
Abstract
TRIO and F-actin-binding protein (TRIOBP) also referred to as Tara, was originally isolated as a cytoskeleton remodeling protein. TRIOBP-1 is important for regulating F-actin filament reorganization. TRIOBP variants are broadly classified as variant-1 or − 4 and do not share exons. TRIOBP variant-5 contains all exons. Earlier studies indicated that TRIOBP-4/5 mutation is a pivotal element of autosomal recessive nonsyndromic hearing loss. However, recent studies provide clues that TRIOBP variants are associated with other human diseases including cancer and brain diseases. In this review, recent functional studies focusing on TRIOBP variants and its possible disease models are described.
Similar content being viewed by others


Background
TRIO and F-actin-binding protein (TRIOBP) was originally identified as a cytoskeleton-associated protein that prompts actin cytoskeletal reorganization and cell proliferation and migration [1]. To date, TRIOBPs have been studied as two distinct variants, TRIOBP-1 or TRIOBP-4/5. In this review, the categories of three major TRIOBP variants for human diseases and subcellular functions are discussed as well as recent associations between TRIOBP and cancer.
TRIOBP variants
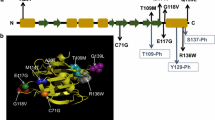
TRIOBP is expressed through two promoters and can be classified into three variants (Fig. 1a). The first variant is TRIOBP-5 (218 kDa), the longest transcript in humans, the first promoter that encodes from exon 1 to exon 24. The second variant is TRIOBP-4 (107 kDa), a shorter protein product, that contains the repeat motifs of exon 6 but none of the carboxyl-terminal domains of TRIOBP-5. The third variant, represented by Tara/TRIOBP-1 (72 kDa) [1], is initiated from the second promoter downstream of exon 6. TRIOBP-1 encodes a protein that does not contain the N-terminal repeat motifs but does include the C-terminal domains of TRIOBP-5 encoded by exons 11–24. Therefore, TRIOBP-1 and TRIOBP-4 do not share any exons or amino acid sequences. Overexpression of TRIOBP-1 (also known as TAP68 or Tara) interacts with and stabilizes the actin cytoskeleton in human HeLa cells [1]. TRIOBP-1 is ubiquitously expressed in all tissues, whereas TRIOBP-4 and TRIOBP-5 are predominantly expressed in the retina and inner ear [2, 3].

Schematic Representation of TRIOBP Variants. a Structure of TRIO and F-actin-binding protein (TRIOBP) variants via alternative splicing. TRIOBP-1 and TRIOBP-4 do not share any genetic sequences. b Genetically deafness frequently was mutated in exons 6–9 of TRIOBP-4. c The first and second coiled-coil regions contain multimerization sites in TRIOBP-1
Role of TRIOBP-4/5 in hearing loss
The phenotype of Deafness, autosomal recessive 28 (DFNB28), is characterized by prelingual, severe to profound sensorineural hearing impairment. In 2006, two research groups found that DFNB28 maps to chromosome 22q13.1 and is associated with TRIOBP-4 and TRIOBP-5 (TRIOBP-4/5) mutations in hearing loss patients in 15 families [2, 3]. To date, in 22 families, all TRIOBP mutations causing human deafness are frequently located in exons 6–9 only in TRIOBP-4/5, but do not affect TRIOBP-1 [4,5,6,7,8] (Fig. 1b).
Inner ear hair cells detect sound through deflection of hearing sensor stereocilia. The stereocilia are linked by rootlets in the cuticular plate of the inner ear (Fig. 2a). The C-terminus of TRIOBP-5, corresponding to coiled-coil domain of TRIOBP-1, is localized at rootlets [9]. Exon 6 of TRIOBP-4/5 encodes several copies of two repeated motifs, R1 and R2; only the R1 motif is the major actin-binding domain of TRIOBP-4/5 [2, 10]. TRIOBP-4/5 is the protein that plays a pivotal role in the formation of rootlets, because Triobp-4 knockout mice cannot form rootlets. In addition, stereociliary fusion in both inner hair and outer hair cells was observed in Triobp-4 knockout mice [11, 12]. The mutations of TRIOBP-4/5 in human hereditary deafness DFNB28 leads to stereociliary fusion caused by disruption of actin networks in the apical region of inner ear hair cells.

