
Abstract
Background
Kiwifruit (Actinidia chinensis var. Chinensis) is abundant with vitamin C and is a rapidly developing crop in China, New Zealand, and other countries. It has been widely used as a raw material for food and kiwifruit wine. Among these, A. chinensis var. chinensis and A. chinensis var. deliciosa are the most valuable kiwifruit in production. Kiwifruit is a typical dioecious plant and its female and male plants have different economic values. Therefore, sex identification, especially at the seedling stage, has important implications for the scientific planning of its production and economic benefits. However, the kiwifruit sex regulation mechanism is very complex and molecular studies are in the initial stages. Currently, there is not a universal and effective sex identification method for A. chinensis.
Methods
In this study, we used a label-free quantitative proteomics approach to investigate differentially accumulated proteins, including their presence/absence and significantly different levels of abundances during A. chinensis var. chinensis male and female flower bud development.
Results
A total of 6485 proteins were identified, among which, 203 were identified in male buds, which were mainly associated with phenylalanine metabolism, tyrosine metabolism, and plant hormone signal transduction. In female buds, 241 were identified, which were mainly associated with the ErbB signaling pathway, growth hormone synthesis, secretion and action, and mRNA surveillance pathway. A total of 373 proteins were significantly differentially accumulated proteins (fold change > 2; P < 0.05), of which, 168 were upregulated and 205 were downregulated. Significant differences between proteins involved 13 signaling pathways, most of which were involved in flavonoid biosynthesis, phenylpropanoid biosynthesis, and starch and sucrose metabolism. Protein interaction analysis showed that enriched protein nodes included cell division cycle 5-like protein, 40S ribosomal protein S8, ribosomal protein, and 40S ribosomal protein like, which interact with 35, 25, 22, and 22 proteins, respectively.
Conclusions
This study provide valuable information for cloning key genes that control sex traits and functionally analyze their roles, which lay a foundation to the development of molecular markers for male and female kiwifruit identification.
Similar content being viewed by others


