
Abstract
T cell-based adoptive cell therapy (ACT) has exhibited excellent antitumoral efficacy exemplified by the clinical breakthrough of chimeric antigen receptor therapy (CAR-T) in hematologic malignancies. It relies on the pool of functional T cells to retain the developmental potential to serially kill targeted cells. However, failure in the continuous supply and persistence of functional T cells has been recognized as a critical barrier to sustainable responses. Conferring stemness on infused T cells, yielding stem cell-like memory T cells (TSCM) characterized by constant self-renewal and multilineage differentiation similar to pluripotent stem cells, is indeed necessary and promising for enhancing T cell function and sustaining antitumor immunity. Therefore, it is crucial to identify TSCM cell induction regulators and acquire more TSCM cells as resource cells during production and after infusion to improve antitumoral efficacy. Recently, four common cytokine receptor γ chain (γc) family cytokines, encompassing interleukin-2 (IL-2), IL-7, IL-15, and IL-21, have been widely used in the development of long-lived adoptively transferred TSCM in vitro. However, challenges, including their non-specific toxicities and off-target effects, have led to substantial efforts for the development of engineered versions to unleash their full potential in the induction and maintenance of T cell stemness in ACT. In this review, we summarize the roles of the four γc family cytokines in the orchestration of adoptively transferred T cell stemness, introduce their engineered versions that modulate TSCM cell formation and demonstrate the potential of their various combinations.
Video Abstract
Similar content being viewed by others


Background
Cancer is the leading cause of death globally and a predominant obstacle to increasing life expectancy [1, 2]. The emergence of immunotherapy has revolutionized cancer treatment and offers more treatment options for patients with cancer [3, 4]. Tumor immunotherapy mainly consists of immune checkpoint blockade (ICB) and adoptive T-cell therapy (ACT). ACT, including chimeric antigen receptor therapy (CAR-T) and engineered T-cell receptor T cell therapy (TCR-T), relies on targeted destruction of cancer cells by potent antitumor T cells associated with the CD8(+) T cell state [5,6,7,8,9]. Despite the substantial antitumor activity in hematological tumors, adoptively transferred CAR-T cells have a limited effect in solid tumors, mainly due to poor expansion and persistence in vivo [8, 10,11,12,13,14,15,16]. During chronic stimulation of tumor antigens, adoptively transferred T cells are inevitably exhausted and exhibit an anergic state of cytotoxicity loss by virtue of the progressive expression of co-inhibitory molecules such as PD-1, TIM-3, LAG-3, CTLA-4, and TIGIT [6, 13]. As a result, approaches to acquiring long-lived functional T cells with stem cell-like properties, termed stem cell-like memory T cells (TSCM), should be urgently developed for CAR-T therapy.
T cell stemness is termed to describe the stem cell-like behavior of T cells, including self-renewal, multipotency, and functional persistence. TSCM cells, featured by CD45RA+ CD45RO− CD27+ CD28+ CCR7+ CD62L+ CD95+ CD122+ CD127+, were first discovered in mouse models of human graft versus host disease (GVHD) in 2005 and isolated in vitro in 2013 [5, 17,18,19,20,21]. As a unique subset of memory T cells, apart from memory traits, TSCM cells receive the stem cell-like attributes, that are the self-renewal and multipotent ability to continually generate all memory and effector T cell subsets. To identify TSCM cells with analogous properties, Gattinoni et al. stimulated naïve T cells by triggering Wnt signaling with Wnt3A or inhibitors of glycogen synthase kinase-3β (GSK-3β) TWS119 [20, 21].
At present, steady progress has been made with respect to the induction of T cell stemness. Of note, many factors involved in the generation and maintenance of TSCM cells are known, such as Notch [22,23,24], Wnt [25,26,27], mTOR [28], and cGAS-STING [29] signaling pathways, cytokines, and transcriptional factor c-Myb [5, 30, 31]. Thereinto, cytokines like IL-7 and IL-15 were listed among the top twelve immunotherapeutic agents with wide appeal to the immunotherapy and, by consensus, held particular promise for use in cancer therapy, as shown by the US National Cancer Institute in 2008 [32]. With the emergence of cytokine therapy for cancer, the four cytokines of the common cytokine receptor γ chain (γc, CD132) family, containing interleukin-2 (IL-2), IL-7, IL-15, and IL-21 are dictated to regulate the T cell stemness formation and maintenance. They serve as the third signal that triggers the antigen-specific immunological response and are theoretically demonstrated as essential factors to coordinate the differentiation and the cytotoxicity of CD8(+) T cells via the formation of the tight immunological synapse [33]. Thus, the cytokine milieu plays a fate-defining role for T cells. The four γc family cytokines alone or their different combination may substantially affect the modulation of adoptively transferred T cell stemness. Applying these four γc family cytokines to adoptively transferred T cell cultivation in vitro with various combination protocols will promote the expansion of TSCM cells with enhanced capacities to engraft, persist and mediate prolonged immune attacks against tumor masses. In addition to acquiring TSCM during the manufacturing phase ex vivo, they are expected to maintain and expand TSCM after co-administration with autologous T cells. Nevertheless, the administration of these wild-type cytokines is associated with some obstacles such as non-specific toxicities, off-target effect, and inefficiency, and their engineered versions may make up for these deficiencies to a large extent. In this review, we outline the potential of the stemness of the transferred T cells and summarize the roles of wild-type IL-2, IL-7, IL-15 and IL-21 belonging to γc family cytokines in the production, maintenance, and expansion of T cells with stemness in ACT. We also introduce their engineered types prompting TSCM induction and discuss the limitations and future directions of incorporating the four cytokines in stemness induction for T cell-based cancer immunotherapy.
Potential of T cell stemness in ACT
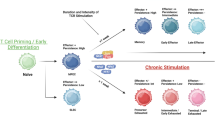
The efficacy of ACT largely depends on the status of adoptively transferred T cells. Low-differentiated T cells with stemness have elicited a significant superiority over conventionally activated T cells in tumor control, owing to the capability of enhanced self-renewal and persistence, as well as the rapid generation of effector subsets in vivo [34,35,36]. Regardless of the status, adoptive TSCM cells and conventionally activated T cells without stemness are in the same suppressive tumor microenvironment (TME) and share a typical terminal response process, i.e., “transient cytotoxicity, consistent exhaustion, and ultimate apoptosis.” After infusion, adoptively transferred T cells are activated completely and dominantly differentiate into cytotoxic T lymphocytes (CTL), followed by the release of a bulk of cytotoxic molecules, such as perforin, granzyme B, and γ-interferon (γ-IFN), into the targeted synapse to achieve an ideal hit of tumor cell eradication. However, under an immunosuppressive microenvironment and continuous tumor antigen stimulation, most activated CD8(+) T cells upregulate the expression of co-inhibitory markers and exhibit an exhausted phenotype, culminating in a stepwise loss of cytotoxicity and non-response to immunotherapy [6, 13, 37] (Fig. 1). Beyond that, functional CTLs are subjected to activation-induced cell death (AICD) via high expression of the Fas/FasL axis, which further prevents an excessive immune response [16]. Remarkably, TSCM cells with memory traits mediate a faster and stronger recall response on a lower threshold of antigen restimulation. Hence, a limited number of engineered T cells without stemness execute the limited tumor-killing effect and unavoidably go forward to exhaustion and apoptosis without sufficient supplements, whereas TSCM cells preserve the ability of self-renewal and differentiation into better effector T cells to mount a robust antitumor response after substantial expansion (Fig. 1).

The potential of TSCM for ACT. The limited adoptively transferred T cells inevitably experience apoptosis and exhaustion. Conventional infused effector T cells demonstrate transient cytotoxicity due to lack of complement, whereas infused TSCM cells induced by the four γc family cytokines continually self-renew and produce enough effectors to mount a robust and persistent immune attack. ACT, adoptively cell therapy; TEF, effector T cells; TSCM, stem cell-like T cells; Tex, exhausted T cells
TSCM cells have been elucidated to trigger complete tumor regression and durable response in hematopoietic malignancies and solid cancers [27, 34, 38,39,40,41,42]. In human B-cell malignancies, CD19-CAR-modified TSCM cells present enhanced metabolic fitness and mediate long-lasting antitumor responses [39]. In patients with non-small cell lung cancer (NSCLC), TSCM cells are located in peripheral blood, producing antitumor molecules. Relatively fewer TSCM cells are found in lymph nodes, contributing to faster recall responses against cancer cells [43]. Therefore, conferring stemness to antitumor T cells might unleash the full potential of immunotherapies based on CD8(+) T cells. Disappointingly, terminally differentiated T cells are commonly enriched in the TME after ACT, and a lower TSCM state of tumor-infiltrating T lymphocytes (TILs) exists in vivo [42]. Intra-tumor immune niches in which TSCM cells reside are commonly deficient in patients with progressive tumors [44]. Thus, augmenting the pool of TSCM cells, either by isolating and expanding intrinsic stem-like neoantigen-specific T cells or by engineering T cells to acquire stem-like attributes in vitro might provide promising opportunities for developing more effective T cell-based immunotherapies. Harnessing the generation of more TSCM cells via γc family cytokines might lead to the development of potent cancer immunotherapy.
Four cytokines of the γc family regulate T cell stemness
Cytokines containing the γc family serve as a communicative bridge among immune cells and non-immune cells in the TME, providing a crucial signal to regulate the ultimate differentiation of antigen-specific T cells and critically impact their cytotoxicity. The γc family of cytokines, including IL-2, IL-4, IL-9, IL-15, and IL-21, is a specific group of cytokines that share a common cytokine receptor γ chain. IL-4 is primarily recognized to promote humoral immunity and regulatory T cell (Treg) development, and IL-9 is thought to improve TH9 differentiation [45, 46]. In particular, it is four γc family cytokines, IL-2, IL-7, IL-15 and IL-21, that are crucial regulators of T cell-based cellular immunity and involved in orchestrating T cell stemness, contributing to enhanced antitumor activity in CAR-T therapy. Because of the great potential of TSCM cells for tumor control, further understanding of how these cytokines orchestrate the induction of persistent TSCM cells will contribute to the optimization of infused TSCM cell production before transfer. Here, we respectively clarify the role of the four γc family cytokines in regulating the formation and expansion of TSCM cells, as well as the underlying mechanisms.
IL-2 contributes to terminal differentiation of CD8(+) T cells
IL-2 is discovered as a pleiotropic T cell growth factor [47, 48] mainly derived from CD4(+) T cells, and plays a major role in cellular immunity. Cellular immune responses are triggered by antigen encounter and TCR-CD3 activation and then amplified by the interaction of IL-2 and its receptors as the third signal. The high-affinity IL-2 receptor (IL-2R) comprised of α, β, and γc subunits is mainly distributed in activated effector T cells and Tregs. In contrast, memory CD8(+) T cells express intermediate-affinity heterodimeric IL-2R, which only includes IL-2Rβ/γc chains [49]. Regardless of affinity, both receptors can transmit signals through the recruitment of JAK1 and JAK3 by the intracellular domains of IL-2Rβ and IL-2Rγ respectively, as well as through the phosphorylation of tyrosine residues [50,51,52]. These transmitted signals can further activate several pathways in T cells, including the JAK1/3-STAT5, JAK-RAS-MAPK cascade, PI3K-mTORC1, and PI3K-AKT pathways [49, 52]. Among them, STAT5 signaling has been shown to promote the formation of terminal effectors [53,54,55] (Fig. 2).