Roles of TRIOBP Variants for Human Diseases. a Classification of cellular functions and human diseases based on the major TRIOBP variants 1 or 4/5. b Human TRIOBP expression in different tumor types from The Cancer Genome Atlas database. Adapted from cBioPortal: http://www.cbioportal.org/index.do
Role of TRIOBP-1/Tara/TAP68 in disease
TRIOBP-1 (referred to as Tara or TAP68) is associated with regulating actin cytoskeletal organization. To date, the disease relevance of TRIOBP-1 is less clear than that of TRIOBP-4/5. TRIOBP-1 consists of an N-terminal pleckstrin homology (PH) domain and a C-terminal coiled-coil region which is responsible for its homo-multimerization [1, 13]. TRIOBP-1 is ubiquitously expressed in mammalians [2, 3]. To reorganize the actin cytoskeleton, TRIOBP-1 recruits Nuclear distribution element-like 1 (Ndel1) to F-actin structure [14]. Nde1 interacts with TRIOBP-1 to regulate cell migration. TRIOBP-1 is associated with telomeric repeat binding factor 1 (TRF1) and recruits TRF1 and Tankyrase to the centrosome during mitosis [15]. Thr457 of TRIOBP-1 is phosphorylated by Polo-like kinase 1 (Plk1), which plays important regulatory functions during mitosis. The centrosomal localization of TRIOBP-1 depends on the Thr457 phosphorylation. Centrosomal localization of phosphorylated TRIOBP-1 by Plk1 is important for normal chromosome segregation [16]. Moreover, TRIOBP-1 is ubiquitinated by the E3 ubiquitin ligase HECTD3. The ubiquitination of TRIOBP-1 promotes the degradation of TRIOBP-1. The degradation of TRIOBP-1 caused by HECTD3 knockdown mediates genomic instability [17]. These results indicate that regulation of TRIOBP is important for mitotic processes and the cell cycle and may be associated with cancer.
Accumulation of TRIOBP-1 in the brain
Protein aggregations have been shown in the brain of patients with chronic mental illness caused by disruption of protein degradation, specifically, in schizophrenia studies. A monoclonal antibody was found to detect TRIOBP-1 in brain aggregomes exclusively of schizonphrenia patients, and not controls. TRIOBP-1 has a high propensity to aggregate when overexpressed in neuroblastoma cells, unlike TRIOBP-4. Endogenous TRIOBP-1 may also spontaneously form aggregomes in post-mitosis, consistently causing aggregation of TRIOBP-1 in the differentiated neurons [13, 18]. The aggregation property of TRIOBP-1 is caused by the homo-multimerization site (amino acids 324–348) between the first coiled-coil and second coiled-coil domain (Fig. 1c) [13]. These results suggest that TRIOBP-1 aggregation can lead to mental illness.
Oncogenic TRIOBPs in Cancer
A frameshift deletion mutation of TRIOBP was found in one family with gastric and rectal cancer [19]. Although the mutation could not be determined as contributing to cancer, TRIOBP may be a hereditary factor in cancer. TRIOBP-4/5 is significantly upregulated in pancreatic cancer cells and human cancer tissues. TRIOBP-4/5 facilitates the motility of pancreatic cancer cells by regulating actin cytoskeletal reorganization in the filopodia of the cells [20]. TRIOBP-1 is also abundant in adherent junctions and regulates the organization of epithelial cell sheets and integrity by upregulating E-cadherin transcription leading to carcinogenesis [21] (Fig. 2a). Both transcripts and proteins of TRIOBP-4/5 are upregulated in glioma and glioblastoma multiform (GBM) cell lines. TRIOBP-1/5 is significantly upregulated in GBM patients. Specific siRNAs for TRIOBP-4/5 reduced cell proliferation and migration in GBM cells [22]. Interestingly, specific siRNAs for TRIOBP-1 reduced the cell migration and proliferation in human mesenchymal stem cells [23, 24]. The Cancer Genome Atlas (TCGA) analysis also confirms that TRIOBP mRNA is enhanced in most cancer patients (Fig. 2b).
Taken together, these results contribute to the understanding of TRIOBP function in broad tumors. Consequently, TRIOBPs may be a novel diagnostic marker and therapeutic target for glioma and other cancers.
Further focus for TRIOBPs
The oncogenic function of TRIOBPs is poorly understood. Biochemical evidence is lacking to encourage studies regarding this gene. However, recent studies have shown that all TRIOBP variants may contribute to most cancers including GBM. Based on the available data regarding TRIOBPs, in this review we described and associated their characteristics with cellular biology topics, therefore providing several promising aspects for future TRIOBP studies.
