
Abstract
Adult stem cells have a unique ability to self-renew and to generate differentiated daughter cells that are required in the body tissues. The identity of adult stem cells is maintained by extrinsic signals from other cell types, known as niche cells. Thus, the niche is required for appropriate tissue homeostasis. Niche is formed and recruits stem cells during tissue development; therefore, it is essential to establish niche cells and stem cells in proper numbers during development. A small niche may recruit too few stem cells and cause tissue degeneration, while a large niche may maintain too many stem cells and lead to tumorigenesis. Given that vertebrate tissues are not suitable for large-scale forward genetics studies, the Drosophila ovary stands out as an excellent model for studying how multiple niche cell types and germ cells (GCs) are coordinately regulated in vivo. Recent studies are beginning to reveal how various signaling molecules regulate niche formation and how niche cells non-autonomously influence GC number. In this review, we summarize the ovarian niche structure, the key signaling pathways for niche formation, and how niche cells generate extrinsic factors to control GC proliferation during ovarian development.
Video abstract
Similar content being viewed by others


Introduction
Stem cells and their niches constitute functional units that maintain body tissue homeostasis [1,2,3,4]. In 1978, the concept of the niche was first proposed by Schofield [5] to describe the physiological microenvironment that supports stem cells; since then, niche structures have been identified in a host of groups, from invertebrates to mammals [6,7,8,9,10]. A stem cell niche allows the establishment of stem cells and the maintenance of a balance between stem cell self-renewal and differentiation [11,12,13,14]. Many studies have focused on stem cell maintenance in the adult stage [15,16,17,18]. The niche is established and anchors stem cells during tissue development [19]; therefore, it is essential to establish niche and stem cells in suitable numbers during development. However, very little is understood about how to form fixed numbers of both niche cells and stem cells during development. Understanding the underlying regulatory mechanisms behind this process will help to identify genetic developmental defects that disrupt adult tissue formation.
The mammalian ovary is not appropriate for large-scale systematic screening of genes involved in niche formation and function; however, Drosophila melanogaster overcomes this limitation. In this context, the Drosophila ovary provides an effective model for studying the genetic and molecular mechanisms behind niche formation and function during ovary development. Herein, we discuss recent knowledge on how niche formation is regulated by signaling molecules and how stem cell number is controlled by extrinsic factors coming from the niche.
Formation of the Drosophila ovarian niche
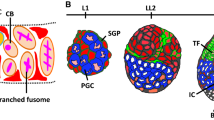
The embryonic ovary in Drosophila consists of two primary cell types: the primordial germ cells (PGCs) and the somatic gonadal precursors (SGPs) (Fig. 1) [20, 21]. During early larval stages, PGCs, which are the precursors of GCs, and somatic precursors proliferate [22]. In the second instar larval (L2) stage, somatic cell-derived intermingled cells (ICs) occupy the central region of the larval gonad and closely interact with GCs (Fig. 1) [23]. By the mid-third instar larval (ML3), somatic swarm cells (SwCs) are located dorsolaterally [24]. Also, terminal filament cells (TFCs) first appear and start to proliferate (Fig. 1) [25, 26]. After 24 h, these TFCs finish flattening, sorting, intercalation and stacking, and finally form 16–20 regularly arranged terminal filaments (TFs) [26]. By that time, most SwCs have also completed their movements and have formed a new posterior domain (Fig. 1) [24]. During the larval–pupal transition (LP transition), ICs adjacent to the basal TFCs differentiate into cap cells (CCs) [27, 28]. Although TFCs and CCs are adjacent to each other, they can be distinguished by morphologies: TFCs are cuboid-shaped whereas CCs are disc-shaped [26, 29]. Once TFs and CCs are formed, GCs can attach to them to become adult GSCs. At the prepupal stage, the newly formed GSC niche, composed of somatic cell TFCs, CCs, and ICs, becomes functional [28].

