
Conclusion
As chemokines regulate the migration of immune cells, targeting chemokines may provide a possible solution to remodel the TME, activate immunoreaction, and promote the clearance of malignant cells. Studies have tried to block the interaction of pro-tumor chemokine ligands and receptors to alleviate the complications and progress in MM. However, chemokines can interact with multiple receptors to take effect. When antagonists or inhibitors block specific receptors, chemokines may bind to alternative receptors to activate related signaling pathways. Thus, some inhibitors or antagonists targeting single chemokine ligands or receptors may not achieve the expected effect. Combining these inhibitors or antagonists with other drugs is a promising strategy to improve their efficacy. By contrast, increasing anti-tumor chemokines to enhance the clearance of tumor cells is another practical strategy. Chemokines can recruit and activate immune cells, which makes it a possible solution for the exhaustion of CAR-T cells. Arming CAR-T cells with anti-tumor chemokines reveals feasibility and is worth further exploration [16]. Considering exosomes or proteinases are also associated with chemokines [13, 31], research on chemokines can inspire the development of other inhibitors in MM, which may contribute to more potential targets. Finally, probing into chemokines will supplement the mechanism of the current treatment, such as immunomodulators, and better guide the clinical practice. To utilize chemokines to treat MM, more research focused on the related signaling pathways of chemokines should be done. With a comprehensive knowledge of chemokine/chemokine receptor axes, researchers can better take advantage of chemokines to relieve and treat MM.
CXCR4 inhibitor plerixafor is the only chemokine modulator put into clinical use, while motixafortide has passed phase III trial recently. Modulators targeting CXCL12(SDF-1)-CXCR4 have promising potential in development and clinical use. Although F50067, a CXCR4 antagonist, was observed to be toxic in the phase I study, other modulators targeting CXCR4 and CXCL12 reveal safety and efficacy in clinical tests. Other modulators targeting BCMA, CD38, and SLAMF7 are also approved for treating MM patients. Talquetamab, a GPRC5D inhibitor, is in the phase III trial now. By comparison, there are some difficulties in developing VEGF inhibitors with limited efficacy. Inhibition of targets such as CCL3-CCR1, CCL5-CCR1, CXCL10-CXCR3, CXCL13-CXCR5, MMPs, and CCL2-CCR2 may also be developed and applied.
Availability of data and materials
Not applicable.
Abbreviations
- MM:
-
Multiple myeloma
- PCs:
-
Plasma cells
- C:
-
Cysteine
- TME:
-
Tumor microenvironment
- MΦs:
-
Macrophages
- Tregs:
-
Regulatory T cells
- DCs:
-
Dendritic cells
- TAMs:
-
Tumor-associated macrophages
- MDSCs:
-
Myeloid-derived suppressor cells
- MCP-1:
-
Monocyte chemoattractant protein-1
- MCPIP1:
-
MCP-1 induced protein
- MIP-1α:
-
Macrophage inflammatory protein-1α
- PBMCs:
-
Peripheral blood mononuclear cells
- LEN:
-
Lenalidomide
- HSPCs:
-
Hematopoietic stem and progenitor cells
- MMP13:
-
Matrix metalloproteinase-13
- MSCs:
-
Mesenchymal stromal cells
- IL-8:
-
Interleukin-8
- AREG:
-
Amphiregulin
- EGFR:
-
Epidermal growth factor receptor
- BMSCs:
-
Bone marrow stromal cells
- BTK:
-
Bruton’s tyrosine kinase
- RANKL:
-
Receptor activator of nuclear kappa B ligand
- SDF-1:
-
Stromal cell-derived factor-1
- PI3K:
-
Phosphoinositide 3-kinase
- PKB:
-
Protein kinase B
- IL-6:
-
Interleukin-6
- HSPGs:
-
Heparan sulfate proteoglycans
- cPCs:
-
Circulating plasma cells
- PF-4:
-
Platelet factor 4
- NK:
-
Natural killer
- ACT:
-
Adoptive cellular therapy
- IRF8:
-
Interferon regulatory factor 8
- PBMCs:
-
Peripheral blood mononuclear cells
- CAR:
-
Chimeric antigen receptor
- BCMA:
-
B-cell maturation antigen
- Tscms:
-
Stem cell-like memory T cells
- MIF:
-
Macrophage migratory inhibitory factor
- RA:
-
Rheumatoid arthritis
- MS:
-
Multiple sclerosis
- mAb:
-
Monoclonal antibody
- Mo-DCs:
-
Monocyte-derived dendritic cells
- SC-DCs:
-
Stem cell-derived dendritic cells
- HMCLs:
-
Human myeloma-derived cell lines
- VEGF:
-
Vascular endothelial growth factor
- MMPs:
-
Matrix metalloproteinases
- GPRC5D:
-
G protein-coupled receptor class C group 5 member D
- SLAMF7:
-
Signaling lymphocytic activation molecule F7
- CDC:
-
Complement-dependent cytotoxicity
- ADCC:
-
Antibody-dependent cellular cytotoxicity
- aHSCs:
-
Autologous hematopoietic stem cells
- VEGFR:
-
Vascular endothelial growth factor receptor
- RTKI:
-
Tyrosine kinase inhibitor
- PDGFR:
-
Platelet-derived growth factor receptor
- ADPC:
-
Antibody-dependent cellular cytotoxicity
- ADC:
-
Antibody-drug conjugate
References
van de Donk NWCJ, Pawlyn C, Yong KL. Multiple myeloma. Lancet. 2021;397(10272):410–27.