Based on previous studies, TRIOBP variants 1/4/5 have different functions. Our recent study showed that all TRIOBP variants are upregulated in GBM [22]. The molecular mechanisms of TRIOBP variants that regulate tumorigenesis are unclear. Interestingly, TCGA data indicate a relationship between TRIOBPs and most cancers. Tissue-specific TRIOBP-4/5 gene is expressed in brain cancers but not in normal brain tissues. Further studies focusing on TRIOBPs as potential cancer biomarkers or regulators of cancer metabolite are necessary [25].
The accumulation of TRIOBP-1 associated with brain diseases such as schizophrenia has been suggested. Thus, further neurobiological studies regarding the effects of TRIOBPs on brain cancer are necessary.
The cellular localization of TRIOBPs is required to determine the specific function of TRIOBPs in subcellular organelles such as centrosomes, telomeres, filopodia, and adherent junctions. In addition, understanding TRIOBP function during the cell cycle such as mitosis or interphase could provide anti-cancer strategies for tumorigenesis.
Conclusions
The first known gene, TRIOBP, to be associated with hearing loss has recently been linked to cancer. TRIOBP variant 1 and TRIOBP variant 4 have been reported to have completely different functions, but both have been found to be associated with cancer. Finally, a new anti-cancer strategy would be identified through a study of the cancer mechanism of TRIOBP variants.
References
Seipel K, O'Brien SP, Iannotti E, Medley QG, Streuli M. Tara, a novel F-actin binding protein, associates with the Trio guanine nucleotide exchange factor and regulates actin cytoskeletal organization. J Cell Sci. 2001;114:389–99.
Riazuddin S, Khan SN, Ahmed ZM, Ghosh M, Caution K, Nazli S, Kabra M, Zafar AU, Chen K, Naz S, et al. Mutations in TRIOBP, which encodes a putative cytoskeletal-organizing protein, are associated with nonsyndromic recessive deafness. Am J Hum Genet. 2006;78:137–43.
Shahin H, Walsh T, Sobe T, Abu Sa'ed J, Abu Rayan A, Lynch ED, Lee MK, Avraham KB, King MC, Kanaan M. Mutations in a novel isoform of TRIOBP that encodes a filamentous-actin binding protein are responsible for DFNB28 recessive nonsyndromic hearing loss. Am J Hum Genet. 2006;78:144–52.
Diaz-Horta O, Duman D, Foster J, Sirmaci A, Gonzalez M, Mahdieh N, Fotouhi N, Bonyadi M, Cengiz FB, Menendez I, et al. Whole-exome sequencing efficiently detects rare mutations in autosomal recessive Nonsyndromic hearing loss. PLoS One. 2012;7
Gu X, Guo L, Ji H, Sun S, Chai R, Wang L, Li H. Genetic testing for sporadic hearing loss using targeted massively parallel sequencing identifies 10 novel mutations. Clin Genet. 2015;87:588–93.
Naz S, Imtiaz A, Mujtaba G, Maqsood A, Bashir R, Bukhari I, Khan MR, Ramzan M, Fatima A, Rehman AU, et al. Genetic causes of moderate to severe hearing loss point to modifiers. Clin Genet. 2017;91:589–98.
Wesdorp M, van de Kamp JM, Hensen EF, Schraders M, Oostrik J, Yntema HG, Feenstra I, Admiraal RJC, Kunst HPM, Tekin M, et al. Broadening the phenotype of DFNB28: mutations in TRIOBP are associated with moderate, stable hereditary hearing impairment. Hear Res. 2017;347:56–62.
Yan D, Tekin D, Bademci G, Foster J, Cengiz FB, Kannan-Sundhari A, Guo SR, Mittal R, Zou B, Grati M, et al. Spectrum of DNA variants for non-syndromic deafness in a large cohort from multiple continents. Hum Genet. 2016;135:953–61.
Kazmierczak M, Kazmierczak P, Peng AW, Harris SL, Shah P, Puel JL, Lenoir M, Franco SJ, Schwander M. Pejvakin, a candidate Stereociliary rootlet protein, regulates hair cell function in a cell-autonomous manner. J Neurosci. 2017;37:3447–64.
Bao JJ, Bielski E, Bachhawat A, Taha D, Gunther LK, Thirumurugan K, Kitajiri S, Sakamoto T. R1 motif is the major actin-binding domain of TRIOBP-4. Biochemistry. 2013;52:5256–64.
Kitajiri S, Sakamoto T, Belyantseva IA, Goodyear RJ, Stepanyan R, Fujiwara I, Bird JE, Riazuddin S, Riazuddin S, Ahmed ZM, et al. Actin-bundling protein TRIOBP forms resilient rootlets of hair cell Stereocilia essential for hearing. Cell. 2010;141:786–98.