Introduction
Actinidia chinensis var. chinensis and A. chinensis var. deliciosa are the most valuable kiwifruit in production. As a typical dioecious plant, research has shown that using male plants as rootstocks has strong grafting advantages, but most rootstocks currently used in production consist of 1–2-year-old seedlings. Because there is not a good method for distinguishing male and female plants at the seedling stage, there are both female and male plants being used as rootstocks. Thus, there is inconsistent growth of fruit trees, which complicates the later stages of management and leads to inconsistent kiwifruit quality. Additionally, the relatively long juvenile kiwifruit phase lengthens its breeding time. Therefore, it is important to identify the gender of kiwifruit at the seedling stage. Since the 1980s, researchers have studied genetic markers between male and female plants, including morphological markers, flavonoid content [1], soluble sugar content [2], chlorophyll content [3], and protective enzyme activity [4]. However, these genetic markers are unstable due to their sensitivity to the environment and have not been used effectively in practical applications. Thus, the goal of this study was to resolve the sex-determining mechanism of kiwifruit, which lay a foundation to develop molecular markers to identify gender at the seedling stage.
The sex determination mechanism of dioecious plants is very complex. Sex chromosomes, sex genes, and transcription factors are the key genetic factors that affect the development of male and female individuals and sex organs. Sex determination and sex differentiation are two processes of male and female plant development, but the differentiation process and regulation mechanism remain unclear. Thus far, only a few reports exist on kiwifruit sex development. In A. chinensis var. chinensis, the literature suggests that chromosome 25 may be the sex-chromosome [5] and a male related gene, friendly boy (Frby), has been identified [6]. Frby was cloned and the sequence was mapped to chromosome 8 in female A. chinensis var. Chinensis [7], indicating that the homologous sequences on the X chromosome that correspond with Frby vary considerably. Clearly, the sex differentiation mechanism in kiwifruit is very complex and requires further investigation.
Recently, the sex development mechanism has been studied in dioecious and monoecious plants via proteomics technology on the differential expression of genes/proteins, and provide a theoretical basis for the mechanism of flower bud development in different sexed plants. Sex-related differential proteins of Pistacia chinensis were identified using the proteomic method and it was speculated that phosphoglycerate kinase and temperature-induced lipid delivery proteins may be molecular markers related to gender differences in Pistacia chinensis [8]. Moreover, in the dioecious and monoecious plants of Pistacia chinensis, differentially expressed proteins were detectable during antioxidant stress, ribosome activity, and photosynthesis [9]. According to research on differentially expressed genes in male and female flower buds of Ginkgo biloba (Xiaowen), it was speculated that some female flower buds are located in the female specific region of the W chromosome and homologous gene sequences are present on the Z chromosome [10].
Until now, kiwifruit sex differentiation has rarely been studied. In order to explore the sex differentiation mechanism of A. chinensis var. chinensis, this study employed the labor-free quantitative proteomics method to compare and analyze related differential proteins between male and female flower buds of A. chinensis var. chinensis for the first time. The purpose of this study was to provide valuable information for cloning key genes that control sex traits and functionally analyze their roles, as well as lay a foundation to develop molecular markers to identify gender at the seedling stage.
Materials
Proteins extraction and peptide digestions
The male and female flower buds (about 2 cm in diameter) were collected from the kiwifruit experimental farm during the 2019 growing season in Ankang Municipaty Agricultural Sciences Research Institute, China, (E:108°47′47″, N:32°43′56″) on April 25th, with three biological replicates for each sample, and sample was ground individually in liquid nitrogen.
For cell lysis, cell pellets were suspended on ice in 200 μL SDT (4%SDS, 100 mM Tris-HCl, 1 mM DTT, pH 7.6) lysis buffer. The supernatant were collected and quantified with a BCA Protein Assay Kit (Bio-Rad, USA).
Digestion of protein was performed according to the FASP (filter-aided sample preparation) procedure described by Wisniewski J.R [11].. The peptide of each sample was desalted on C18 Cartridges (Empore™ SPE Cartridges C18 (standard density), bed I.D. 7 mm, volume 3 ml, Sigma), then concentrated by vacuum centrifugation and reconstituted in 40 μl of 0.1% (v/v) formic acid. The peptide content was estimated by UV light spectral density at 280 nm.
Liquid chromatography (LC) - electrospray ionization (ESI) tandem MS (MS/MS) data acquisition
Each sample was separated by HPLC with an Easy-nLC system (Thermo Fisher Scientific), which was coupled to a Q-Exactive Mass Spectrometer (Thermo Scientific). The samples was loaded onto the column (Thermo Scientific Acclaim PepMap100, 100 μm*2 cm, nanoViper C18) in buffer A (0.1% (v/v) formic acid), and separated by analytical column (Thermo Scientific EASY column, 10 cm long, 75 μm inner diameter, 3 μm resin, C18-A2) with a linear gradient of buffer B (84% (v/v) acetonitrile and 0.1%(v/v) formic acid)at a flow rate of 300 nl/min, the column was re-equilibrated with 95% buffer A.
MS data was acquired using a data-dependent top10 method dynamically choosing the most abundant precursor ions from the survey scan (300–1800 m/z). Determination of the target value is based on predictive Automatic Gain Control (pAGC). Target value for the full scan MS spectra was 3 × 106 charges in the 300–1800 m/z range with a maximum IT(injection time) of 50 ms and a resolution of 70,000 at m/z 200, dynamic exclusion duration was 60s. The mass charge ratio of polypeptide and polypeptide fragment was determined according to the following method set: 20 fragments (MS2 scan) were collected after each full scan, and the MS2 activation type was HCD, isolation window was 2 m /z, the resolution of secondary mass spectrometry was 17,500 at 200 m / z, the Normalized Collision Energy was 30 eV and the underfill ratio, which specifies the minimum percentage of the target value likely to be reached at maximum fill time, was defined as 0.1%.
Protein identification and quantification analysis
Raw files were processed with MaxQuant (v 1.5.3.17) using the standard settings against a Actinidia chinensis protein database (uniprot_Actinidia_chinensis_33232.fasta, 76,417 total entries, downloaded 2014/12/12). An initial search was set at a precursor mass window of 6 ppm. The search followed an enzymatic cleavage rule of Trypsin/P and allowed maximal two missed cleavage sites and a mass tolerance of 20 ppm for fragment ions. The cutoff of global false discovery rate (FDR) was set to 1% for protein and peptide identifications. Protein aboundance was calculated on the basis of the normalized spectral protein intensity (LFQ intensity) [12].The differentially accumulated proteins, including proteins of presence or absence abundant (with two or more null values in one group samples), and proteins were defined as regulated with at least 2 fold changes and a p-value ≤0.05 between male flower buds and female flower buds.
Identification of differentially expressed proteins
The differentially expressed proteins, including proteins of presence or absence expressed (with two or more null values in one group samples), and significant differentially expressed proteins (up-regulated more than 2 fold or down regulated less than 0.5 and p-value< 0.05) were screened by UniProt database (https://www.uniprot.org/). The differentially expressed proteins were analyzed; follow-up bioinformatics.
Bioinformatic analysis
Gene ontology (GO) term
Bast2Go (HTTPS:0/www.blast2go.com/) software was employed to annotate the GO term of all the proteins identified in this study. The process of GO annotation includes Blast, Mapping, Annotation, and Annotation Augmentation was conducted in InterProScan (https://www.ebi.ac.uk/interpro/search/sequence/).
Kyoto encyclopedia of genes and genomes (KEGG) pathway
KAAS (KEGG automatic annotation server) was employed to annotate the KEGG pathway.
GO and KEGG pathway enrichment analysis
The enrichment analysis of GO term or KEGG pathway was performed by Fisher’s exact test.
Protein clustering analysis
Hierarchical clustering method was used to cluster the differentially accumulated proteins in the male and female flower buds, and the results were displayed in the heat map.
Protein-protein interactions (PPIs) analysis
To investigate how these significantly differentially accumulated proteins (A total of 373 proteins) are functionally associated with each other, PPI analysis was conducted by using the String (https://string-db.org/), the corresponding results from the String database were downloaded in STV format, then visualized and edit by Cytoscape software (http://www.cytoscape.org/, v3.7.1), as well as further analyze the degree of each targeted protein within the PPI network, which estimate its corresponding significance.
Results
Protein identification
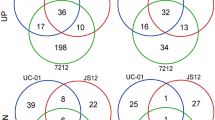
A total of 6485 proteins were identified. Among these, 404 proteins were found in female plants, 384 in male plants, and 5526 identical proteins were found both males and females (Fig. 1c).