Schematic of ovarian development in Drosophila from the embryo to the larval–pupal (LP) transition stage. The embryonic gonad consists of primordial germ cells (PGCs: blue) and somatic gonadal precursors (SGPs: red). The L2 larval ovary is composed of germ cells (GCs: blue) surrounded by intermingled cells (ICs: red). By the mid-third instar larval (ML3), terminal filament cells (TFCs: yellow) first appear and somatic swarm cells (SwCs: purple) are located dorsolaterally. During the LP transition, SwCs form a new posterior domain and the ICs adjacent to basal TFCs differentiate into cap cells (CCs: green); the TFCs are organized into 16–20 TF stacks
The availability of enriched genetic tools, including many cell type-specific Gal4 drivers (Table 1) for performing targeted gene knockdown, rescue, or overexpression manipulation in niche cells makes Drosophila ovarian GSC niche an attractive system for studying niche formation and function.
Signal regulation during the formation of the ovarian niche
The ovarian niche consists of three different types of somatic cells. These cells start to proliferate at different times and several different signaling pathways are involved in this process. Below, we summarize the signaling pathways that regulate the formation of CCs, TFCs, and ICs.
Pathways that regulate CC formation
Activation of Notch signaling helps to promote CC formation [29, 33]. Xie and colleagues showed that newly formed TFCs express the Notch ligand Delta, which activates Notch signaling in adjacent somatic cells and induces them to become CCs [29]. Overexpression of Delta or forced expression of activated Notch results in an increase in CC number [29, 33]. Another key signaling pathway that plays a role in niche formation is related to Ecdysone signaling [22, 34]. Reduction of Ecdysone signaling activity in somatic cells results in expansion of CCs [34]. Moreover, Ecdysone prevents precocious CC differentiation. In Ecdysone Receptor (EcR) RNAi ovaries, CCs appear at ML3 instead of the prepupal stage [22]. Given that both the Notch and Ecdysone signaling pathways are involved in CC formation, it is important to know whether and how these two pathways interact to regulate the CC formation process. Yatsenko and Shcherbata explored this question and found that miR-125 acts as an intermediary between Notch and Ecdysone signaling in the process of CC formation [35]. Specifically, Ecdysone signaling induces expression of miR-125, which targets a Notch signaling antagonist called Tom. Downregulation of Tom activates Delta, which activates Notch signaling in adjacent somatic cells and converts them into CCs [35].
In addition to the Notch and Ecdysone signaling pathways, protein Traffic jam (Tj) affects the specification of CC [27], but whether it affects CC number is not yet clear.
Pathways that regulate TFC formation
TFC proliferation is controlled by several signaling pathways, including the Notch, Hippo, Janus kinase/signal transduction and activator of transcription (JAK/STAT), Insulin and Target of rapamycin (Tor) pathways (Table 2) [36,37,38,39,40]. Yatsenko and Shcherbata showed that Notch signaling is required for TFC formation. In Notch mutant ovaries, TFs are abnormally shaped and TFC number is reduced, indicating that Notch signaling is essential for TF morphogenesis and TFC formation [36]. Hippo signaling pathway components Hippo (Hpo) and Yorkie (Yki) are both expressed in TFCs [37]. Altering Hippo pathway activity in soma notably affects TFC proliferation and TF stack formation: RNAi of hpo or warts (wts) or overexpressing yki significantly increases TFC and TF stack number. Conversely, knocking down yki or overexpressing hpo in the soma results in a remarkable decrease in TFC and TF stack number [37]. In addition, knocking down hpo in somatic cells leads to notably increased Stat92E expression in TFCs and ICs, suggesting that Hippo pathway activity limits JAK/STAT pathway activity in the soma, as Stat92E is a readout of JAK/STAT activity [37]. In addition, decreasing the activity of Dome, a JAK/STAT receptor, in the soma significantly decreases TFC number while double knockdown of dome and hpo completely suppresses the phenotype of hpo knockdown alone [37]. This genetic evidence suggests that Hippo signaling regulates TFC proliferation through interactions with the JAK/STAT pathway.