Huang J, et al. The epidemiological landscape of multiple myeloma: a global cancer registry estimate of disease burden, risk factors, and temporal trends. Lancet Haematol. 2022;9(9):e670–7.
Blimark CH, et al. Outcome and survival of myeloma patients diagnosed 2008–2015. Real-world data on 4904 patients from the Swedish Myeloma Registry. Haematologica. 2018;103(3):506–13.
Libby E, et al. Disease-specific survival for patients with multiple myeloma: significant improvements over time in all age groups. Leuk Lymphoma. 2014;55(12):2850–7.
Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. 2014;32:659–702.
Ghobrial IM, et al. Phase I/II trial of the CXCR4 inhibitor plerixafor in combination with bortezomib as a chemosensitization strategy in relapsed/refractory multiple myeloma. Am J Hematol. 2019;94(11):1244–53.
Ghobrial IM, et al. A phase Ib/II trial of the first-in-class anti-CXCR4 antibody ulocuplumab in combination with lenalidomide or bortezomib plus dexamethasone in relapsed multiple myeloma. Clin Cancer Res. 2020;26(2):344–53.
DiPersio J, et al. A phase III, multicenter, randomized, double-blind, placebo-controlled, comparative trial of AMD3100 (Plerixafor)+G-CSF vs. G-CSF+Placebo for mobilization in multiple myeloma (MM) patients for autologous hematopoietic stem cell (aHSC) transplantation. Blood. 2007;110(11):445.
Crees ZD, et al. Motixafortide and G-CSF to mobilize hematopoietic stem cells for autologous transplantation in multiple myeloma: a randomized phase 3 trial. Nat Med. 2023;29(4):869–79.
Xue D, et al. Role of chemokines in hepatocellular carcinoma (Review). Oncol Rep. 2021;45(3):809–23.
Dobroch J, et al. The exploration of chemokines importance in the pathogenesis and development of endometrial cancer. Molecules. 2022;27(7):2041.
Zou Q, et al. Chemokines in progression, chemoresistance, diagnosis, and prognosis of colorectal cancer. Front Immunol. 2022;13:724139.
Lo CH, et al. Host-derived matrix metalloproteinase-13 activity promotes multiple myeloma-induced osteolysis and reduces overall survival. Can Res. 2021;81(9):2415–28.
Liu L, et al. Multiple myeloma hinders erythropoiesis and causes anaemia owing to high levels of CCL3 in the bone marrow microenvironment. Sci Rep. 2020;10(1):20508.
Xu R, et al. CCL2 promotes macrophages-associated chemoresistance via MCPIP1 dual catalytic activities in multiple myeloma. Cell Death Dis. 2019;10(10):781.
Duan D, et al. The BCMA-targeted fourth-generation CAR-T cells secreting IL-7 and CCL19 for therapy of refractory/recurrent multiple myeloma. Front Immunol. 2021;12:609421.
Liu T, et al. IP-10 enhances the amplification capacity and antitumor activity of CAR-T cells in vitro and could influence positive outcomes in MM patients treated with CAR-T cell therapy. Int Immunopharmacol. 2022;112:109253.