The signaling pathways mediated by the four γc family cytokines regulate T cell stemness. The stemness of T cells is cooperated by several signaling pathways. The four γc family cytokines trigger the JAK-STAT, RAS-MAPK, and PI3K-AKT signaling pathways to collectively modulate TSCM phenotype induction. IL-2 combines with high-affinity IL-2R via dimeric STAT5 to induce terminal differentiation whereas reduced STAT5 signaling by intermediate-affinity IL-2R can increase the expression of memory- and antiapoptotic-associated molecules. IL-7 and the trans-presented IL-15 also activate STAT5 phosphorylation to initiate the expression of the stem-like markers such as CD95, TCF1, and CD62L, for persistent survival and TSCM cell induction. IL-21 mainly activates the phosphorylation of STAT1 and STAT3, the latter of which induces the expression of Scal-1, CD95, TCF1 and CD62L, thereby contributing to TSCM phenotype formation. The activation of P70S6K via PI3K-AKT and mTOR is involved in T cell differentiation; therefore, inhibitors of the AKT pathway, such as AKT inhibitors and mTOR inhibitor Rapamycin, provide opportunities to regulate stemness. WNT inhibits GSK3β to release β-catenin into the nucleus to regulate the expressions of Bcl-2, c-Myc, c-Jun, CD62L and TCF1 to promote TSCM formation, the same as the GSK3β inhibitor TWS119. In addition, the activation of the Notch and cGAS-STING pathways can also promote T cell stemness
With IL-2Rα (CD25) being upregulated by TCR-CD3 activation, the binding of IL-2 to high-affinity IL-2R enables CD8(+) T cells to become effector T cells and release cytotoxic molecules. To maintain immune homeostasis and prevent overactivation, some negative feedback molecules such as Fas/FasL and other inhibitory molecules gradually present on the effector T cells. Subsequently, they mediate T cell anergy and apoptosis at the advanced stage of the immune response, which is partially owing to the Treg cell-mediated immune suppression. With the constitutive expression of high-affinity IL-2Rα, Treg cells competitively deprive effector T cells of IL-2 to support their expansion and suppress the T cell response [56, 57]. A murine model with Treg cell-specific conditional knockout of high-affinity IL-2R was established, which showed that the deficiency of IL-2 consumption by Treg cells impaired their suppression of CD8(+) T cell proliferation [56], particularly that of memory T cells. In general, IL-2 combined with high-affinity IL-2R enhances the expansion of CD8(+) effector T cells and promotes their terminal differentiation both directly [57, 58] and indirectly by maintaining the suppressive function of Treg cells [52, 57, 59, 60]. In addition, IL-2 binds to the intermediate-affinity IL-2R to mediate a low level of IL-2 signaling for facilitating the expression of IL-7α and CD62 ligand (CD62L), and preferentially bring about memory T cells [52, 54]. These findings suggest that IL-2 has a dual and opposing function in regulating the CD8(+) T cell phenotype. High levels of IL-2 signaling drive CD8(+) T cells to differentiate into short-lived effector cells, while low levels of IL-2 promote the differentiation of long-lived memory T cells [52, 54]. An appropriate affinity of IL-2 for IL-2R may raise the possibility to develop and maintain a subset of memory T cells with persistent survival and self-renewal capacity, known as TSCM.
With this regard, by reducing IL-2 signal strength, CD8(+) T cells can be successfully cultivated in vitro to acquire stemness and mediate persistent tumor suppression in CAR-T therapy. Of note, IL-2-producing CD8(+) T cells demonstrated attenuated IL-2-dependent STAT5 signaling, probably resulting in the restriction of terminal differentiation. This finding was supported by the observation that a specific subset of CD8(+) T cells capable of synthesizing IL-2 during the effector phase attained stem-like memory traits and resisted exhaustion at the effector phase [55]. Similar to the lower signaling mediated by intrinsically generated IL-2, short-term culture with exogenous IL-2 promoted the CD62L+CCR7+ memory CAR-T cells possessing stronger propagating ability and better tumor control in vivo. In contrast, long-term culture drove terminal differentiation and dampened, rather than boosted the antitumoral function of CAR-T cells [61].
Beyond the lower dose and shorter incubation time of wild-type IL-2, engineered IL-2 and receptors may provide another feasible strategy for precise and efficient TSCM induction. Wild-type IL-2 administration for cancer receives low complete response rates and poor tumor control due to its short half-life, which requires a very high amount of intravenous IL-2 associated with severe non-specific toxicities, and off-target effects on Treg cells [62,63,64,65,66,67]. Thus, some engineered IL-2 proteins with prolonged half-life are designed to improve cell targeting and selectivity for dimeric intermediate-affinity IL-2R, typically entailing the reduced interaction of IL-2 with the CD25 subunit or enhanced binding to CD122 [68,69,70,71,72]. They can be engineered by introducing mutations that shift the selectivity towards cells expressing intermediate-affinity IL-2R, yielding IL-2 muteins, orthogonal IL-2-IL-2R mutein pairs or fusion with other proteins including polyethylene glycol (Peg) (PEGylated IL-2), antibodies (IL-2 immune complexes), and the extracellular domain of CD25 (IL-2-CD25 fusion proteins) [71,72,73]. These engineered IL-2 proteins have the potential to preferentially target antigen-experienced memory T cells and NK cells that express dimeric intermediate-affinity IL-2R and manifest enhanced antitumoral responses in T cell-based therapy [74, 75], some of which partially benefit from the increased formation of TSCM. H9T, an engineered IL-2 partial agonist obtained via a single mutation Q126T in ‘superkine’ H9 that reduced the binding of H9 to IL-2Rγ, promoted the expansion of transferred CD8(+) T cells in vitro without terminal differentiation, and maintained a stem-cell-like state, which was attributed to reduced STAT5 signaling and increased T cell transcription factor 1 [76]. Intriguingly, much lower expression of exhaustion markers PD-1, TIM-3, and LAG-3 on infused T cells was induced by co-culturing with H9T in comparison with IL-2 or H9, which impaired the impediment to antitumor response and prolonged survival. As a result, the appropriately reduced binding of IL-2 to dimeric IL-2R may be a potential approach to promoting and maintaining the stem-cell-like phenotype of CD8(+) T cells without compromising the function of inducing amplification. To further reduce systemic toxicity due to IL-2 pluripotency in vivo, IL-2 and its receptor were engineered as an orthogonal cytokine-cytokine receptor pair, in which orthogonal IL-2 selectively interacts with its orthogonal receptor expressed on CAR-T cells capable of delivering an appropriate IL-2 signal in vivo [77]. IL-2 cytokine-receptor orthogonal pairs promote the specific expansion of orthogonal IL-2Rβ-modified T cells in vivo with negligible toxicity and improved antitumor response against leukemia and B16-F10 melanoma [77, 78]. Developed as orthogonal human IL-2, STK-009 selectively expanded orthogonal IL-2Rβ (hoRb)-expressing CAR-T cells and maintained the presence of TSCMin vivo, which delivered complete responses in refractory lymphomas [79]. Another orthogonal IL-2-IL-2R mutein pair, human chimeric orthogonal IL-2Rβ-ECD–IL-9R-ICD (O9R), fused orthogonal IL-2 receptor extracellular domain (ECD) with the intracellular domain (ICD) of IL-9R such that the orthogonal IL-2 elicited the corresponding γc cytokine signal [10]. Mediating a reduced STAT5 signal compared to O2R, orthogonal IL-2 drove stemness and superior effector capacity in O9R-expressing TCR-or CAR-T cells in mouse solid tumor models of melanoma and pancreatic cancer. Furthermore, compared with the direct co-administration of wild-type or engineered IL-2 in vivo with T cell transfer, synthetic cytokine circuits such as tumor-specific synNotch receptors and synthetic zinc finger transcription regulators (synZiFTRs) on engineered T cells allowed the precise production of IL-2 in time and space to achieve less systemic toxicity [74, 80]. In contrast to the lower dose and shorter incubation time of IL-2 during the manufacturing phase in vitro, the suitable alteration of IL-2 and IL-2R in an engineered manner may inspire a more efficient way to not only precisely improve the induction of targeted T cell stemness in vivo but also alleviate the side effects caused by its pleiotropy, which gives rise to adoptively transferred TSCM cells and mediates continual responses in ACT.
IL-7 induces TSCM cell differentiation and long-term longevity
Unlike IL-2, IL-7 was first identified as a stromal cell-derived factor and was encoded from human cDNA in vitro [81, 82]. It signals through IL-7R containing IL-7Rα (CD127) and γc subunits with activation of the JAK-STAT and PI3K-AKT pathways. Intriguingly, IL-7 is essential for T cell development and for maintaining and restoring CD8(+) memory T cell homeostasis alone or together with IL-15, another γc family cytokine illuminated later [83]. During thymopoiesis, IL-7R is present on double-negative (DN) T cells, absent on double-positive (DP) T cells, restored on single-positive (SP) T cells, and retained on mature T cells in the thymus [84], indicating that T cell development is closely related to the controlled expression of IL-7R. During mature T cell differentiation in peripheral lymphoid organs, in contrast to other γc family cytokines, IL-7R is highly expressed on naïve T cells but lost on the most effector T cells after TCR activation, and then re-expressed on memory T cells. Exceptionally, a small minority of effector T cells with increased IL-7R expression are predisposed to differentiation into memory cell subsets that persist for a long time in vivo, implicating that IL-7/IL-7R serves as a critical regulator of memory T cell transition and maintenance.
The mechanism underlying IL-7-mediated regulation of the survival of long-lived memory T cells is explicitly associated with the reprogramming of energy metabolism to some extent, including lipid metabolism and oxidative phosphorylation. The glycerol channel aquaporin 9 (AQP9)-dependent triglyceride (TAG) synthesis driven by IL-7 is indispensable for promoting the longevity of memory CD8(+) T cells [85]. Likewise, IL-7 increases glucose uptake by TSCM cells via overexpression GLUT1 and upregulation of the glycolytic enzyme hexokinase 2 (HK2), as illustrated by the inhibition of TSCM cell generation and expansion using the selective glucose uptake inhibitor WZB117 [86, 87]. Additionally, sustained expression of the antiapoptotic proteins BCL-2 and Mcl-1 in response to IL-7 is also involved in the survival of memory T cells [88,89,90] (Fig. 2). Moreover, IL-7 is controlled by a negative regulatory feedback loop to maintain homeostasis of memory T cells [91, 92]. Therefore, IL-7 highlights the considerable potential for an efficient transition to long-lived TSCM cells and enhanced antitumor responses by elevating their expansion.