Nutritional cues provided by the Insulin and Tor pathways also affect TF number (Table 2) [38,39,40,41]. In Drosophila, Insulin binding to the Insulin-like receptor (InR) induces recruitment and phosphorylation of the Insulin receptor substrate (encoded by the chico gene) and subsequent activation of Phosphoinositide 3-kinase (PI3K) and Akt [42, 43]. Gancz and Gilboa reported that overexpression of InR in somatic cells results in a significant increase in TF number. Conversely, knocking down chico, Akt, or Tor by RNAi leads to a remarkable reduction in the number of TFs [38].
Activin signaling is transmitted by Baboon (Babo), a type I receptor of the TGF-β (transforming growth factor-β) superfamily in Drosophila [44]. Lengil et al. found that Activin signaling promotes TF formation in developing ovaries [45]. Gancz et al. showed that Ecdysone can also accelerate TF formation [22]. In wild-type ML3 ovaries, only a few short TFs can be detected. In contrast, a significant increase in TF stack number is observed in ovaries of Babo mutant and EcR knockdown line. In addition, in the wild-type, TFs are well organized, whereas in the ovaries with low activity of Ecdysone or Activin receptor, they are unevenly spaced [22, 45], suggesting that precocious TF formation results in morphogenesis defects.
In addition to these signaling pathways, a few genes, including twinstar (tsr), bric-à-brac (bab1/bab2), LIM homeobox transcription factor 1 alpha (Lmx1a) and longitudinals lacking (lola), have been reported to be involved in TFC formation [46,47,48,49,50]. Chen et al. found that in tsr mutant ovaries, the TFC number decreased and all TFCs have a rounded appearance. Importantly, the TF structure is not present, suggesting that tsr is essential for the formation of TFC and TF [46]. In 1995, Godt and Laski discovered that bab affects TF formation [25]. The Bab locus encodes Bab1 and Bab2. In 2020, a study found that a reduction of both Bab1 and Bab2, but not each separately, impedes TF formation [48]. Another study found that the transcription factor Lmx1a is required for the formation of TF. In Lmx1a mutant ovaries, TFCs fail to organize in individualized stacks, leading to aberrant TF structures [49]. That previous study also showed that bab1/bab2 is essential for Lmx1a expression and function, which raised the possibility that bab1/bab2 affects TF formation by regulating Lmx1a expression. However, this requires further confirmation, to answer questions such as whether overexpression of Lmx1a can rescue bab1/bab2 mutant phenotype in vivo. Zhao et al. reported that lola is also essential for TF formation. In lola RNAi ovaries, either no TF stacks are formed or they are disordered [50]. Coincidentally, both lola and bab/bab2 have a BTB domain, suggesting that this domain may be necessary for TF formation.
Pathways that regulate IC formation
ICs are in direct contact with GCs and are thought to give rise to escort cells in adult ovaries [22, 29, 51]. The Hippo signaling pathway is involved in IC proliferation [37]. Lowering Hippo pathway activity by knocking down hpo or wts or overexpressing yki results in a remarkable increase in IC number. On the contrary, overexpression of hpo or depletion of yki by RNAi significantly reduces IC number (Table 2) [37]. Sarikaya and Extavour found that knockdown of hpo in the soma significantly increased pMAPK (a readout of epidermal growth factor receptor [EGFR] activity) expression in ICs, suggesting that Hippo activity limits EGFR activity in ICs. Furthermore, knockdown of hpo and egfr partially rescues hpo RNAi-induced overgrowth of ICs [37]. These results suggest that the Hippo pathway interacts with the EGFR pathway to regulate IC number. That study also found that the JAK/STAT signaling activity is very high in ICs, and that RNAi against JAK/STAT receptor dome or the ligand unpaired (upd1) can rescue the increased IC number resulting from hpo RNAi. These results suggest that the Hippo pathway interacts with the JAK-STAT and EGFR signals in regulating IC proliferation.