Zeissig MN, et al. Expression of the chemokine receptor CCR1 promotes the dissemination of multiple myeloma plasma cells in vivo. Haematologica. 2020;106(12):3176–87.
Bianconi V, et al. The regulation and importance of monocyte chemoattractant protein-1. Curr Opin Hematol. 2018;25(1):44–51.
Xu M, et al. Role of the CCL2-CCR2 signalling axis in cancer: mechanisms and therapeutic targeting. Cell Prolif. 2021;54(10):e13115.
Tsubaki M, et al. The MIP-1alpha autocrine loop contributes to decreased sensitivity to anticancer drugs. J Cell Physiol. 2018;233(5):4258–71.
Kuwahara-Ota S, et al. Lenalidomide and pomalidomide potently interfere with induction of myeloid-derived suppressor cells in multiple myeloma. Br J Haematol. 2020;191(5):784–95.
Vandyke K, et al. HIF-2alpha promotes dissemination of plasma cells in multiple myeloma by regulating CXCL12/CXCR4 and CCR1. Cancer Res. 2017;77(20):5452–63.
Ricart BG, et al. Dendritic cells distinguish individual chemokine signals through CCR7 and CXCR4. J Immunol. 2011;186(1):53–61.
Haessler U, et al. Dendritic cell chemotaxis in 3D under defined chemokine gradients reveals differential response to ligands CCL21 and CCL19. Proc Natl Acad Sci U S A. 2011;108(14):5614–9.
Shinde P, et al. Autologous hematopoietic stem cells are a preferred source to generate dendritic cells for immunotherapy in multiple myeloma patients. Front Immunol. 2019;10:1079.
Teijeira A, et al. IL8, neutrophils, and NETs in a collusion against cancer immunity and immunotherapy. Clin Cancer Res. 2021;27(9):2383–93.
Beider K, et al. CXCL13 chemokine is a novel player in multiple myeloma osteolytic microenvironment, M2 macrophage polarization, and tumor progression. J Hematol Oncol. 2022;15(1):144.
Ren Z, et al. The CXCL12gamma chemokine immobilized by heparan sulfate on stromal niche cells controls adhesion and mediates drug resistance in multiple myeloma. J Hematol Oncol. 2021;14(1):11.
Geng S, et al. Single-cell RNA sequencing reveals chemokine self-feeding of myeloma cells promotes extramedullary metastasis. FEBS Lett. 2020;594(3):452–65.
Raimondo S, et al. Multiple myeloma-derived exosomes are enriched of amphiregulin (AREG) and activate the epidermal growth factor pathway in the bone microenvironment leading to osteoclastogenesis. J Hematol Oncol. 2019;12(1):2.
Schenk BI, et al. Platelet-derived chemokines CXC chemokine ligand (CXCL)7, connective tissue-activating peptide III, and CXCL4 differentially affect and cross-regulate neutrophil adhesion and transendothelial migration1. J Immunol. 2002;169(5):2602–10.
Goto Y, et al. CXCR4+CD45− cells are niche forming for osteoclastogenesis via the SDF-1, CXCL7, and CX3CL1 signaling pathways in bone marrow. Stem Cells. 2016;34(11):2733–43.
Allegra A, et al. Changes in serum interleukin-8 and sRAGE levels in multiple myeloma patients. Anticancer Res. 2020;40(3):1443–9.
Kamalakar A, et al. Circulating interleukin-8 levels explain breast cancer osteolysis in mice and humans. Bone. 2014;61:176–85.
Hussain SK, et al. Serum levels of the chemokine CXCL13, genetic variation in CXCL13 and its receptor CXCR5, and HIV-associated non-hodgkin B-cell lymphoma risk. Cancer Epidemiol Biomarkers Prev. 2013;22(2):295–307.
Bürkle A, et al. Overexpression of the CXCR5 chemokine receptor, and its ligand, CXCL13 in B-cell chronic lymphocytic leukemia. Blood. 2007;110(9):3316–25.
Kurtova AV, et al. Mantle cell lymphoma cells express high levels of CXCR4, CXCR5, and VLA-4 (CD49d): importance for interactions with the stromal microenvironment and specific targeting. Blood. 2009;113(19):4604–13.
Zhang G, et al. Mesenchymal stem cells from bone marrow regulate invasion and drug resistance of multiple myeloma cells by secreting chemokine CXCL13. Bosn J Basic Med Sci. 2020;20(2):209–17.