IL-15 promotes TSCM cell phenotype expansion in vitro
IL-15 is commonly produced by a wide range of cells and acts on various immune cells like T cells through IL-15 receptors (IL-15R) to serve as a T cell growth factor [93, 94]. IL-15R is comprised of three subunits: IL-15Rα (CD215), IL-15/IL-2Rβ (CD122), and γc (CD132). Since two members of the γc family cytokines, IL-2 and IL-15, share IL-2Rβ, they consequently share some common biological properties, which was proven by evidence that IL-2Rβ deficiency impeded T cell proliferation induced by IL-15 [93]. By contrast with IL-2, which induces effector T cell terminal differentiation through high-affinity IL-2R, IL-15 remarkably tends to promote the maintenance and expansion of memory CD8(+) T cells owing to the unique IL-15Rα [95,96,97,98]. More strikingly, the distinct subunit IL-15Rα mainly presents IL-15 in trans to neighboring cells, including memory T cells, by antigen-presenting cells (APCs) [99]. As an autonomous and antigen-independent process [99], the trans-presentation of IL-15 provides sufficient signals to sustain antigen-specific memory CD8(+) T cell survival and expansion in the absence of antigens [95, 100,101,102,103]. The underlying mechanism is that membrane IL-15Rα on APCs captures IL-15 with high affinity and trans-presents IL-15 to activate the IL-2/15Rβγ heterodimer of memory CD8(+) T cells, subsequently activating the same JAK-STAT pathway as IL-2/IL-2R [104]. The phosphorylated STAT5 proteins form heterodimers to regulate the expression of downstream target genes, involving the upregulation of the antiapoptotic protein Bcl-2 and NF-kB signaling and the downregulation of the expression of pro-apoptotic molecules Bim and Puma [105, 106]. Beyond the JAK-STAT pathway, IL-15-induced proliferation of memory CD8(+) T cells partially relies on activation of the RAS-RAF-MAPK cascade and PI3K signalings [95, 106]. Besides, the increased activity of mTORC1 [101] and FKBP12 (FK506-binding protein 1 A, also known as FKBP1A) driven by IL-15 activates P70 S6 kinase and is responsible for promoting the cell cycle progression of memory T cells [95, 100] (Fig. 2).
As IL-15 mediates a lower mTORC1 activity than IL-2 to prevent T cell terminal differentiation, co-culture with IL-15 alone is superior to that with IL-2 alone in preserving the TSCM phenotype during the expansion process of CAR-T cells ex vivo [107]. Upon tumor challenge, CAR-T cells exposed to IL-15 exhibited fewer apoptotic features, higher proliferative capacity, and a superior antitumor response than those exposed to IL-2 in vivo. In addition, memory CD8(+) T cells were found to expand significantly following 3-day administration of recombinant human IL-15 (rhIL-15), among which TSCM cells were also observed a profound tendency to expand in phase I clinical trial [108]. However, its short half-life and low availability limit its application in vivo. Thus, different IL-15 derivatives by fusion with soluble IL-15Rα, Fc domain, or Peg have been engineered for stable bioactivity to overcome these barriers [109,110,111,112,113]. To further prolong the persistence of infused T cells, stable IL-15/IL-15Rα complexes were incorporated to induce and sustain the expansion of the CD62L+ and CCR7+ central memory T cell (Tcm) phenotype ex vivo resulting in augmented efficacy of adoptive immunotherapy [114]. As a result, efforts have been made to optimize the IL-15/IL-15Rα structure to achieve a higher proportion of infused T cells with stemness and greater antitumor potency in ACT [115]. In addition to its incorporation in ex vivo precultures, IL-15 and its receptor complex have been integrated into CAR engineering to maintain TSCM expansion in vivo for durable responses [116,117,118,119,120,121,122]. To mimic the trans-presentation of IL-15 in the context of IL-15Rα, membrane-bound chimeric IL-15 (mbIL15) was generated by the fusion of native IL-15 to IL-15Rα via a flexible linker. It was co-expressed with second-generation CAR, yielding mbIL15-CAR-T cells that retained long-term persistence and memory potential with a TSCM-like phenotype [116]. Furthermore, to attenuate IL-15-induced off-target toxicity, a next-generation tumor-conditional IL-15, called pro-IL15, was developed to fulfill tumor-targeted delivery by fusing the extracellular domain of IL-15Rβ into the N-terminus of IL-15-IL-15Rα-Fc (super IL-15-Fc) using a peptide linker specifically cleaved by matrix metalloproteinase (MMP) inside the TME [123]. In mouse tumor models, pro-IL-15 significantly increased the proportion of stem-like CD8(+) T cells in tumor tissue and enhanced sensitivity to immune checkpoint inhibitors [123].
IL-21 drives the development of naïve T cells into TSCM cells
IL-21 is mainly derived from activated CD4(+) T cells and NK cells and demonstrates broad pleiotropic effects on the immune system [83, 124, 125]. Similar to IL-2, the binding of IL-21 to a functional IL-21 receptor (IL-21R) consisting of heterodimers of γc (CD132) and specific chain IL-21Rα (CD360) stimulates the phosphorylation of tyrosine residues to activate several downstream signals covering the JAK-STAT1/3, PI3K-AKT and MAPK signaling pathways (Fig. 2). Among these, STAT3 phosphorylation is involved in IL-21-induced TSCM cell formation by upregulating the expression of the memory-associated transcriptional factors notch, TBX21, and SOCS1, and downregulation of mature effector markers Eomesodermin (EOMES) and GATA Binding Protein 3 (GATA3) [38]. Besides, metabolic reprogramming by IL-21 is also responsible for orchestrating memory CD8(+) T cell differentiation with stemness. When cultured with IL-21 in vitro, CD8(+) T cells elicit a metabolic skewing away from aerobic glycolysis towards a naïve-like metabolically quiescent state characterized by oxidative phosphorylation and fatty acid oxidation (FAO) with increased mitochondrial fitness and biogenesis [126, 127].
Therefore, IL-21 modulates the differentiation of memory CD8(+) T cell subsets as a critical threshold for the generation of memory stem-like CD8(+) T cells from naïve T cells [38, 39, 128,129,130]. IL-21 alone preferentially impedes terminal differentiation and improves memory subset formation of T cells [38, 128]. In the exploration of the conditions used to raise TSCM cellsfrom naïve T cells ex vivo, the addition of IL-21 helped transferred TSCM cells maintain their differentiation stage and potential for an increased response after short-time anti-CD3/CD28 co-stimulation in adoptive immunotherapy [129]. When synergized with other factors, IL-21 exerts a pivotal role in TSCM cell attainment and expansion, exemplified by the synergy with lactate dehydrogenase (LDH) inhibitor in a mouse model of pmel-1 specific TCR-T cell adoptive cancer immunotherapy [127]. When treated with a combination of LDH inhibitor and IL-21 in vitro, naïve pmel-1 CD8(+) T cells showed a naïve-like metabolic immunophenotype similar to that with IL-21 alone, which promoted the production of CD44low CD62high Sca1high cells and induced the suppression of exhaustion markers LAG3, PD1, 2B4, and TIM3, typically of TSCM cells [127]. Furthermore, IL-21 augmented rapamycin in the maintenance and expansion of AFP peptide-specific TSCM cells in vitro [39] and induced the CD19-CAR-modified TSCM cells from naïve precursors with the GSK-3β inhibitor TWS119 and IL-7, which showed superior elimination of tumors [39]. Strikingly, PD-1Ab21, a fusion protein of anti-PD-1 antibody and IL-21, which was successfully developed to block the interaction of PD-1 on T cells with PD-L1 and targeted IL-21 on PD-1+ T cells simultaneously, is expected to further stimulate the differentiation of activated T cells back to TSCM cells mediated by the IL-21 receptor in vitro [130]. In tumor-bearing mice, stronger tumor remission was observed with PD-1Ab21 treatment than that with the combination of PD-1 blockade and IL-21 infusion, which was attributed to the increased frequency of TSCM cells and robust expansion of tumor-specific CD8(+) T cells with a memory phenotype. In summary, IL-21 plays a crucial role in the induction and maintenance of transferred TSCM cells with enhanced antitumor and self-renewal capacities, which has significant implications for adoptive T cell-based immunotherapy.
Different combinations of the four cytokines for an optimal protocol to acquire efficient TSCM cells
Taken together, IL-2, IL-7, IL-15 and IL-21 are jointly involved in T cell differentiation and play different roles in T cell stemness. IL-2 drives T cell activation and terminal differentiation, while IL-21 prompts naïve T cells to TSCM cells phenotype. Meanwhile, IL-7 enables TSCM cells to form and maintain and IL-15 primarily stimulates their robust expansion [131]. Their different functions imply a multi-target strategy that different combinations might be an appropriate way to manufacture TSCM cells in vitro for ACT. From naïve precursor CD8(+) T cells, TSCM cells were generated by culturing with IL-7 and IL-15 in vitro [132, 133] or in the presence of IL-7/IL-21 and GSK-3β inhibitor TWS119 [39]. Except for promoting the generation of TSCM cells, adding reduced TCR stimulation to IL-7/IL-15 prevented terminal differentiation to efficiently maintain the stemness phenotype for a long time [134]. Moreover, effector T cells were demonstrated to convert into TSCM-like cells by IL-7 and IL-15 [23]. In a preclinical model, autologous reoriented CD19-CAR-T cells were incubated with IL-7 and IL-15 to obtain and preserve the TSCM cell subpopulation, which was transferred into patients with B-cell malignancies and produced stronger antitumor responses [135]. In many clinical trials, IL-7 combined with IL-15 has been used to induce adoptively transferred T cell stemness in order to prolong survival in vivo and mediate continual responses against tumors in adoptive cell immunotherapy (Table 1). As mentioned above, the transferred TSCM subset was efficiently induced and maintained by the combined utilization of IL-15 and IL-7, showing a better antitumoral effect for adoptive T-cell therapy [133, 135]. To characterize an applicable and efficient combination of different cytokines, Gargett et al. showed that it was more accessible to the acquisition of the TSCM phenotype by co-culture with IL-7/IL-15 than that with IL-2/IL-21 [136]. Nevertheless, Alizadeh et al. [107] demonstrated that inclusion of IL-7 and/or IL-21 impaired the effect of IL-15 on stem-like phenotype maintenance and antitumor potency [107]. Therefore, the optimal protocol of the four γc family cytokines, which produces effective infused TSCMin vitro to mediate a robust clinical outcome for ACT remains opaque and needs further investigation. Interestingly, targeted inducible delivery of the four crucial cytokines into the tumor focus may be another strategy for T cells to directly induce intrinsic stemness formation in vivo and reprogram the TME [137, 138].
Other cytokines and signalings that regulate T cell fate and promote TSCM.
In addition to the four γc family cytokines, there are other cytokines related to regulating T-cell fate, such as proinflammatory IL-1β, IL-18 and anti-inflammatory transfer growth factor β (TGF-β). A study [144] demonstrated that as effective proinflammatory cytokines, the increased production of IL-1β and IL-18 upon TIM-3 loss by dendritic cells (DCs) facilitated the maintenance of stem-like cells. To further identify the limited efficacy of recombinant IL-18 in clinical trials, an engineered “decoy resistant” IL-18 (DR-18) was designed to be impervious to IL-18BP inhibition [145], a high-affinity IL-18 receptor upregulated in various tumors and impedes the antitumor activity of IL-18. As a result, DR-18 not only maintained signaling potential but also exerted a robust antitumor activity by expanding the pool of stem-like TCF1+ precursor CD8(+) memory T cells and decreasing T cell exhaustion [145]. In addition, pre-stimulation with the combination of IL-12 and IL-18 contributes to memory T cells proliferation [146], and engineering T cells with scIL-12 and DR-18 demonstrates potent antitumoral effects [147]. Subsequently, membrane-bound form of IL-12 (mbIL12) engineered T cells were designed to improves potency of CAR-T cells both in vitro and in vivo [148]. Served as a typical anti-inflammatory cytokine, TGF-β is accepted for suppressing T cell activation and expansion [149,150,151]. As expected, TGF-β prominently impaired IL-7-induced memory T cell proliferation including TSCM cells [150]. Inhibition of TGF-β signaling by either a TGF-β antibody or a small molecule inhibitor augmented the generation of CD62highCD44high central memory CD8(+) T cells effectively [151]. However, exposure to TGF-β ex vivo resulted in the augmentation of early memory T cells through the downregulated expression of Blimp-1 and upregulated of the memory-associated transcription factor ID3 [149].