Insulin and Tor signaling play a role in the proliferation of ICs [38]. Somatic overexpression of InR significantly increases IC number. By contrast, RNAi against Tor, chico, or Akt leads to a decrease in IC number (Table 2) [38]. Whether the EGFR pathway regulates IC number is controversial. One study found that blocking Egfr signaling in ICs results in fewer ICs [52]. However, another reported that IC number was unaltered by knockdown of egfr or spi alone [37].
According to the signaling pathways that regulate the formation of TFs and ICs, it can be noted that the Hippo signaling and Insulin/Tor signaling have the same effect (Table 2). In addition, simultaneous reduction of the Hippo and JAK/STAT pathway activity has the same effect on IC and TFC numbers. Moreover, these studies used Tj-Gal4, a driver that is only expressed in ICs, not in TFCs, to drive gene knockdown or overexpression in soma. Thus, these data indicate that signals in ICs non-autonomously regulate TFC proliferation.
Niche function: extrinsic cues for GC proliferation
In the Drosophila embryonic gonad, there are initially ~ 12 PGCs [53]. During the larval stage, the PGCs remain undifferentiated and proliferate to more than 100 [54, 55]. This proliferation process is regulated by extrinsic signaling molecules, which are released from niche cells.
Signaling molecules in the niche cells regulate GC proliferation non-autonomously (Table 2) [22, 37, 38, 56]. The Hippo pathway regulates the proliferation of GCs: hpo or wts RNAi or overexpression of yki in the soma leads to a significant increase in GC number. On the contrary, overexpression of hpo in the soma decreases GC number [37]. Lowering EGFR activity in the soma also increases GC number [52, 57]. Moreover, double knockdown of hpo/egfr results in fewer GCs, which completely rescues the hpo RNAi-induced increase in GC number [37]. In addition, knockdown of JAK/STAT receptor dome or the ligand upd1 completely rescues hpo RNAi-induced GC overproliferation [37]. These results indicate that Hippo signaling regulates GC proliferation non-autonomously via interactions with the EGFR and JAK/STAT pathways.
The Insulin and Tor signaling also play a role in GC proliferation [38]. Somatic expression of Tor, chico, or Akt RNAi non-autonomously reduces GC number, and somatic overexpression of InR results in precocious GC differentiation [38]. RNAi constructs against EcR lead to precocious GC differentiation as well [22]. Recently, Lehmann lab utilized a single-cell RNA atlas of LL3 ovaries and identified the SwCs as mediators of the Ecdysone signal for GC differentiation: once SwCs reach the posterior of the ovary, the Ecdysone induces expression of Torso-like (Tsl) in SwCs, which acts as a soma-to-germline signal to stimulate PGC differentiation [24, 58]. Of note, Gancz and Gilboa found that the Insulin and Ecdysone pathways act in parallel to regulate GC differentiation [38].
It can be noted that Insulin and Ecdysone signaling pathways are required in parallel for GC differentiation and niche formation, which raises an interesting question of how these two different processes are connected.
Signaling crosstalk in ovarian niche and germ cells
When organized niches that contain a fixed number of stem cells are established during organ development, the proliferation rate of the niche cells and stem cells should be coordinated. How such coordination is achieved and how systemic factors might affect these processes remain largely unknown. The developing Drosophila ovary, which contains proper number of niche cells and germ cells, is an excellent model system with which to investigate this problem. Table 2 and Fig. 2 summarize the available data on the signaling pathways that regulate the formation of niche cells and GCs. The Ecdysone-miR-125-Notch signaling is required for CC formation. The Hippo signaling regulates TFC and GC proliferation through interactions with the JAK/STAT pathway. In addition, the Hippo pathway interacts with the JAK-STAT and EGFR signals to regulate IC proliferation. The Insulin and Tor signaling pathways function in the soma and regulate proliferation, each autonomously in ICs and TFCs and non-autonomously in GCs.