Li L, et al. Chemokine receptor CXCR4: an important player affecting the molecular-targeted drugs commonly used in hematological malignancies. Expert Rev Hematol. 2020;13(12):1387–96.
Peled A, et al. Role of CXCL12 and CXCR4 in the pathogenesis of hematological malignancies. Cytokine. 2018;109:11–6.
Liu Y, et al. Blockade of SDF-1/CXCR4 reduces adhesion-mediated chemoresistance of multiple myeloma cells via interacting with interleukin-6. J Cell Physiol. 2019;234(11):19702–14.
Rueda P, et al. The CXCL12γ chemokine displays unprecedented structural and functional properties that make it a paradigm of chemoattractant proteins. PLoS One. 2008;3(7):e2543.
Bladé J, de Larrea CF, Rosiñol L. Extramedullary involvement in multiple myeloma. Haematologica. 2012;97(11):1618–9.
Pei L, et al. Platelet factor 4 induces cell apoptosis by inhibition of STAT3 via up-regulation of SOCS3 expression in multiple myeloma. Haematologica. 2013;98(2):288–95.
Bai J, et al. Serum platelet factor 4 is a promising predictor in newly diagnosed patients with multiple myeloma treated with thalidomide and VAD regimens. Hematology. 2019;24(1):387–91.
Tokunaga R, et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - a target for novel cancer therapy. Cancer Treat Rev. 2018;63:40–7.
Qian C, et al. TLR agonists induce regulatory dendritic cells to recruit Th1 cells via preferential IP-10 secretion and inhibit Th1 proliferation. Blood. 2006;109(8):3308–15.
Ponzetta A, et al. Multiple myeloma impairs bone marrow localization of effector natural killer cells by altering the chemokine microenvironment. Can Res. 2015;75(22):4766–77.
Gilchrist A, Echeverria SL. Targeting chemokine receptor CCR1 as a potential therapeutic approach for multiple myeloma. Front Endocrinol (Lausanne). 2022;13:846310.
Markl F, et al. Utilizing chemokines in cancer immunotherapy. Trends Cancer. 2022;8(8):670–82.
Piddock RE, et al. Myeloma-derived macrophage inhibitory factor regulates bone marrow stromal cell-derived IL-6 via c-MYC. J Hematol Oncol. 2018;11(1):66.
Chen H, et al. JAK1/2 pathway inhibition suppresses M2 polarization and overcomes resistance of myeloma to lenalidomide by reducing TRIB1, MUC1, CD44, CXCL12, and CXCR4 expression. Br J Haematol. 2020;188(2):283–94.
Okabe S, et al. Copanlisib, a novel phosphoinositide 3-kinase inhibitor, combined with carfilzomib inhibits multiple myeloma cell proliferation. Ann Hematol. 2019;98(3):723–33.
Bonanni V, et al. Targeting of CXCR3 improves anti-myeloma efficacy of adoptively transferred activated natural killer cells. J Immunother Cancer. 2019;7(1):290.
Brana I, et al. Carlumab, an anti-C-C chemokine ligand 2 monoclonal antibody, in combination with four chemotherapy regimens for the treatment of patients with solid tumors: an open-label, multicenter phase 1b study. Target Oncol. 2015;10(1):111–23.
Ludwig H, et al. Olaptesed pegol, an anti-CXCL12/SDF-1 Spiegelmer, alone and with bortezomib-dexamethasone in relapsed/refractory multiple myeloma: a phase IIa study. Leukemia. 2017;31(4):997–1000.
Bilusic M, et al. Phase I trial of HuMax-IL8 (BMS-986253), an anti-IL-8 monoclonal antibody, in patients with metastatic or unresectable solid tumors. J Immunother Cancer. 2019;7(1):240.
Alraouji NN, Aboussekhra A. Tocilizumab inhibits IL-8 and the proangiogenic potential of triple negative breast cancer cells. Mol Carcinog. 2021;60(1):51–9.
Mahler DA, et al. Efficacy and safety of a monoclonal antibody recognizing interleukin-8 in COPD: a pilot study. Chest. 2004;126(3):926–34.
Normanno N, et al. Gefitinib inhibits the ability of human bone marrow stromal cells to induce osteoclast differentiation: implications for the pathogenesis and treatment of bone metastasis. Endocrine-Related Cancer Endocr Relat Cancer. 2005;12(2):471–82.