Differentially accumulated proteins (DAPs) analysis: a Venn diagram of protein identified in female group. b Venn diagram of protein identified in male group. c Venn diagram of DAPs in multiple pairwise comparison. d The volcano map of DAPs analyzed in male vs female. Note: In the Fig. 1d, the red dot is the protein with significant differentially accumulated proteins, and the black dot is the protein without difference change
Differential accumulated proteins screening
The number of differentially accumulated proteins in each comparisons are shown in Table 1. Consistent presence/absence abundant profile means the differential proteins with two or more non null values in one group of samples and null values in the other group. Significantly changing in abundance means fold change between the two groups of samples and p-value obtained by T test, were used to draw the volcano map (Fig. 1d).
Cluster analysis of differentially accumulated proteins
Using the hierarchical cluster to cluster the differentially accumulated proteins in the comparison group, the genes usually classified as one group have practical relationship in some biological processes, or in some metabolism and signal pathway. In this study, proteins with similar accumulated patterns were clustered together and displayed in the form of heat map by differentially accumulated proteins (Fig. 2). It were identified 373 proteins that were significantly differentially accumulated between male and female buds. These differentially accumulated proteins were mainly divided into two clusters, of the 205 proteins which were up-regulated in female and down-regulated in male, 168 which were up-regulated in male and down-regulated in female. It can be seen that there is a significant difference in protein expression between male and female flower buds of A. chinensis, indicating that different proteins have different functions and ways of action between the development and differentiation of male and female flower buds.

Heatmap of the abundances levels of the 373 proteins in male vs female buds using the laber-free quantitative protein test. Note: In Fig. 2, each row represents a protein, each column represents a group of samples, in which red represents a significant up-regulated protein, blue represents a significant down-regulated protein, and gray represents no protein quantitative information
GO enrichment analysis of differentially accumulated proteins
GO functional enrichment analysis showed that 1257 terms were annotated including 710 terms in the biological process (BP), 365 terms in the molecular function (MF) and 185 terms in the cellular component (CC).
GO enrichment analysis of proteins of presence or absence
The top 20 functional enrichment proteins were analyzed (Fig. 3). For biological processes, mitochondrial fission is a representative GO term, followed by organelle fission. Among the molecular functions, the nutrient reservoir activity is highly representative. For cell components, nuclear part, histone methyltransferase complex, set1c/Compass complex are highly representative.

The top 20 GO enrichment of proteins of presence/absence in male vs female buds
GO enrichment analysis of significant differentially accumulated proteins
For biological processes, carbohydrate metabolic process is a representative GO term, followed by lipid localization and lipid transport. Among the molecular functions, the proteins with redox activity and catalytic activity were highly representative. For cell components, protein DNA complex and DNA packaging complex (DNA packaging) were highly representative. Complex and nucleosome were also highly representative (Fig. 4).