Apart from cytokines, several signaling pathways in T cells play a crucial role in the formation of the TSCM phenotype, including the Notch, Wnt-β-catenin, mTOR, and cGAS-STING signaling pathways, as well as c-Myb (Fig. 2). A long time has witnessed that Notch signaling could influence the lineage commitment of T cells as well as maintain the memory T cell survival [152, 153]. Expressing a Notch ligand, Delta-like 1 (OP9-hDLL1), stromal OP9 cells were used to generate TSCM-like cells in vitro from activated T cells successfully and converted conventional human CAR-T cells into TSCM-like CAR-T cells through Notch-FOXM1 axis [22,23,24]. Similarly, the utilization of Wnt-β-catenin signaling suppression, which includes inhibitors of GSK-3β or the Wnt protein family member Wnt3α, arrested T cell differentiation into terminal effector cells and promoted the generation of self-renewing multipotent CD8(+) memory stem cells characterized by CD44lowCD62LhighSca-1highCD122highBcl-2high [25,26,27]. Additionally, Stoycheva et al. discovered that deficiency of IFN-γR signaling promoted the formation of long-lived memory CD8(+) T cells and their sensitivity to weak TCR stimulation, which was correlated with reduced activation of mTOR and the accumulation of long-lived CD62LhighBcl-2highEomeshigh stem-like memory T cell precursors [154]. Simultaneously, the inhibition of mTORC1 in human naïve T cells after stimulation contributes to the induction of TSCM cells [28]. In addition, the cGAS-STING mediated DNA sensing pathway in T cells is essential for antitumor immune responses and promotes the maintenance of stem cell-like CD8(+) T cells mechanistically by regulating transcription factor TCF1 expression [29]. Another factor involved in T cell stemness is the transcription factor c-Myb. It can promote stemness by inducing pro-memory and survival programs via TCF7 and Bcl2 and restricting the terminal differentiation [30]. Collectively, multiple signaling pathways in T cells work together to regulate the fate of T cell differentiation and play a key role in antitumor immunity.
Combination of stemness promotion and exhaustion inhibition of T cells achieves further tumor eradication
Leveraging γc cytokines to induce T cell stemness contributes to a stable TSCM pool to give rise to sufficient quantities of T cells for sustained immune elimination, partially alleviating the poor persistence of T cells in the TME. However, T cells stemmed from TSCM will inevitably become dysfunctional and exhausted when continuously exposed to antigens in the TME. Therefore, integrated efforts of preserving TSCM pool using γc cytokines and reinvigorating exhausted T cell are promising strategies to enhance antitumor immunity.
Exhaustion is cell adaptation of T cells in response to chronic antigen stimulation in chronic viral infection and tumors [155,156,157,158,159], with the aim of maintaining moderate levels of inflammatory responses while obviating excessive tissue damage. Exhausted T cells (TEX) are heterogenous and comprised of progenitor exhausted T cells (TPEX), intermediate TEX, and irreversible terminally differentiated TEX [158, 160]. TEX are phenotypically different from memory/effector T cells and hallmarked by upregulation of multiple inhibitory receptors, altered transcriptional and epigenetic profiles, and progressive loss of effector functions, capabilities of proliferation, and cytokine secretions [156, 158, 161,162,163,164] (Fig. 3). Proinflammatory cytokines featured by IL-2, interferon-γ (IFN-γ), and tumor necrosis factor-α (TNF-α) are fundamentally important for the survival, proliferation, and cytotoxicity of T cells. Particularly, IL-2 production in the TME is required for T cells to proliferate and elicit potent antitumor immune responses, while impaired secretion of IL-2 greatly dampens T cell activity [165, 166]. Besides the deficiencies in cytokine release, TEX harbors increased expression of inhibitory receptors including PD-1, CTLA-4, LAG-3, TIM-3, TIGIT, CD160, and 2B4, which play pivotal roles in modulating the length and magnitude of immune responses and avoiding unrestrained cytotoxicity of T cells. Tumor cells excel in taking advantages of elevated inhibitory ligand/receptor axis to enable immune escape [155, 157, 159, 167, 168]. Activation of inhibitory ligand/receptor cascade signaling not only competes with target receptors or ligands that mediate activation signaling [169], but also attenuates signals from activated receptors via intracellular regulations [170]. Furthermore, the patterns of co-expression of inhibitory receptors and the quantities of inhibitory receptors on T cells substantially determine the intensity of T cell exhaustion [155, 169, 171,172,173].

The combination of TSCM formation by the γc family cytokines and ICB. A irreversibly terminal TEX formation and exhaustion in the TME. B the combination of the γc cytokines IL-2, 7, 15, 21 and ICB efficiently promotes abundance of TEF for tumor eradication
The advent of T cell exhaustion is a major obstacle to complete elimination of target cells in adoptive cell therapy (ACT) [157]. Immune checkpoint blockade (ICB) targeting inhibitory receptors and their ligands have revolutionized antitumor therapies by “releasing the brakes” and potentiating T cell activities [165, 168, 174, 175]. Blockade of PD-1/PD-L1 interactions restored T cell functions and demonstrated impressive efficacy in controlling tumor outgrowth [176, 177], which could be attributable to reversed T cell exhaustion. Recent studies have revealed that PD-1/PD-L1 blockade preferentially expands “stem-like” TPEX with self-renewal capacity and “effector-like” transitioning intermediate TEX rather than irreversible terminally differentiated TEX [170, 178,179,180,181]. Furthermore, combined targeting of PD-1 and CTLA-4 displayed better efficacy than the monotherapy [182,183,184,185], though the combination caused concerns of increased toxicity. More inhibitory receptors, such as TIM-3, LAG-3, and TIGIT, have gradually been leveraged as targets for therapeutic intervention [173, 186,187,188,189]. IL-2 replenishment has also been explored to alleviate T cell exhaustion ex vivo [165], and IL-2 replenishment plus PD-1/PD-L1 blockade has enriched better-functioning T cells in vivo and reprogrammed T cell exhaustion [190] via the orchestration that IL-2 stimulates antigen-specific T cells with stemness to expand and differentiate into effector cells and PD-1/PD-L1 blockade mitigates exhaustion and improves the antitumor capacity.
The elegant combination of IL-2 replenishment and PD-1/PD-L1 blockade raises the hypothesis that while “stepping on the accelerator” of immunity by increasing the sources of T cells, exhaustion should be mitigated to “release the brake”. Thus, it is both the quantities and quality of functional T cells that contribute to superior response in immunotherapy. The four γc family cytokines, IL-2, IL-7, IL-15, and IL-21, co-operate to form TSCM pool, thereby improving T cell reserve. ICB are well-established agents that polarize T cells to more cytotoxic phenotype. Collectively, the synergy of γc family cytokines with ICB might be a paradigm-shifting combination for immunotherapy. For example, anti-PD-1 antibody has been fused with IL-2, IL-15, or IL-21 to expand functional T cells and result in evident tumor remission [73, 130, 191]. Fused protein PD1-IL-2v bound PD-1 and IL-2βγ in cis on the same cell, enabled T cells to differentiate into stem-like cells and effector cells, alleviated terminally differentiated TEX formation, and exhibited superior efficacy in the mouse pancreatic adenocarcinoma models [73]. Engineered αPD-1-IL-15R protein, fused anti-PD-1 antibody with IL-15-IL-15Rα, navigated sIL-15 to intra-tumoral T cells via cis-delivery [191]. It not only significantly expanded tumor-specific CD8(+) T cells for tumor rejection but also reduced systematic toxicity by concealing activity region with immunoglobulin Fc region [191]. In addition, the fusion protein PD-1Ab21 successfully reinvigorated tumor-specific T cells and promoted TSCM proliferation [130].
Conclusion
TSCM belongs to a unique memory phenotype between naïve T cells and Tcm cells, possessing both stem-like memory and naïve phenotypic characteristics. Meanwhile, the number and proliferative capacity of TSCM in the TME have gradually become valuable predictors of reactivity in tumor immunotherapy. A robust antitumoral immunity is dependent on the activation of immune cells, including CD8(+) T cells and CD4(+) T cells. Both cell types are activated by antigen presentation and play crucial roles in antitumor immune responses, particularly CD8(+) T cells. CD8(+) effector T cells, cytotoxic lymphocytes, serve as dominant killers of tumor cells directly, making the stemness of CD8(+) T cells an area of increasing interest. Theoretically, the more antigen-specific TSCM in the TME, the stronger and more durable antitumor responses occur. Limited to the few intrinsic TSCMin vivo, the pursuit of inducing more “everlasting” TSCM from naïve or effector T cells has gradually become a crucial goals of tumor immunotherapy research. At this point, it has been elucidated that the four γc family cytokines, including IL-2, IL-7, IL-15, and IL-21, could regulate the fate of T cell differentiation into TSCM after antigen stimulation, and different combinations could make a different impact on TSCM production and amplification. However, systemic utilization of these cytokines could bring some side effects, such as non-specific inflammatory toxicities, cytokine release syndrome, and off-target adverse. For example, high-dose IL-2 has been known to induce fatal capillary leak syndrome [192], while IL-15 has been associated with hypotension, thrombocytopenia, and other adverse effects included [193]. To further achieve high-quality and quantity TSCM generation to maximize the antitumor effect and minimize the adverse effects, the development of gene editing techniques facilitates the production of multifunctional engineered T cells and optimal structure of the four γc family cytokines and their receptors [10, 76, 116, 123, 130]. Engineered versions have been developed to realize their full potential to promote the formation and persistence of TSCM and to reduce efficacy-independent toxicities (Fig. 4) (Table 2). Furthermore, their delivery into the TME by engineering synthetic gene circuits on engineered T cells also lowers systemic toxicities and precisely controls infused T cell function [74, 80]. Despite their great potential for stemness maintenance, the immunogenicity of engineered proteins is a concern. Many clinical trials are currently conducted to search for an efficient protocol of the four cytokines for TSCM formation and maintenance during ACT (Table 1). In addition to regulating T cell functions, they also exert an influence on other immune cells mainly dependent on the various expressions of their receptors, which play a pivotal role in different aspects of immunity. For example, IL-15 also promotes the proliferation and survival of NK cells, NKT cells, and mucosal associated invariant T (MAIT) cells that express IL-15R [193,194,195,196], and enhances the efficacy of NK cell associated transfer therapy [197, 198].