Schematic diagram of signaling crosstalk that regulates the formation of ovarian niche cells and germ cells. The Hippo, Insulin and Tor signaling pathways function in the soma to regulate proliferation both autonomously in TFCs and ICs and non-autonomously in GCs. The Notch signaling is required for TFC and CC formation. The EGFR pathway regulates homeostatic growth of both IC and GC numbers
It is noteworthy that GCs can influence the survival of IC conversely [52]. Gilboa and Lehmann found that EGFR functions in ICs to inhibit GC proliferation, and in turn, GCs express Spitz (an EGFR ligand), which is required for IC survival [52]. Thus, coordination of growth between the soma and germ line in the developing ovary is achieved.
Conclusions and future perspectives
It has been more than 40 years since the concept of the niche was proposed. During this time, we have learned that the function of the niche is to provide extrinsic signals for stem cells to maintain their identity in adult tissue. Stem cells self-renewal is crucial for development and tissue homeostasis in various organisms. For example, in the testes of Drosophila, hub cells (stem cell niche) are implicated as a source of JAK-STAT signaling that promotes self-renewal of both GSCs and CySCs (somatic cyst stem cells) [59,60,61]. Brawley and Matunis showed that loss of Stat92E leads to a loss of all GSCs from the niche [59]. In CySCs, zinc finger homeodomain 1 (zfh1) and chronologically inappropriate morphogenesis (chinmo) are direct target genes of Stat92E [60, 61]. Flaherty et al. reported that CySCs lacking either zfh1 or chinmo rapidly differentiate [60, 61]. In fact, during tissue development, the niche has already begun to provide growth factors for germ cells. Dennd1a, which is mainly expressed in somatic cells in the ovaries of mouse fetuses, is essential for oogenesis. Its disruption results in a significant reduction in germ cell number [62]. Thus, although the structure of the mammalian ovary is different from that of Drosophila, they share a common phenomenon wherein the niche forms and controls GC proliferation during ovary development.
Some studies have reported that some genes (e.g., tsr, bab1/bab2, lola, and Lmx1a) affect niche formation, but it is unclear whether these genes function through the known signaling pathways to control niche formation. At present, only one study has demonstrated that GCs can control niche cell survival; whether there are more mechanisms through which GCs regulate proliferation of niche cell remains unknown. Most importantly, how niche cells are specified and how their fates are stabilized remain unclear.
The answers to the above questions will certainly provide a better understanding of how niche and germ cells develop coordinately at the molecular and cellular levels. As stem cells and their niches have many similarities across species, the knowledge gained from the Drosophila system will provide insight into niche and stem cell regulation in mammalian systems.
Availability of data and materials
Not applicable.
Abbreviations
- GCs:
-
Germ cells
- PGCs:
-
Primordial germ cells
- SGPs:
-
Somatic gonadal precursors
- L2:
-
Second instar larval
- ML3:
-
Mid-third instar larval
- LP transition:
-
Larval–pupal transition
- ICs:
-
Intermingled cells
- TFCs:
-
Terminal filament cells
- TFs:
-
Terminal filaments
- CCs:
-
Cap cells
- SwCs:
-
Somatic swarm cells
- EcR:
-
Ecdysone Receptor
- InR:
-
Insulin-like receptor
- Tj:
-
Traffic jam
- JAK/STAT:
-
Janus kinase/signal transduction and activator of transcription
- Tor:
-
Target of rapamycin
- EGFR:
-
Epidermal growth factor receptor
- Lmx1a:
-
LIM homeobox transcription factor 1 alpha
References
Arwert EN, Hoste E, Watt FM. Epithelial stem cells, wound healing and cancer. Nat Rev Cancer. 2012;12:170–80.
Nakada D, Levi BP, Morrison SJ. Integrating physiological regulation with stem cell and tissue homeostasis. Neuron. 2011;70:703–18.
Spradling AC, Nystul T, Lighthouse D, Morris L, Fox D, Cox R, Tootle T, Frederick R, Skora A. Stem cells and their niches: integrated units that maintain Drosophila tissues. Cold Spring Harb Symp Quant Biol. 2008;73:49–57.
Blanpain C, Fuchs E. Epidermal homeostasis: a balancing act of stem cells in the skin. Nat Rev Mol Cell Biol. 2009;10:207–17.