Seki Y, Miyasaka Y, Suzuki S, Wada K, Yasuda SP, Matsuoka K, Ohshiba Y, Endo K, Ishii R, Shitara H, et al. A novel splice site mutation of myosin VI in mice leads to stereociliary fusion caused by disruption of actin networks in the apical region of inner ear hair cells. PLoS One. 2017;12
Bradshaw NJ, Yerabham ASK, Marreiros R, Zhang T, Nagel-Steger L, Korth C. An unpredicted aggregation-critical region of the actin-polymerizing protein TRIOBP-1/Tara, determined by elucidation of its domain structure. J Biol Chem. 2017;292:9583–98.
Hong JH, Kwak Y, Woo Y, Park C, Lee SA, Lee H, Park SJ, Suh Y, Suh BK, Goo BS, et al. Regulation of the actin cytoskeleton by the Ndel1-Tara complex is critical for cell migration. Sci Rep. 2016;6
Lan JP, Zhu YY, Xu LL, Yu HJ, Yu J, Liu X, Fu CH, Wang XG, Ke YW, Huang H, Dou Z. The 68-kDa Telomeric repeat binding factor 1 (TRF1)-associated protein (TAP68) interacts with and recruits TRF1 to the spindle pole during mitosis. J Biol Chem. 2014;289:14145–56.
Zhu YY, Wang C, Lan JP, Yu J, Jin CJ, Huang H. Phosphorylation of Tara by Plk1 is essential for faithful chromosome segregation in mitosis. Exp Cell Res. 2012;318:2344–52.
Yu J, Lan JP, Zhu YY, Li XX, Lai XY, Xue Y, Jin CJ, Huang H. The E3 ubiquitin ligase HECTD3 regulates ubiquitination and degradation of Tara. Biochem Biophys Res Commun. 2008;367:805–12.
Bradshaw NJ, Bader V, Prikulis I, Lueking A, Mullner S, Korth C. Aggregation of the protein TRIOBP-1 and its potential relevance to schizophrenia. PLoS One. 2014;9
Thutkawkorapin J, Picelli S, Kontham V, Liu T, Nilsson D, Lindblom A. Exome sequencing in one family with gastric- and rectal cancer. BMC Genet. 2016;17
Bao JJ, Wang S, Gunther LK, Kitajiri S, Li CY, Sakamoto T. The actin-bundling protein TRIOBP-4 and-5 promotes the motility of pancreatic cancer cells. Cancer Lett. 2015;356:367–73.
Yano T, Yamazaki Y, Adachi M, Okawa K, Fort P, Uji M, Tsukita S, Tsukita S. Tara up-regulates E-cadherin transcription by binding to the Trio RhoGEF and inhibiting Rac signaling. J Cell Biol. 2011;193:319–32.
Lee H, Kim M, Park J, Tran Q, Hong Y, Cho H, Park S, Hong S, Brazil DP, Kim SH, Park J. The roles of TRIO and F-actin-binding protein in glioblastoma cells. Mol Med Rep. 2018;17:4540–6.
Lee SH, Lee YJ, Park SW, Kim HS, Han HJ. Caveolin-1 and integrin beta 1 regulate embryonic stem cell proliferation via p38 MAPK and FAK in high glucose. J Cell Physiol. 2011;226:1850–9.
Yun SP, Ryu JM, Jang MW, Han HJ. Interaction of Profilin-1 and F-actin via a beta-Arrestin-1/JNK signaling pathway involved in prostaglandin E-2-induced human Mesenchymal stem cells migration and proliferation. J Cell Physiol. 2011;226:559–71.
Tran Q, Lee H, Park J, Kim SH, Park J. Targeting Cancer metabolism - revisiting the Warburg effects. Toxicol Res. 2016;32:177–93.
Funding
This work was financially supported by research fund of Chungnam National University in 2015 (Jongsun Park) and by the National Research Foundation of Korea (NRF) grant funded by the Korea Government (MEST) (NRF-2017R1A6A3A11031556, NRF-2016K1A3A1A08953546, NRF-2015R1A2A2A01003597, NRF-2015R1D1A3A01015694).
Author information
Authors and Affiliations
Contributions
SP and JP were involved in the conception, design and drafting of the manuscript. HL and MK performed the illustration of the fig. JP contributed to manuscript preparation. S-HK and JP jointly supervised the review. The authors discussed and reviewed the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consent for publication.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Park, S., Lee, H., Kim, M. et al. Emerging roles of TRIO and F-actin-binding protein in human diseases. Cell Commun Signal 16, 29 (2018). https://doi.org/10.1186/s12964-018-0237-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-018-0237-y