The engineered IL-2/IL-2R or IL-15/IL-15R for adoptively TSCM induction. A Engineered IL-2 for selectivity to dimeric intermediate-affinity IL-2R and TSCM induction. Activated T cells with high-affinity IL-2R finally go towards apoptosis by IL-2, and Treg with constitutively IL-2Rα deprive T cells of IL-2 to impair cytotoxicity. Reducing the affinity of IL-2 for IL-2Rα or increasing affinity for IL-2Rβ could target dimeric intermediate-affinity IL-2R and generate an appropriate affinity for TSCM induction by further lowering the signal level. Applying to CAR-T engineering, engineered orthogonal IL-2 and IL-2Rβ system or fused protein will be expressed to function CAR-T cells to induce TSCM. B Engineered mIL-15 armored CAR-T cell for stemness maintenance. The trans-presentation of IL-15 by IL-15Ra on APC to T cells contributes to TSCM induction independent of antigen stimulation. As a result, co-expressing IL-15 and IL-15Ra phenocopy the special presentation by a linker, yielding membrane-bounding IL-15. The mIL-15- CAR-T cells are able to differentiate into TSCM in vivo
After infusion, self-renewed TSCM rapidly differentiated into effector T cells, yet the latter unavoidably experienced exhaustion and apoptosis in the immunosuppressive TME, another obstacle for sustained antitumor responses. Thus, robust and durable immune responses in vivo require not only sufficient resources of TSCM cells to differentiate into better effector T cells efficiently, but also avoid infused T cell exhaustion after infusion for ACT. Administration of T cell growth factor IL-2 and PD-1/PD-L1 blockade may achieve the dual purpose of modifying the T cell exhaustion program and yield better effector T cells from TSCM cells after the transfer, resulting in complete tumor remission [73, 190]. The underlying mechanism of synergy is that IL-2 stimulates antigen-specific T cells with stemness to differentiate into effectors and expand intensely, and PD-1 blockade inhibits exhaustion and improves the antitumor capacity. Similarly, IL-15 and IL-21 were designed to be fused with the anti-PD-1 antibody to yield fusion proteins αPD-1-IL-15R and PD-1Ab21, respectively, for tumor rejection [130, 191]. To our knowledge, the application of TSCM has a bright future, as well as the orchestration of four γc family cytokines on adoptively transfer TSCM cells in ACT, but the road is long and difficult.
Availability of data and materials
Not applicable.
Abbreviations
- ACT:
-
Adoptive cell therapy
- CAR:
-
Chimeric antigen receptor
- TME:
-
Tumor microenvironment
- TSCM :
-
Stem cell-like memory T cells/stem cell memory T cells
- Tcm:
-
Central memory T cells
- γc:
-
The common cytokine receptor γ chain
- IL-2/7/15/21:
-
Interleukin-2/7/15/21
- PD-1/PD-L1:
-
Programmed cell death protein 1/ligand 1
- ICB:
-
Immune checkpoint blockage
- CTL:
-
Cytotoxic T lymphocyte
- AICD:
-
Activation induced cell death
- NSCLC:
-
Non-small cell lung cancer
- TILs:
-
Tumor infiltering T lymphocytes
- GVHD:
-
Graft versus host disease
- GSK-3β:
-
Glycogen synthase kinase-3β
- Treg:
-
Regulatory T cell
- CD62L:
-
CD62 ligand
- AQP9:
-
Aquaporin 9
- TAG:
-
Triglyceride
- EOMES:
-
Eomesodermin
- GATA3:
-
GATA Binding Protein 3
- FAO:
-
Fatty acid oxidation
- LDH:
-
Lactate dehydrogenase
- mTORC1:
-
mammalian target of rapamycin complex1
References
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72(1):7–33.
Ferlay J, Colombet M, Soerjomataram I, Parkin DM, Pineros M, Znaor A, et al. Cancer statistics for the year 2020: an overview. Int J Cancer. 2021;149(4):778–89.
MacKay M, Afshinnekoo E, Rub J, Hassan C, Khunte M, Baskaran N, et al. The therapeutic landscape for cells engineered with chimeric antigen receptors. Nat Biotechnol. 2020;38(2):233–.
Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-engineered T cells in patients with refractory cancer. Science. 2020;367(6481):1001–.
Wang Y, Qiu F, Xu Y, Hou X, Zhang Z, Huang L, et al. Stem cell-like memory T cells: the generation and application. J Leukoc Biol. 2021;110(6):1209–23.
van der Leun AM, Thommen DS, Schumacher TN. CD8(+) T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer. 2020;20(4):218–32.
Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T Cell States Associated with response to Checkpoint Immunotherapy in Melanoma. Cell. 2018;175(4):998–.
June CH, Sadelain M. Chimeric Antigen receptor therapy. N Engl J Med. 2018;379(1):64–73.
June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361–5.
Kalbasi A, Siurala M, Su LL, Tariveranmoshabad M, Picton LK, Ravikumar P, et al. Potentiating adoptive cell therapy using synthetic IL-9 receptors. Nature. 2022;607(7918):360–5.
Munshi NC, Anderson LD, Shah N, Madduri D, Berdeja J, Lonial S, et al. Idecabtagene Vicleucel in Relapsed and Refractory Multiple Myeloma. N Engl J Med. 2021;384(8):705–16.
Berdeja JG, Madduri D, Usmani SZ, Jakubowiak A, Agha M, Cohen AD, et al. Ciltacabtagene autoleucel, a B-cell maturation antigendirected chimeric antigen receptor T-cell therapy in patients with relapsed or refractory Multiple Myeloma (CARTITUDE-1): a phase 1b/2 open-label study. Lancet. 2021;398(10297):314–24.
Newick K, O’Brien S, Moon E, Albelda SM. CAR T cell therapy for solid tumors. Annu Rev Med. 2017;68:139–52.
Larson RC, Maus MV. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer. 2021;21(3):145–61.
Fraietta JA, Lacey SF, Orlando EJ, Pruteanu-Malinici I, Gohil M, Lundh S, et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic Leukemia. Nat Med. 2018;24(5):563–71.
Finck AV, Blanchard T, Roselle CP, Golinelli G, June CH. Engineered cellular immunotherapies in cancer and beyond. Nat Med. 2022;28(4):678–89.
Lugli E, Gattinoni L, Roberto A, Mavilio D, Price DA, Restifo NP, et al. Identification, isolation and in vitro expansion of human and nonhuman primate T stem cell memory cells. Nat Protoc. 2013;8(1):33–42.
Stemberger C, Neuenhahn M, Gebhardt FE, Schiemann M, Buchholz VR, Busch DH. Stem cell-like plasticity of naïve and distinct memory CD8 + T cell subsets. Semin Immunol. 2009;21(2):62–8.
Zhang Y, Joe G, Hexner E, Zhu J, Emerson SG. Host-reactive CD8 + memory stem cells in graft-versus-host Disease. Nat Med. 2005;11(12):1299–305.
Papatriantafyllou M. T cell memory: the stem of T cell memory. Nat Rev Immunol. 2011;11(11):716.
Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17(10):1290–7.
Kondo T, Morita R, Okuzono Y, Nakatsukasa H, Sekiya T, Chikuma S, et al. Notch-mediated conversion of activated T cells into stem cell memory-like T cells for adoptive immunotherapy. Nat Commun. 2017;8:15338.
Kondo T, Imura Y, Chikuma S, Hibino S, Omata-Mise S, Ando M, et al. Generation and application of human induced-stem cell memory T cells for adoptive immunotherapy. Cancer Sci. 2018;109(7):2130–40.
Kondo T, Ando M, Nagai N, Tomisato W, Srirat T, Liu B, et al. The NOTCH-FOXM1 Axis plays a key role in mitochondrial Biogenesis in the induction of human stem cell memory-like CAR-T cells. Cancer Res. 2020;80(3):471–83.
Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, et al. Wnt signaling arrests effector T cell differentiation and generates CD8 + memory stem cells. Nat Med. 2009;15(7):808–13.
Forget MA, Huon Y, Reuben A, Grange C, Liberman M, Martin J, et al. Stimulation of Wnt/ss-catenin pathway in human CD8 + T lymphocytes from blood and lung tumors leads to a shared young/memory phenotype. PLoS ONE. 2012;7(7):e41074.
Yan C, Chang J, Song X, Yan F, Yu W, An Y, et al. Memory stem T cells generated by wnt signaling from blood of human renal clear cell carcinoma patients. Cancer Biol Med. 2019;16(1):109–24.
Scholz G, Jandus C, Zhang L, Grandclement C, Lopez-Mejia IC, Soneson C, et al. Modulation of mTOR signalling triggers the formation of stem cell-like memory T cells. EBioMedicine. 2016;4:50–61.
Li W, Lu L, Lu J, Wang X, Yang C, Jin J, et al. cGAS-STING-mediated DNA sensing maintains CD8(+) T cell stemness and promotes antitumor T cell therapy. Sci Transl Med. 2020;12(549):eaay9013.
Gautam S, Fioravanti J, Zhu W, Le Gall JB, Brohawn P, Lacey NE, et al. The transcription factor c-Myb regulates CD8(+) T cell stemness and antitumor immunity. Nat Immunol. 2019;20(3):337–49.
Tsui C, Kretschmer L, Rapelius S, Gabriel SS, Chisanga D, Knopper K, et al. MYB orchestrates T cell exhaustion and response to checkpoint inhibition. Nature. 2022;609(7926):354–60.
Cheever MA. Twelve immunotherapy Drugs that could cure cancers. Immunol Rev. 2008;222:357–68.
Briukhovetska D, Dorr J, Endres S, Libby P, Dinarello CA, Kobold S. Interleukins in cancer: from biology to therapy. Nat Rev Cancer. 2021;21(8):481–99.
Gattinoni L, Klebanoff CA, Restifo NP. Paths to stemness: building the ultimate antitumour T cell. Nat Rev Cancer. 2012;12(10):671–84.
Gattinoni L, Speiser DE, Lichterfeld M, Bonini C. T memory stem cells in health and Disease. Nat Med. 2017;23(1):18–27.
Flynn JK, Gorry PR. Stem memory T cells (TSCM)-their role in cancer and HIV immunotherapies. Clin Transl Immunology. 2014;3(7):e20.
Thommen DS, Schumacher TN. T cell dysfunction in Cancer. Cancer Cell. 2018;33(4):547–62.
Chen Y, Yu F, Jiang Y, Chen J, Wu K, Chen X, et al. Adoptive transfer of Interleukin-21-stimulated human CD8 + T memory stem cells efficiently inhibits Tumor Growth. J Immunother. 2018;41(6):274–83.
Sabatino M, Hu J, Sommariva M, Gautam S, Fellowes V, Hocker JD, et al. Generation of clinical-grade CD19-specific CAR-modified CD8 + memory stem cells for the treatment of human B-cell malignancies. Blood. 2016;128(4):519–28.
Sheng SY, Gu Y, Lu CG, Tang YY, Zou JY, Zhang YQ, et al. The characteristics of naive-like T cells in Tumor-infiltrating lymphocytes from human Lung Cancer. J Immunother. 2017;40(1):1–10.
Li Y, Wu D, Yang X, Zhou S. Immunotherapeutic potential of T memory stem cells. Front Oncol. 2021;11:723888.
Krishna S, Lowery FJ, Copeland AR, Bahadiroglu E, Mukherjee R, Jia L, et al. Stem-like CD8 T cells mediate response of adoptive cell immunotherapy against human cancer. Science. 2020;370(6522):1328–34.
Hong H, Gu Y, Sheng SY, Lu CG, Zou JY, Wu CY. The distribution of human stem cell-like memory T cell in Lung Cancer. J Immunother. 2016;39(6):233–40.