Schofeld R. The relationship between the spleen colony forming cell and the haemopoietic stem cell. Blood Cells. 1978;4:7–25.
Xie T, Spradling AC. A niche maintaining germ line stem cells in the Drosophila ovary. Science. 2000;290:328–30.
Crittenden SL, Bernstein DS, Bachorik JL, Thompson BE, Gallegos M, Petcherski AG, Moulder G, Barstead R, Wickens M, Kimble J. A conserved RNA-binding protein controls germline stem cells in Caenorhabditis elegans. Nature. 2002;417:660–3.
Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, Martin RP, Schipani E, Divieti P, Bringhurst FR, Milner LA, Kronenberg HM, Scadden DT. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425:841–6.
Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramova N, Vincent P, Pumiglia K, Temple S. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science. 2004;304:1338–40.
Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, Ross J, Haug J, Johnson T, Feng JQ, Harris S, Wiedemann LM, Mishina Y, Li L. Identification of the haematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–41.
Hayashi Y, Yoshinari Y, Kobayashi S, Niwa R. The regulation of Drosophila ovarian stem cell niches by signaling crosstalk. Curr Opin Insect Sci. 2020;37:23–9.
Gilboa L. Organizing stem cell units in the Drosophila ovary. Curr Opin Genet Dev. 2015;32:31–6.
Liu Z, Zhong G, Chai PC, Luo L, Liu S, Yang Y, Baeg GH, Cai Y. Coordinated niche-associated signals promote germline homeostasis in the Drosophila ovary. J Cell Biol. 2015;211:469–84.
Singh A, Yadav CB, Tabassum N, Bajpeyee AK, Verma V. Stem cell niche: Dynamic neighbor of stem cells. Eur J Cell Biol. 2019;98:65–73.
Xie T. Control of germline stem cell self-renewal and differentiation in the Drosophila ovary: concerted actions of niche signals and intrinsic factors. Wiley Interdiscip Rev Dev Biol. 2013;2:261–73.
Kahney EW, Snedeker JC, Chen X. Regulation of Drosophila germline stem cells. Curr Opin Cell Biol. 2019;60:27–35.
Zhang H, Cai Y. Signal transduction pathways regulating Drosophila ovarian germline stem cells. Curr Opin Insect Sci. 2020;37:1–7.
Tu R, Duan B, Song X, Chen S, Scott A, Hall K, Blanck J, DeGraffenreid D, Li H, Perera A, Haug J, Xie T. Multiple Niche Compartments Orchestrate Stepwise Germline Stem Cell Progeny Differentiation. Curr Biol. 2021;31:827–39.
Hsu HJ, Bahader M, Lai CM. Molecular control of the female germline stem cell niche size in Drosophila. Cell Mol Life Sci. 2019;76:4309–17.
Dansereau DA, Lasko P. The development of germline stem cells in Drosophila. Methods Mol Biol. 2008;450:3–26.
Williamson A, Lehmann R. Germ cell development in Drosophila. Annu Rev Cell Dev Biol. 1996;12:365–91.
Gancz D, Lengil T, Gilboa L. Coordinated regulation of niche and stem cell precursors by hormonal signaling. PLoS Biol. 2011;9: e1001202.
Li MA, Alls JD, Avancini RM, Koo K, Godt D. The large Maf factor Traffic Jam controls gonad morphogenesis in Drosophila. Nat Cell Biol. 2003;5:994–1000.
Banisch TU, Slaidina M, Gupta S, Ho M, Gilboa L, Lehmann R. A transitory signaling center controls timing of primordial germ cell differentiation. Dev Cell. 2021;56:1742–55.
Godt D, Laski FA. Mechanisms of cell rearrangement and cell recruitment in Drosophila ovary morphogenesis and the requirement of bric à brac. Dev Camb Engl. 1995;121:173–87.
Sahut-Barnola I, Godt D, Laski FA, Couderc JL. Drosophila ovary morphogenesis: analysis of terminal filament formation and identification of a gene required for this process. Dev Biol. 1995;170:127–35.