The top 20 GO enrichment of significant differentially accumulated proteins in male vs female buds
KEGG enrichment analysis of differentially accumulated proteins
KEGG enrichment analysis of proteins of presence or absence
KEGG enrichment pathway of proteins only in female buds or in male buds was showed in Fig. 5, among which ErbB signaling pathway, growth hormone synthesis, secretion and action, mRNA surveillance pathway are occured mainly in female plants (Fig. 5a), and phenylalanine metabolism, tyrosine metabolism and plant hormone signal transduction are occured mainly in male plants (Fig. 5b). Phenylalanine metabolism is one of the most important pathways of plant secondary metabolism [13], which includes mainly two metabolic pathways, namely phenylpropanoid pathway and flavonoid biosynthesis pathway [14,15,16].

The top 20 KEGG pathway enrichment of differentially accumulated proteins in male vs female buds; a. Proteins accumulated only in female buds. b. Proteins accumulated only in male buds
KEGG enrichment pathway of all proteins that contain expressed only in female and in male buds was showed in Fig. 6, five metabolic pathways have been found by analyzing whether there are differential proteins enriched in KEGG pathway, in order of P-value from small to large, including ErbB signaling pathway, plant hormone signal transduction and so on.

The KEGG pathway enrichment of identified proteins on presence/absence in male vs female buds
Among all pathways, the plant hormone signal transduction was the most important, in which, five proteins were identified, including jasmonic acid-amido synthetase and auxin transporter-like protein, which were expressed only in male buds, and three proteins that were expressed only in female buds (Table 2). jasmonic acid-amido synthetase is an enzyme in the jasmonates metabolism pathway [17]. Jasmonate is an important class of lipid-derived plant hormone that regulates multiple aspects of plant growth and development, as well as plays key roles in stamen development [18,19,20,21,22,23]. jasmonic acid-amido synthetase (Jar1) is a regulatory gene that regulates the metabolism of jasmonic acid (JA) to jasmonic acid-isoleucine conjugate (Ja-ile). Wang Jing et al. [24] showed that the reproductive activities of the root knot nematode in a JA deletion mutant and JA-ile deletion mutant (aos1, opr3, Jar1) were significantly lower than the wild type. Exogenous Ja-ile restored the reproductive capacity of root knot nematodes in Jar1 roots to the same level as wild type roots. Dai Liangying et al. [15] analyzed Jasmonate functioning and its biosynthesis and metabolic pathway, and found that some jasmine are involved in growth and pollen development, while some are resistant to insects, disease, and stress through the participation of Jar1. It was also proved that auxin regulates stamen development [25] and there were no stamen or abnormal anther morphology as the germination rate of anthers decreased in an auxin transporter protein mutant [26]. Once the auxin transporter is destroyed, it affects stamen development, which thereby affects pollination and fertilization, resulting in reduced fruit yield and quality.
KEGG enrichment analysis of significant differentially accumulated proteins
In the KEGG pathway (Fig. 7), the first three pathways were flavonoid biosynthesis, arginine biosynthesis, and starch and sucrose metabolism. Flavonoid serves many functions, among which, it can attract pollination and regulate seed germination through flower and fruit pigmentation. Previous studies showed that a Petunia hybrida mutant exhibited pollen sterility, while wild-type stigma possess a certain substance that restores pollen fertility. The analysis found that this substance is a flavonoid compound that can promote pollen tube growth and pollen function [27]. Arginine serves a variety of functions that promotes cell division, seed germination, flower bud differentiation, root growth, and development. In flower bud differentiation, arginine synthesizes polyamine to promote bud and pollen germination and increases the fruit setting rate [28]. During arginine metabolism, putrescine is synthesized first, then polyamine is synthesized.

The KEGG pathway enrichment of significantly differential accumulated proteins in male vs female buds
Network analysis of protein-protein interaction (PPI)
Protein-protein interactions (PPIs) engage in dynamic biological processes and play important roles in these diverse processes, and are essential for understanding life at the system-level. Results revealed 243 nodes (proteins) and 542 edges (interactions), indicating a highly profound network of sex differentiation of A. chinensis var. chinensis buds (Fig. 8). Aoa2r6pkf6, a cell division cycle 5-like protein (Cdc5L), was a node protein that interacted with 35 proteins (yellow); the proteins that interacted with Cdc5L were mainly related to ribosome formation. The expression level of Cdc5L in female buds was 2.22-fold higher than male buds. A previous study found that Cdc5L is a regulator of mitotic progression as a pre-mRNA splicing factor. The deletion of endogenous Cdc5L decreases cell viability via dramatic mitotic arrest [29], which has not been reported in plants, but has been widely studied in animals, especially in the proliferation of cancer cells [30]. Cdc5L is also essential for porcine oocyte maturation [31]. Cdc5L could be a potential molecular marker in neuroblastoma [32] and may play an important role in mitosis and cell activity during female flower bud formation.