Richardson PG, et al. Ibrutinib alone or with dexamethasone for relapsed or relapsed and refractory multiple myeloma: phase 2 trial results. Br J Haematol. 2018;180(6):821–30.
Adachi K, et al. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat Biotechnol. 2018;36(4):346–51.
Raghu G, et al. CC-chemokine ligand 2 inhibition in idiopathic pulmonary fibrosis: a phase 2 trial of carlumab. Eur Respir J. 2015;46(6):1740–50.
Kitamura T, et al. Inactivation of chemokine (C-C motif) receptor 1 (CCR1) suppresses colon cancer liver metastasis by blocking accumulation of immature myeloid cells in a mouse model. Proc Natl Acad Sci U S A. 2010;107(29):13063–8.
Reuss R, et al. No significant effect of orally administered chemokine receptor 1 antagonist on intercellular adhesion molecule-3 expression in relapsing–remitting multiple sclerosis patients. Mult Scler. 2010;16(3):366–9.
Paul PT, et al. Chemokine receptor CCR1 antagonist CCX354-C treatment for rheumatoid arthritis: CARAT-2, a randomised, placebo controlled clinical trial. Ann Rheum Dis. 2013;72(3):337.
Kivitz A, et al. THU0109 lack of efficacy of CCR1 antagonist BMS-817399 in patients with moderate to severe rheumatoid arthritis: results of 12-week proof-of-concept study. Ann Rheum Dis. 2014;73(Suppl 2):215.
Baum P, et al. THU0128 evaluation of safety, pharmacokinetics and pharmacodynamics of BI 638683, a novel CCR1 antagonist. Ann Rheum Dis. 2014;73(Suppl 2):223.
Brighton TA, et al. Randomized, double-blind, placebo-controlled, multicenter study of siltuximab in high-risk smoldering multiple myeloma. Clin Cancer Res. 2019;25(13):3772–5.
Kovacs MJ, et al. A phase II study of ZD6474 (Zactima, a selective inhibitor of VEGFR and EGFR tyrosine kinase in patients with relapsed multiple myeloma–NCIC CTG IND.145. Invest New Drugs. 2006;24(6):529–35.
Srkalovic G, et al. A phase II trial of BAY 43–9006 (sorafenib) (NSC-724772) in patients with relapsing and resistant multiple myeloma: SWOG S0434. Cancer Med. 2014;3(5):1275–83.
Somlo G, et al. Phase II randomized trial of bevacizumab versus bevacizumab and thalidomide for relapsed/refractory multiple myeloma: a California Cancer Consortium trial. Br J Haematol. 2011;154(4):533–5.
White D, et al. Results from AMBER, a randomized phase 2 study of bevacizumab and bortezomib versus bortezomib in relapsed or refractory multiple myeloma. Cancer. 2013;119(2):339–47.
Zangari M, et al. Phase II study of SU5416, a small molecule vascular endothelial growth factor tyrosine kinase receptor inhibitor, in patients with refractory multiple myeloma. Clin Cancer Res. 2004;10(1 Pt 1):88–95.
Prince HM, et al. Vascular endothelial growth factor inhibition is not an effective therapeutic strategy for relapsed or refractory multiple myeloma: a phase 2 study of pazopanib (GW786034). Blood. 2009;113(19):4819–20.
Chari A, et al. Talquetamab, a T-cell-redirecting GPRC5D bispecific antibody for multiple myeloma. N Engl J Med. 2022;387(24):2232–44.
Usmani SZ, et al. Carfilzomib, dexamethasone, and daratumumab versus carfilzomib and dexamethasone for patients with relapsed or refractory multiple myeloma (CANDOR): updated outcomes from a randomised, multicentre, open-label, phase 3 study. Lancet Oncol. 2022;23(1):65–76.
Martin T, et al. Isatuximab, carfilzomib, and dexamethasone in patients with relapsed multiple myeloma: updated results from IKEMA, a randomized Phase 3 study. Blood Cancer J. 2023;13(1):72.
Dimopoulos MA, et al. Elotuzumab plus lenalidomide/dexamethasone for relapsed or refractory multiple myeloma: ELOQUENT-2 follow-up and post-hoc analyses on progression-free survival and tumour growth. Br J Haematol. 2017;178(6):896–905.