Panchal T, Chen X, Alchits E, Oh Y, Poon J, Kouptsova J, Laski FA, Godt D. Specification and spatial arrangement of cells in the germline stem cell niche of the Drosophila ovary depend on the Maf transcription factor Traffic jam. PLoS Genet. 2017;13: e1006790.
Zhu C-H, Xie T. Clonal expansion of ovarian germline stem cells during niche formation in Drosophila. Dev Camb Engl. 2003;130:2579–88.
Song X, Call GB, Kirilly D, Xie T. Notch signaling controls germline stem cell niche formation in the Drosophila ovary. Development. 2007;134:1071–80.
Lai CM, Lin KY, Kao SH, Chen YN, Huang F, Hsu HJ. Hedgehog signaling establishes precursors for germline stem cell niches by regulating cell adhesion. J Cell Biol. 2017;216:1439–53.
Cabrera GR, Godt D, Fang PY, Couderc JL, Laski FA. Expression pattern of Gal4 enhancer trap insertions into the bric à brac locus generated by P element replacement. Genesis. 2002;34:62–5.
Díaz-Torres A, Rosales-Nieves AE, Pearson JR, Santa-Cruz Mateos C, Marín-Menguiano M, Marshall OJ, Brand AH, González-Reyes A. Stem cell niche organization in the Drosophila ovary requires the ECM component Perlecan. Curr Biol. 2021;31:1744-53.e5.
Ward EJ, Shcherbata HR, Reynolds SH, Fischer KA, Hatfield SD, Ruohola-Baker H. Stem cells signal to the niche through the Notch pathway in the Drosophila ovary. Curr Biol. 2006;16:2352–8.
König A, Yatsenko AS, Weiss M, Shcherbata HR. Ecdysteroids affect Drosophila ovarian stem cell niche formation and early germline differentiation. EMBO J. 2011;30:1549–62.
Yatsenko AS, Shcherbata HR. Stereotypical architecture of the stem cell niche is spatiotemporally established by miR-125-dependent coordination of Notch and steroid signaling. Development. 2018;145:dev159178.
Yatsenko AS, Shcherbata HR. Distant activation of Notch signaling induces stem cell niche assembly. PLoS Genet. 2021;17: e1009489.
Sarikaya DP, Extavour CG. The Hippo pathway regulates homeostatic growth of stem cell niche precursors in the Drosophila Ovary. PLoS Genet. 2015;11: e1004962.
Gancz D, Gilboa L. Insulin and Target of rapamycin signaling orchestrate the development of ovarian niche-stem cell units in Drosophila. Development. 2013;140:4145–54.
Green DA II, Extavour CG. Convergent evolution of a reproductive trait through distinct developmental mechanisms in Drosophila. Dev Biol. 2012;372:120–30.
Green DA II, Extavour CG. Insulin signaling underlies both plasticity and divergence of a reproductive trait in Drosophila. Proc R Soc B Biol Sci. 2014;281:20132673.
Sarikaya DP, Belay AA, Ahuja A, Dorta A, Green DA II, Extavour CG. The roles of cell size and cell number in determining ovariole number in Drosophila. Dev Biol. 2012;363:279–89.
Grewal SS. Insulin/TOR signaling in growth and homeostasis: a view from the fly world. Int J Biochem Cell Biol. 2009;41:1006–10.
Kumar A, Xie L, Ta CM, Hinton AO, Gunasekar SK, Minerath RA, Shen K, Maurer JM, Grueter CE, Abel ED, Meyer G, Sah R. SWELL1 regulates skeletal muscle cell size, intracellular signaling, adiposity and glucose metabolism. Elife. 2020;9: e58941.
Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. 2012;13(10):616–30.
Lengil T, Gancz D, Gilboa L. Activin signaling balances proliferation and differentiation of ovarian niche precursors and enables adjustment of niche numbers. Development. 2015;142:883–92.
Chen J, Godt D, Gunsalus K, Kiss I, Goldberg M, Laski FA. Cofilin/ADF is required for cell motility during Drosophila ovary development and oogenesis. Nat Cell Biol. 2001;3:204–9.