Jansen CS, Prokhnevska N, Master VA, Sanda MG, Carlisle JW, Bilen MA, et al. An intra-tumoral niche maintains and differentiates stem-like CD8 T cells. Nature. 2019;576(7787):465–70.
Owen DL, Mahmud SA, Sjaastad LE, Williams JB, Spanier JA, Simeonov DR, et al. Thymic regulatory T cells arise via two distinct developmental programs. Nat Immunol. 2019;20(2):195–205.
Kaplan MH, Hufford MM, Olson MR. The development and in vivo function of T helper 9 cells. Nat Rev Immunol. 2015;15(5):295–307.
Morgan DA, Ruscetti FW, Gallo R. Selective in vitro growth of T lymphocytes from normal human bone marrows. Science. 1976;193(4257):1007–8.
Gillis S, Smith KA. Long term culture of tumour-specific cytotoxic T cells. Nature. 1977;268(5616):154–6.
Hernandez R, Poder J, LaPorte KM, Malek TR. Engineering IL-2 for immunotherapy of autoimmunity and cancer. Nat Rev Immunol. 2022;22(10):614–28.
Miyazaki T, Kawahara A, Fujii H, Nakagawa Y, Minami Y, Liu ZJ, et al. Functional activation of Jak1 and Jak3 by selective association with IL-2 receptor subunits. Science. 1994;266(5187):1045–7.
Spangler JB, Moraga I, Mendoza JL, Garcia KC. Insights into cytokine-receptor interactions from cytokine engineering. Annu Rev Immunol. 2015;33:139–67.
Ross SH, Cantrell DA. Signaling and function of Interleukin-2 in T lymphocytes. Annu Rev Immunol. 2018;36:411–33.
Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, Rao A. Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity. 2010;32(1):79–90.
Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, Ahmed R. Prolonged interleukin-2Ralpha expression on virus-specific CD8 + T cells favors terminal-effector differentiation in vivo. Immunity. 2010;32(1):91–103.
Kahan SM, Bakshi RK, Ingram JT, Hendrickson RC, Lefkowitz EJ, Crossman DK, et al. Intrinsic IL-2 production by effector CD8 T cells affects IL-2 signaling and promotes fate decisions, stemness, and protection. Sci Immunol. 2022;7(68):eabl6322.
Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An essential role for the IL-2 receptor in Treg cell function. Nat Immunol. 2016;17(11):1322–33.
Abbas AK, Trotta E, Marson DRS, Bluestone A. JA. Revisiting IL-2: Biology and therapeutic prospects. Sci Immunol. 2018;3(25):eaat1482.
Taniguchi T, Matsui H, Fujita T, Takaoka C, Kashima N, Yoshimoto R, et al. Structure and expression of a cloned cDNA for human interleukin-2. Nature. 1983;302(5906):305–10.
Furtado GC, Curotto de Lafaille MA, Kutchukhidze N, Lafaille JJ. Interleukin 2 signaling is required for CD4(+) regulatory T cell function. J Exp Med. 2002;196(6):851–7.
Pol JG, Caudana P, Paillet J, Piaggio E, Kroemer G. Effects of interleukin-2 in immunostimulation and immunosuppression. J Exp Med. 2020;217(1):e20191247.
Zhang X, Lv X, Song Y. Short-term culture with IL-2 is beneficial for potent memory chimeric antigen receptor T cell production. Biochem Biophys Res Commun. 2018;495(2):1833–8.
Rosenberg SA, Lotze MT, Muul LM, Chang AE, Avis FP, Leitman S, et al. A progress report on the treatment of 157 patients with advanced cancer using lymphokine-activated killer cells and interleukin-2 or high-dose interleukin-2 alone. N Engl J Med. 1987;316(15):889–97.
Fyfe G, Fisher RI, Rosenberg SA, Sznol M, Parkinson DR, Louie AC. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J Clin Oncol. 1995;13(3):688–96.
Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K, et al. High-dose recombinant interleukin 2 therapy for patients with metastatic Melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol. 1999;17(7):2105–16.
deLeeuw RJ, Kost SE, Kakal JA, Nelson BH. The prognostic value of FoxP3 + tumor-infiltrating lymphocytes in cancer: a critical review of the literature. Clin Cancer Res. 2012;18(11):3022–9.
Malek TR. The biology of interleukin-2. Annu Rev Immunol. 2008;26:453–79.
Arenas-Ramirez N, Woytschak J, Boyman O. Interleukin-2: Biology, Design and Application. Trends Immunol. 2015;36(12):763–77.
Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, et al. Exploiting a natural conformational switch to engineer an interleukin-2 ‘superkine’. Nature. 2012;484(7395):529–33.
Mitra S, Ring AM, Amarnath S, Spangler JB, Li P, Ju W, et al. Interleukin-2 activity can be fine tuned with engineered receptor signaling clamps. Immunity. 2015;42(5):826–38.
Silva DA, Yu S, Ulge UY, Spangler JB, Jude KM, Labao-Almeida C, et al. De novo design of potent and selective mimics of IL-2 and IL-15. Nature. 2019;565(7738):186–91.
Charych DH, Hoch U, Langowski JL, Lee SR, Addepalli MK, Kirk PB, et al. NKTR-214, an Engineered Cytokine with biased IL2 receptor binding, increased Tumor exposure, and marked efficacy in mouse Tumor models. Clin Cancer Res. 2016;22(3):680–90.
Charych D, Khalili S, Dixit V, Kirk P, Chang T, Langowski J, et al. Modeling the receptor pharmacology, pharmacokinetics, and pharmacodynamics of NKTR-214, a kinetically-controlled interleukin-2 (IL2) receptor agonist for cancer immunotherapy. PLoS ONE. 2017;12(7):e0179431.
Codarri Deak L, Nicolini V, Hashimoto M, Karagianni M, Schwalie PC, Lauener L, et al. PD-1-cis IL-2R agonism yields better effectors from stem-like CD8(+) T cells. Nature. 2022;610(7930):161–72.
Allen GM, Frankel NW, Reddy NR, Bhargava HK, Yoshida MA, Stark SR, et al. Synthetic cytokine circuits that drive T cells into immune-excluded tumors. Science. 2022;378(6625):eaba1624.
Bentebibel SE, Hurwitz ME, Bernatchez C, Haymaker C, Hudgens CW, Kluger HM, et al. A first-in-human study and Biomarker Analysis of NKTR-214, a Novel IL2Rbetagamma-Biased cytokine, in patients with Advanced or metastatic solid tumors. Cancer Discov. 2019;9(6):711–21.
Mo F, Yu Z, Li P, Oh J, Spolski R, Zhao L, et al. An engineered IL-2 partial agonist promotes CD8(+) T cell stemness. Nature. 2021;597(7877):544–8.
Sockolosky JT, Trotta E, Parisi G, Picton L, Su LL, Le AC, et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science. 2018;359(6379):1037–42.
Zhang Q, Hresko ME, Picton LK, Su L, Hollander MJ, Nunez-Cruz S, et al. A human orthogonal IL-2 and IL-2Rbeta system enhances CAR T cell expansion and antitumor activity in a murine model of Leukemia. Sci Transl Med. 2021;13(625):eabg6986.
Aspuria PJ, Vivona S, Bauer M, Semana M, Ratti N, McCauley S, et al. An orthogonal IL-2 and IL-2Rbeta system drives persistence and activation of CAR T cells and clearance of bulky Lymphoma. Sci Transl Med. 2021;13(625):eabg7565.
Li HS, Israni DV, Gagnon KA, Gan KA, Raymond MH, Sander JD, et al. Multidimensional control of therapeutic human cell function with synthetic gene circuits. Science. 2022;378(6625):1227–34.
Namen AE, Lupton S, Hjerrild K, Wignall J, Mochizuki DY, Schmierer A, et al. Stimulation of B-cell progenitors by cloned murine interleukin-7. Nature. 1988;333(6173):571–3.
Goodwin RG, Lupton S, Schmierer A, Hjerrild KJ, Jerzy R, Clevenger W, et al. Human interleukin 7: molecular cloning and growth factor activity on human and murine B-lineage cells. Proc Natl Acad Sci U S A. 1989;86(1):302–6.
Leonard WJ, Lin JX, O’Shea JJ. The Gammac Family of cytokines: Basic Biology to therapeutic ramifications. Immunity. 2019;50(4):832–50.
Mazzucchelli R, Durum SK. Interleukin-7 receptor expression: intelligent design. Nat Rev Immunol. 2007;7(2):144–54.
Cui G, Staron MM, Gray SM, Ho PC, Amezquita RA, Wu J, et al. IL-7-Induced Glycerol Transport and TAG synthesis promotes memory CD8 + T cell longevity. Cell. 2015;161(4):750–61.
Vignali D, Cantarelli E, Bordignon C, Canu A, Citro A, Annoni A, et al. Detection and characterization of CD8(+) Autoreactive Memory Stem T Cells in patients with type 1 Diabetes. Diabetes. 2018;67(5):936–45.
Fry TJ, Mackall CL. The many faces of IL-7: from lymphopoiesis to peripheral T cell maintenance. J Immunol. 2005;174(11):6571–6.
Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426(6967):671–6.
Schluns KS, Kieper WC, Jameson SC, Lefrancois L. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat Immunol. 2000;1(5):426–32.
Rathmell JC, Farkash EA, Gao W, Thompson CB. IL-7 enhances the survival and maintains the size of naive T cells. J Immunol. 2001;167(12):6869–76.
Park JH, Yu Q, Erman B, Appelbaum JS, Montoya-Durango D, Grimes HL, et al. Suppression of IL7Ralpha transcription by IL-7 and other prosurvival cytokines: a novel mechanism for maximizing IL-7-dependent T cell survival. Immunity. 2004;21(2):289–302.
Mackall CL, Fry TJ, Gress RE. Harnessing the biology of IL-7 for therapeutic application. Nat Rev Immunol. 2011;11(5):330–42.
Bamford RN, Grant AJ, Burton JD, Peters C, Kurys G, Goldman CK, et al. The interleukin (IL) 2 receptor beta chain is shared by IL-2 and a cytokine, provisionally designated IL-T, that stimulates T-cell proliferation and the induction of lymphokine-activated killer cells. Proc Natl Acad Sci U S A. 1994;91(11):4940–4.
Grabstein KH, Eisenman J, Shanebeck K, Rauch C, Srinivasan S, Fung V, et al. Cloning of a T cell growth factor that interacts with the beta chain of the interleukin-2 receptor. Science. 1994;264(5161):965–8.
Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol. 2006;6(8):595–601.
Sneller MC, Kopp WC, Engelke KJ, Yovandich JL, Creekmore SP, Waldmann TA, et al. IL-15 administered by continuous infusion to rhesus macaques induces massive expansion of CD8 + T effector memory population in peripheral blood. Blood. 2011;118(26):6845–8.
Becker TC, Wherry EJ, Boone D, Murali-Krishna K, Antia R, Ma A, et al. Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells. J Exp Med. 2002;195(12):1541–8.
Lugli E, Goldman CK, Perera LP, Smedley J, Pung R, Yovandich JL, et al. Transient and persistent effects of IL-15 on lymphocyte homeostasis in nonhuman primates. Blood. 2010;116(17):3238–48.