Dimopoulos MA, et al. Elotuzumab, lenalidomide, and dexamethasone in RRMM: final overall survival results from the phase 3 randomized ELOQUENT-2 study. Blood Cancer J. 2020;10(9):91.
Dimopoulos MA, et al. Addition of elotuzumab to lenalidomide and dexamethasone for patients with newly diagnosed, transplantation ineligible multiple myeloma (ELOQUENT-1): an open-label, multicentre, randomised, phase 3 trial. Lancet Haematol. 2022;9(6):e403–14.
Kang C. Teclistamab: first approval. Drugs. 2022;82(16):1613–9.
Grosicki S, et al. Elranatamab in combination with daratumumab for patients (pts) with relapsed/refractory multiple myeloma (RRMM): results from the Phase 3 Magnetismm-5 study safety lead-in cohort. Blood. 2022;140(Supplement 1):4407–8.
Martino EA, Bruzzese A, Iaccino E, Labanca C, Mendicino F, Mimmi S, Lucia E, Olivito V, Neri A, Morabito F, Vigna E, Gentile M. Belantamab mafodotin in multiple myeloma. Expert Opin Biol Ther. 2023;23(11):1043–7. https://doi.org/10.1080/14712598.2023.2218543.
Roccaro AM, et al. SDF-1 inhibition targets the bone marrow niche for cancer therapy. Cell Rep. 2014;9(1):118–28.
Fouquet G, et al. Phase I dose-escalation study of F50067, a humanized anti-CXCR4 monoclonal antibody alone and in combination with lenalidomide and low-dose dexamethasone, in relapsed or refractory multiple myeloma. Oncotarget. 2018;9(35):23890–9.
Kerstjens HA, et al. Tolerability and efficacy of inhaled AZD4818, a CCR1 antagonist, in moderate to severe COPD patients. Respir Med. 2010;104(9):1297–303.
Santella JB 3rd, et al. Discovery of the CCR1 antagonist, BMS-817399, for the treatment of rheumatoid arthritis. J Med Chem. 2014;57(18):7550–64.
Borregaard J, et al. Evaluation of the effect of the specific CCR1 antagonist CP-481715 on the clinical and cellular responses observed following epicutaneous nickel challenge in human subjects. Contact Dermatitis. 2008;59(4):212–9.
Dairaghi DJ, et al. CCR1 blockade reduces tumor burden and osteolysis in vivo in a mouse model of myeloma bone disease. Blood. 2012;120(7):1449–57.
Vallet S, et al. MLN3897, a novel CCR1 inhibitor, impairs osteoclastogenesis and inhibits the interaction of multiple myeloma cells and osteoclasts. Blood. 2007;110(10):3744–52.
Merritt JR, et al. Novel pyrrolidine ureas as C-C chemokine receptor 1 (CCR1) antagonists. J Med Chem. 2009;52(5):1295–301.
Merritt JR, et al. Novel pyrrolidine heterocycles as CCR1 antagonists. Bioorg Med Chem Lett. 2010;20(18):5477–9.
Amat M, et al. Pharmacological blockade of CCR1 ameliorates murine arthritis and alters cytokine networks in vivo. Br J Pharmacol. 2006;149(6):666–75.
Sabroe I, et al. A small molecule antagonist of chemokine receptors CCR1 and CCR3. Potent inhibition of eosinophil function and CCR3-mediated HIV-1 entry. J Biol Chem. 2000;275(34):25985–92.
Lionakis Michail S, et al. Pharmacological blockade of the chemokine receptor CCR1 protects mice from systemic candidiasis of hematogenous origin. Antimicrob Agents Chemother. 2017;61(3):e02365-e2416.
Acknowledgements
Not applicable.
Funding
This project was funded by Shanghai Shenkang Hospital Development Center (No. SHDC2020CR2070B).
Author information
Authors and Affiliations
Contributions
JD designed this study and guided the entire process. ZL wrote the manuscript and prepared Tables 1-2 and Figures 1-3. JD, XF, and XG edited the original draft, tables, and figures. JD, XF, XG, GL, and JH supervised the project and ultimately approved the manuscript. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Du, J., Lin, Z., Fu, XH. et al. Research progress of the chemokine/chemokine receptor axes in the oncobiology of multiple myeloma (MM). Cell Commun Signal 22, 177 (2024). https://doi.org/10.1186/s12964-024-01544-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-024-01544-7