Bolívar J, Pearson J, López-Onieva L, González-Reyes A. Genetic dissection of a stem cell niche: the case of the Drosophila ovary. Dev Dyn. 2006;235:2969–79.
Miscopein Saler L, Hauser V, Bartoletti M, Mallart C, Malartre M, Lebrun L, Pret AM, Théodore L, Chalvet F, Netter S. The Bric-à-Brac BTB/POZ transcription factors are necessary in niche cells for germline stem cells establishment and homeostasis through control of BMP/DPP signaling in the Drosophila melanogaster ovary. PLoS Genet. 2020;16: e1009128.
Allbee AW, Rincon-Limas DE, Biteau B. Lmx1a is required for the development of the ovarian stem cell niche in Drosophila. Development. 2018;145:dev163394.
Zhao T, Xiao Y, Huang B, Ran MJ, Duan X, Wang YF, Lu Y, Yu XQ. A dual role of lola in Drosophila ovary development: regulating stem cell niche establishment and repressing apoptosis. Cell Death Dis. 2022;13:756.
Reilein A, Kogan HV, Misner R, Park KS, Kalderon D. Adult stem cells and niche cells segregate gradually from common precursors that build the adult Drosophila ovary during pupal development. Elife. 2021;10: e69749.
Gilboa L, Lehmann R. Soma–germline interactions coordinate homeostasis and growth in the Drosophila gonad. Nature. 2006;443:97–100.
Poirié M, Niederer E, Steinmann-Zwicky M. A sex-specific number of germ cells in embryonic gonads of Drosophila. Development. 1995;121:1867–73.
King RC, Aggarwal SK, Aggarwal U. The development of the female Drosophila reproductive system. J Morphol. 1968;124:143–66.
Wang Z, Lin H. Nanos maintains germline stem cell self-renewal by preventing differentiation. Science. 2004;303:2016–9.
Tarikere S, Ylla G, Extavour CG. Distinct gene expression dynamics in germ line and somatic tissue during ovariole morphogenesis in Drosophila melanogaster. G3 (Bethesda). 2022;12:jkab305.
Matsuoka S, Hiromi Y, Asaoka M. Egfr signaling controls the size of the stem cell precursor pool in the Drosophila ovary. Mech Dev. 2013;130:241–53.
Slaidina M, Banisch TU, Gupta S, Lehmann R. A single-cell atlas of the developing Drosophila ovary identifies follicle stem cell progenitors. Genes Dev. 2020;34:239–49.
Brawley C, Matunis E. Regeneration of male germline stem cells by spermatogonial dedifferentiation in vivo. Science. 2004;304:1331–4.
Flaherty MS, Zavadil J, Ekas LA, Bach EA. Genome-wide expression profiling in the Drosophila eye reveals unexpected repression of notch signaling by the JAK/STAT pathway. Dev Dyn. 2009;238:2235–53.
Flaherty MS, Salis P, Evans CJ, Ekas LA, Marouf A, Zavadil J, Banerjee U, Bach EA. chinmo is a functional effector of the JAK/STAT pathway that regulates eye development, tumor formation, and stem cell self-renewal in Drosophila. Dev Cell. 2010;18:556–68.
Shi J, Niu Q, Gao Q, Fu J, Ma J. Initiation of oogenesis and meiosis in the fetal ovary depends on Dennd1a-mediated production of Wnt5a and retinoic acid from the somatic niches. Front Biosci (Landmark Ed). 2021;26:1513–24.
Acknowledgements
Not applicable.
Funding
This work was supported by the doctoral research start-up fund from Anhui Normal University (No. 762256).
Author information
Authors and Affiliations
Contributions
TZ wrote the manuscript and designed the artwork, JJ edited and revised the manuscript. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Jin, J., Zhao, T. Niche formation and function in developing tissue: studies from the Drosophila ovary. Cell Commun Signal 21, 23 (2023). https://doi.org/10.1186/s12964-022-01035-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-022-01035-7