Kenesei A, Volko J, Szaloki N, Mocsar G, Jambrovics K, Balajthy Z, et al. IL-15 trans-presentation is an Autonomous, Antigen-Independent process. J Immunol. 2021;207(10):2489–500.
Dubois S, Shou W, Haneline LS, Fleischer S, Waldmann TA, Muller JR. Distinct pathways involving the FK506-binding proteins 12 and 12.6 underlie IL-2-versus IL-15-mediated proliferation of T cells. Proc Natl Acad Sci U S A. 2003;100(24):14169–74.
Richer MJ, Pewe LL, Hancox LS, Hartwig SM, Varga SM, Harty JT. Inflammatory IL-15 is required for optimal memory T cell responses. J Clin Invest. 2015;125(9):3477–90.
Traitanon O, Gorbachev A, Bechtel JJ, Keslar KS, Baldwin WM 3rd, Poggio ED, et al. IL-15 induces alloreactive CD28(-) memory CD8 T cell proliferation and CTLA4-Ig resistant memory CD8 T cell activation. Am J Transplant. 2014;14(6):1277–89.
Waldmann TA. The shared and contrasting roles of IL2 and IL15 in the life and death of normal and neoplastic lymphocytes: implications for cancer therapy. Cancer Immunol Res. 2015;3(3):219–27.
Patidar M, Yadav N, Dalai SK. Interleukin 15: a key cytokine for immunotherapy. Cytokine Growth Factor Rev. 2016;31:49–59.
Ring AM, Lin JX, Feng D, Mitra S, Rickert M, Bowman GR, et al. Mechanistic and structural insight into the functional dichotomy between IL-2 and IL-15. Nat Immunol. 2012;13(12):1187–95.
Mishra A, Sullivan L, Caligiuri MA. Molecular pathways: interleukin-15 signaling in health and in cancer. Clin Cancer Res. 2014;20(8):2044–50.
Alizadeh D, Wong RA, Yang X, Wang D, Pecoraro JR, Kuo CF, et al. IL15 enhances CAR-T cell antitumor activity by reducing mTORC1 activity and preserving their stem cell memory phenotype. Cancer Immunol Res. 2019;7(5):759–72.
Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol. 2015;33(1):74–82.
Rhode PR, Egan JO, Xu W, Hong H, Webb GM, Chen X, et al. Comparison of the Superagonist Complex, ALT-803, to IL15 as Cancer Immunotherapeutics in Animal models. Cancer Immunol Res. 2016;4(1):49–60.
Wu Z, Xu Y. IL-15R alpha-IgG1-Fc enhances IL-2 and IL-15 anti-tumor action through NK and CD8 + T cells proliferation and activation. J Mol Cell Biol. 2010;2(4):217–22.
Dubois S, Patel HJ, Zhang M, Waldmann TA, Muller JR. Preassociation of IL-15 with IL-15R alpha-IgG1-Fc enhances its activity on proliferation of NK and CD8+/CD44high T cells and its antitumor action. J Immunol. 2008;180(4):2099–106.
Chertova E, Bergamaschi C, Chertov O, Sowder R, Bear J, Roser JD, et al. Characterization and favorable in vivo properties of heterodimeric soluble IL-15.IL-15Ralpha cytokine compared to IL-15 monomer. J Biol Chem. 2013;288(25):18093–103.
Mortier E, Quemener A, Vusio P, Lorenzen I, Boublik Y, Grotzinger J, et al. Soluble interleukin-15 receptor alpha (IL-15R alpha)-sushi as a selective and potent agonist of IL-15 action through IL-15R beta/gamma. Hyperagonist IL-15 x IL-15R alpha fusion proteins. J Biol Chem. 2006;281(3):1612–9.
Hasan AN, Selvakumar A, Shabrova E, Liu XR, Afridi F, Heller G, et al. Soluble and membrane-bound interleukin (IL)-15 Ralpha/IL-15 complexes mediate proliferation of high-avidity central memory CD8(+) T cells for adoptive immunotherapy of cancer and Infections. Clin Exp Immunol. 2016;186(2):249–65.
Zhang Y, Zhuang Q, Wang F, Zhang C, Xu C, Gu A, et al. Co-expression IL-15 receptor alpha with IL-15 reduces toxicity via limiting IL-15 systemic exposure during CAR-T immunotherapy. J Transl Med. 2022;20(1):432.
Hurton LV, Singh H, Najjar AM, Switzer KC, Mi T, Maiti S, et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc Natl Acad Sci U S A. 2016;113(48):E7788–E97.
Hoyos V, Savoldo B, Quintarelli C, Mahendravada A, Zhang M, Vera J, et al. Engineering CD19-specific T lymphocytes with interleukin-15 and a Suicide gene to enhance their anti-lymphoma/leukemia effects and safety. Leukemia. 2010;24(6):1160–70.
Krenciute G, Prinzing BL, Yi Z, Wu MF, Liu H, Dotti G, et al. Transgenic expression of IL15 improves Antiglioma Activity of IL13Ralpha2-CAR T cells but results in Antigen loss variants. Cancer Immunol Res. 2017;5(7):571–81.
Li G, Zhang Z, Cai L, Tang X, Huang J, Yu L, et al. Fn14-targeted BiTE and CAR-T cells demonstrate potent preclinical activity against glioblastoma. Oncoimmunology. 2021;10(1):1983306.
Chen Y, Sun C, Landoni E, Metelitsa L, Dotti G, Savoldo B. Eradication of Neuroblastoma by T cells redirected with an optimized GD2-Specific chimeric Antigen receptor and Interleukin-15. Clin Cancer Res. 2019;25(9):2915–24.
Batra SA, Rathi P, Guo L, Courtney AN, Fleurence J, Balzeau J, et al. Glypican-3-Specific CAR T cells coexpressing IL15 and IL21 have Superior Expansion and Antitumor Activity against Hepatocellular Carcinoma. Cancer Immunol Res. 2020;8(3):309–20.
Sun Y, Su Y, Wang Y, Liu N, Li Y, Chen J, et al. CD19 CAR-T cells with membrane-bound IL-15 for B-Cell Acute Lymphoblastic Leukemia after failure of CD19 and CD22 CAR-T cells: Case Report. Front Immunol. 2021;12:728962.
Guo J, Liang Y, Xue D, Shen J, Cai Y, Zhu J, et al. Tumor-conditional IL-15 pro-cytokine reactivates anti-tumor immunity with limited toxicity. Cell Res. 2021;31(11):1190–8.
Ozaki K, Kikly K, Michalovich D, Young PR, Leonard WJ. Cloning of a type I cytokine receptor most related to the IL-2 receptor beta chain. Proc Natl Acad Sci U S A. 2000;97(21):11439–44.
Parrish-Novak J, Dillon SR, Nelson A, Hammond A, Sprecher C, Gross JA, et al. Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature. 2000;408(6808):57–63.
Loschinski R, Bottcher M, Stoll A, Bruns H, Mackensen A, Mougiakakos D. IL-21 modulates memory and exhaustion phenotype of T-cells in a fatty acid oxidation-dependent manner. Oncotarget. 2018;9(17):13125–38.
Hermans D, Gautam S, Garcia-Canaveras JC, Gromer D, Mitra S, Spolski R, et al. Lactate dehydrogenase inhibition synergizes with IL-21 to promote CD8(+) T cell stemness and antitumor immunity. Proc Natl Acad Sci U S A. 2020;117(11):6047–55.
Hinrichs CS, Spolski R, Paulos CM, Gattinoni L, Kerstann KW, Palmer DC, et al. IL-2 and IL-21 confer opposing differentiation programs to CD8 + T cells for adoptive immunotherapy. Blood. 2008;111(11):5326–33.
Alvarez-Fernández C, Escribà-Garcia L, Vidal S, Sierra J, Briones J. A short CD3/CD28 costimulation combined with IL-21 enhance the generation of human memory stem T cells for adoptive immunotherapy. J Transl Med. 2016;14(1):214.
Li Y, Cong Y, Jia M, He Q, Zhong H, Zhao Y, et al. Targeting IL-21 to tumor-reactive T cells enhances memory T cell responses and anti-PD-1 antibody therapy. Nat Commun. 2021;12(1):951.
Gattinoni L, Restifo NP. Moving T memory stem cells to the clinic. Blood. 2013;121(4):567–8.
Gomez-Eerland R, Nuijen B, Heemskerk B, van Rooij N, van den Berg JH, Beijnen JH, et al. Manufacture of gene-modified human T-cells with a memory stem/central memory phenotype. Hum Gene Ther Methods. 2014;25(5):277–87.
Cieri N, Camisa B, Cocchiarella F, Forcato M, Oliveira G, Provasi E, et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood. 2013;121(4):573–84.
Zanon V, Pilipow K, Scamardella E, De Paoli F, De Simone G, Price DA, et al. Curtailed T-cell activation curbs effector differentiation and generates CD8(+) T cells with a naturally-occurring memory stem cell phenotype. Eur J Immunol. 2017;47(9):1468–76.
Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood. 2014;123(24):3750–9.
Gargett T, Brown MP. Different cytokine and stimulation conditions influence the expansion and immune phenotype of third-generation chimeric antigen receptor T cells specific for Tumor antigen GD2. Cytotherapy. 2015;17(4):487–95.
Birocchi F, Cusimano M, Rossari F, Beretta S, Rancoita PMV, Ranghetti A, et al. Targeted inducible delivery of immunoactivating cytokines reprograms glioblastoma microenvironment and inhibits growth in mouse models. Sci Transl Med. 2022;14(653):eabl4106.
Ren Z, Zhang A, Sun Z, Liang Y, Ye J, Qiao J, et al. Selective delivery of low-affinity IL-2 to PD-1 + T cells rejuvenates antitumor immunity with reduced toxicity. J Clin Invest. 2022;132(3):e153604.
Chen F, Zou Z, Du J, Su S, Shao J, Meng F, et al. Neoantigen identification strategies enable personalized immunotherapy in refractory solid tumors. J Clin Invest. 2019;129(5):2056–70.
Rohaan MW, Borch TH, van den Berg JH, Met O, Kessels R, Geukes Foppen MH, et al. Tumor-infiltrating lymphocyte therapy or Ipilimumab in Advanced Melanoma. N Engl J Med. 2022;387(23):2113–25.
Pang N, Shi J, Qin L, Chen A, Tang Y, Yang H, et al. IL-7 and CCL19-secreting CAR-T cell therapy for tumors with positive glypican-3 or mesothelin. J Hematol Oncol. 2021;14(1):118.
Zurko JC, Xu H, Chaney K, Schneider D, Szabo A, Hari P, et al. Bispecific targeting of CD20 and CD19 increases polyfunctionality of chimeric antigen receptor T-cell products in B-cell malignancies. Cytotherapy. 2022;24(8):767–73.
Kochenderfer JN, Dudley ME, Carpenter RO, Kassim SH, Rose JJ, Telford WG, et al. Donor-derived CD19-targeted T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood. 2013;122(25):4129–39.
Dixon KO, Tabaka M, Schramm MA, Xiao S, Tang R, Dionne D, et al. TIM-3 restrains anti-tumour immunity by regulating inflammasome activation. Nature. 2021;595(7865):101–6.
Zhou T, Damsky W, Weizman OE, McGeary MK, Hartmann KP, Rosen CE, et al. IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature. 2020;583(7817):609–14.
Raue HP, Beadling C, Haun J, Slifka MK. Cytokine-mediated programmed proliferation of virus-specific CD8(+) memory T cells. Immunity. 2013;38(1):131–9.
Olivera I, Bolanos E, Gonzalez-Gomariz J, Hervas-Stubbs S, Marino KV, Luri-Rey C, et al. mRNAs encoding IL-12 and a decoy-resistant variant of IL-18 synergize to engineer T cells for efficacious intratumoral adoptive immunotherapy. Cell Rep Med. 2023;4(3):100978.
Lee EHJ, Murad JP, Christian L, Gibson J, Yamaguchi Y, Cullen C, et al. Antigen-dependent IL-12 signaling in CAR T cells promotes regional to systemic Disease targeting. Nat Commun. 2023;14(1):4737.
Dahmani A, Janelle V, Carli C, Richaud M, Lamarche C, Khalili M, et al. TGFβ programs central memory differentiation in Ex vivo-stimulated human T cells. Cancer Immunol Res. 2019;7(9):1426–39.
Nguyen TP, Sieg SF. TGF-β inhibits IL-7-induced proliferation in memory but not naive human CD4(+) T cells. J Leukoc Biol. 2017;102(2):499–506.
Takai S, Schlom J, Tucker J, Tsang KY, Greiner JW. Inhibition of TGF-β1 signaling promotes central memory T cell differentiation. J Immunol. 2013;191(5):2299–307.
Laky K, Fleischacker C, Fowlkes BJ. TCR and notch signaling in CD4 and CD8 T-cell development. Immunol Rev. 2006;209:274–83.
Maekawa Y, Ishifune C, Tsukumo S, Hozumi K, Yagita H, Yasutomo K. Notch controls the survival of memory CD4 + T cells by regulating glucose uptake. Nat Med. 2015;21(1):55–61.
Stoycheva D, Deiser K, Stärck L, Nishanth G, Schlüter D, Uckert W, et al. IFN-γ regulates CD8 + memory T cell differentiation and survival in response to weak, but not strong, TCR signals. J Immunol. 2015;194(2):553–9.
Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8 + T cell exhaustion by multiple inhibitory receptors during chronic viral Infection. Nat Immunol. 2009;10(1):29–37.
Schietinger A, Greenberg PD. Tolerance and exhaustion: defining mechanisms of T cell dysfunction. Trends Immunol. 2014;35(2):51–60.
Blank CU, Haining WN, Held W, Hogan PG, Kallies A, Lugli E, et al. Defining ‘T cell exhaustion’. Nat Rev Immunol. 2019;19(11):665–74.
Dolina JS, Van Braeckel-Budimir N, Thomas GD, Salek-Ardakani S. CD8(+) T cell exhaustion in Cancer. Front Immunol. 2021;12:715234.
Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15(8):486–99.
Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, et al. Progenitor and terminal subsets of CD8 + T cells cooperate to contain chronic viral Infection. Science. 2012;338(6111):1220–5.
Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, Wherry EJ. Network analysis reveals centrally connected genes and pathways involved in CD8 + T cell exhaustion versus memory. Immunity. 2012;37(6):1130–44.
Belk JA, Daniel B, Satpathy AT. Epigenetic regulation of T cell exhaustion. Nat Immunol. 2022;23(6):848–60.
Franco F, Jaccard A, Romero P, Yu YR, Ho PC. Metabolic and epigenetic regulation of T-cell exhaustion. Nat Metab. 2020;2(10):1001–12.
Wherry EJ. T cell exhaustion. Nat Immunol. 2011;12(6):492–9.
Korman AJ, Garrett-Thomson SC, Lonberg N. The foundations of immune checkpoint blockade and the ipilimumab approval decennial. Nat Rev Drug Discov. 2022;21(7):509–28.
Spolski R, Li P, Leonard WJ. Biology and regulation of IL-2: from molecular mechanisms to human therapy. Nat Rev Immunol. 2018;18(10):648–59.
Wykes MN, Lewin SR. Immune checkpoint blockade in infectious Diseases. Nat Rev Immunol. 2018;18(2):91–104.
Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, Mattei MG, et al. A new member of the immunoglobulin superfamily–CTLA-4. Nature. 1987;328(6127):267–70.
Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim-3 and PD-1 expression is associated with Tumor antigen-specific CD8 + T cell dysfunction in Melanoma patients. J Exp Med. 2010;207(10):2175–86.
Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: an overview of FDA-approved immune checkpoint inhibitors. Int Immunopharmacol. 2018;62:29–39.
Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T, et al. Tumor-infiltrating NY-ESO-1-specific CD8 + T cells are negatively regulated by LAG-3 and PD-1 in human Ovarian cancer. Proc Natl Acad Sci U S A. 2010;107(17):7875–80.
Grosso JF, Goldberg MV, Getnet D, Bruno TC, Yen HR, Pyle KJ, et al. Functionally distinct LAG-3 and PD-1 subsets on activated and chronically stimulated CD8 T cells. J Immunol. 2009;182(11):6659–69.
Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207(10):2187–94.
Salas-Benito D, Pérez-Gracia JL, Ponz-Sarvisé M, Rodriguez-Ruiz ME, Martínez-Forero I, Castañón E, et al. Paradigms on Immunotherapy combinations with Chemotherapy. Cancer Discov. 2021;11(6):1353–67.
Wu M, Huang Q, Xie Y, Wu X, Ma H, Zhang Y, et al. Improvement of the anticancer efficacy of PD-1/PD-L1 blockade via combination therapy and PD-L1 regulation. J Hematol Oncol. 2022;15(1):24.
Postow MA, Callahan MK, Wolchok JD. Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol. 2015;33(17):1974–82.
Zhou F, Qiao M, Zhou C. The cutting-edge progress of immune-checkpoint blockade in Lung cancer. Cell Mol Immunol. 2021;18(2):279–93.
Blackburn SD, Shin H, Freeman GJ, Wherry EJ. Selective expansion of a subset of exhausted CD8 T cells by alphaPD-L1 blockade. Proc Natl Acad Sci U S A. 2008;105(39):15016–21.
Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, et al. Developmental relationships of four exhausted CD8(+) T cell subsets reveals underlying transcriptional and epigenetic Landscape Control mechanisms. Immunity. 2020;52(5):825–41. e8.
Hudson WH, Gensheimer J, Hashimoto M, Wieland A, Valanparambil RM, Li P, et al. Proliferating transitory T cells with an effector-like transcriptional signature emerge from PD-1(+) stem-like CD8(+) T cells during chronic Infection. Immunity. 2019;51(6):1043–58. e4.
Budimir N, Thomas GD, Dolina JS, Salek-Ardakani S. Reversing T-cell exhaustion in Cancer: lessons learned from PD-1/PD-L1 Immune Checkpoint Blockade. Cancer Immunol Res. 2022;10(2):146–53.
Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus Ipilimumab in advanced Melanoma. N Engl J Med. 2013;369(2):122–33.
Carlino MS, Larkin J, Long GV. Immune checkpoint inhibitors in Melanoma. Lancet. 2021;398(10304):1002–14.
Yap TA, Parkes EE, Peng W, Moyers JT, Curran MA, Tawbi HA. Development of Immunotherapy combination strategies in Cancer. Cancer Discov. 2021;11(6):1368–97.
Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined Nivolumab and Ipilimumab or Monotherapy in untreated Melanoma. N Engl J Med. 2015;373(1):23–34.
Chiang EY, Mellman I. TIGIT-CD226-PVR axis: advancing immune checkpoint blockade for cancer immunotherapy. J Immunother Cancer. 2022;10(4):e004711.
Liang R, Zhu X, Lan T, Ding D, Zheng Z, Chen T, et al. TIGIT promotes CD8(+)T cells exhaustion and predicts poor prognosis of Colorectal cancer. Cancer Immunol Immunother. 2021;70(10):2781–93.
Grebinoski S, Zhang Q, Cillo AR, Manne S, Xiao H, Brunazzi EA, et al. Autoreactive CD8(+) T cells are restrained by an exhaustion-like program that is maintained by LAG3. Nat Immunol. 2022;23(6):868–77.
Tao J, Han D, Gao S, Zhang W, Yu H, Liu P, et al. CD8(+) T cells exhaustion induced by myeloid-derived suppressor cells in myelodysplastic syndromes patients might be through TIM3/Gal-9 pathway. J Cell Mol Med. 2020;24(1):1046–58.
Hashimoto M, Araki K, Cardenas MA, Li P, Jadhav RR, Kissick HT, et al. PD-1 combination therapy with IL-2 modifies CD8(+) T cell exhaustion program. Nature. 2022;610(7930):173–81.
Shen J, Zou Z, Guo J, Cai Y, Xue D, Liang Y et al. An engineered concealed IL-15-R elicits tumor-specific CD8 + T cell responses through PD-1-cis delivery. J Exp Med. 2022;219(12).
Dougan M, Luoma AM, Dougan SK, Wucherpfennig KW. Understanding and treating the inflammatory adverse events of cancer immunotherapy. Cell. 2021;184(6):1575–88.
Zhang S, Zhao J, Bai X, Handley M, Shan F. Biological effects of IL-15 on immune cells and its potential for the treatment of cancer. Int Immunopharmacol. 2021;91:107318.
Ma S, Caligiuri MA, Yu J. Harnessing IL-15 signaling to potentiate NK cell-mediated cancer immunotherapy. Trends Immunol. 2022;43(10):833–47.
Rha MS, Han JW, Kim JH, Koh JY, Park HJ, Kim SI, et al. Human liver CD8(+) MAIT cells exert TCR/MR1-independent innate-like cytotoxicity in response to IL-15. J Hepatol. 2020;73(3):640–50.
Ellis-Connell AL, Balgeman AJ, Kannal NM, Hansen Chaimson K, Batchenkova A, Safrit JT, et al. IL-15 superagonist N-803 enhances IFN-gamma production of MAIT cells in SIV(+) Macaques. Infect Immun. 2022;90(10):e0025922.
Christodoulou I, Ho WJ, Marple A, Ravich JW, Tam A, Rahnama R et al. Engineering CAR-NK cells to secrete IL-15 sustains their anti-AML functionality but is associated with systemic toxicities. J Immunother Cancer. 2021;9(12).
Chiu E, Felices M, Cichocki F, Davis Z, Wang H, Tuninga K, et al. Anti-NKG2C/IL-15/anti-CD33 killer engager directs primary and iPSC-derived NKG2C(+) NK cells to target Myeloid Leukemia. Mol Ther. 2021;29(12):3410–21.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Key Technology Research and Development Program of China (2022YFC2704200 and 2022YFC2704205), National Natural Science Foundation of China (82272707, 81874109), and Innovation Group Project of Hubei Province (2023AFA036).
Author information
Authors and Affiliations
Contributions
MS L drafted the manuscript and prepared the figures. YW Z, WJ G and HY L collected the related references and participated in the discussion. DM, KM W and QL G revised the manuscript. MS L and YF designed this review and revised the manuscript. All authors contributed to this manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Luo, M., Gong, W., Zhang, Y. et al. New insights into the stemness of adoptively transferred T cells by γc family cytokines. Cell Commun Signal 21, 347 (2023). https://doi.org/10.1186/s12964-023-01354-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-023-01354-3