PPI networks of proteins from differentially identified proteins in the bud
Discussion
In production, dioecious plants are utilized differently based on sex. The reproductive function of female plants is higher than male plants, but the growth potential of female plants is lower than male plants [33]. The different evolutionary directions of male and female plants determine their growth patterns [34], and plants exhibit different stress responses to environmental stress based on sex [35]. Studies on kiwifruit production using male plants as rootstocks showed that they have a strong advantage. Significant differences were detected mainly between molecular functional proteins, of which, most were involved in catalytic activities. In the KEGG enrichment analysis, the plant hormone signaling pathway was mainly involved in the male and female plants comparison, which indicated that the signaling pathways of male and female flower bud development and differentiation were different, while the pathway of significantly different proteins mainly concentrated on flavor biosynthesis, phylopanoid biosynthesis, and sucrose metabolism. Studies on the content differences of key substances in the significant pathways between male and female plants and sex changes induced by these key substances in kiwifruit have been reported. Among them, the effects of hormones and polyamines on sex differentiation in horticultural plants are more frequently reported [36]. Zheng Hanyu et al. [1] reported that the flavonoid content in female kiwifruit leaves was higher than male leaves. Xu Linyue et al. [37] found that the content of putrescine and spermidine in male leaves was significantly higher than female leaves, and the spermine content in female leaves was significantly higher than male leaves. Studies have also shown that exogenous putrescine and spermidine increase the number of plant flower buds [28]. Hale et al. [38] used genotyping-by-sequencing (GBS) technology to locate a sex-linked marker, ac36306, in the natural population of Chinese gooseberry in the United States. After analysis, it was found that the marker was closely linked to arginine decarboxylase, a key enzyme in the polyamine biosynthesis pathway from arginine to putrescine. Zhu Daye et al. [2] found that the soluble sugar content in male leaves was significantly higher than female leaves. Understanding which genes and metabolic pathways are involved in the sex developmental process of Chinese kiwifruit is important for developing androgynous markers suitable for different natural populations of kiwifruit varieties.
DNA molecular markers have also been reported for kiwifruit [5, 38,39,40]. Accordingly, we identified male and female plants in natural populations of A. chinensis var. chinensis and A. chinensis var. deliciosa at the seedling stage, however, none of the aforementioned DNA markers were 100% accurate. This may be due to the fact that most of the mapping populations are specific populations (most were F2) and the DNA markers are only suitable for specific materials. Therefore, it may be that these DNA markers do not co-segregate with sex-determining genes during meiosis; the exchange between markers and sex-determining genes will occur, which will lead to false positives or negatives.
By combining the protein interaction network analysis and pathway annotation results, we obtained a more comprehensive and systematic view of male and female development at the molecular level, which will aid further research and mining for molecular mechanisms. The protein interaction network analysis showed that most proteins interacted with Cdc5L, which promotes cell activities. According to the GO annotations, the molecular function of the protein family is DNA binding and the biological process is cellular response to fibroblast growth factor stimulus (FGF). The genes involved in microspore development and pollen formation in Arabidopsis are cellulose loss deposition [41]. Cdc5L stimulates fibroblast growth and may participate in pollen formation. Through the GO, KEGG, and PPI analyses, the differences between male and female plants ultimately resulted in sex differentiation.
The sex determination mechanism of dioecious plants is very complex. Sex chromosomes, sex genes, and transcription factors are the key genetic factors that affect the development of male and female individuals or organs. Sex determination and sex differentiation are two processes of male and female plant development. Plant sex can be determined only at the reproductive growth stage. However, the differentiation process and regulation mechanism remain unclear, and research on kiwifruit sex development is limited. DNA molecular markers, which are used to distinguish male and female A. chinensis var. chinensis, were all located at different positions on chromosome 25 [5]. Further analysis indicated that most genes on chromosome 25 are associated with the metabolic pathways of plant hormone signal transduction. Thus, it was inferred that chromosome 25 may play a key role as a sex chromosome in A. chinensis var. chinensis sex development and differentiation. Additionally, among the differentially accumulated proteins that were identified, we found that the number of stress resistance proteins in male plants was greater than in female plants, indicating that male plants are more stress resistant. This finding was consistent with the results of previous studies, which found that male plants were more salt and drought tolerant than female plants [42]. Our findings are also consistent with practical applications, wherein male plants used as rootstocks possess higher growth dominance in production.
Conclusion
Taken together, This study employed the labor-free quantitative proteomics method to compare and analyze related differentially accumulated proteins between male and female flower buds of A. chinensis var. chinensis for the first time, which provide valuable information for cloning key genes that control sex traits and functionally analyze their roles, and lay a foundation to the development of molecular markers for male and female kiwifruit identification. Combined with genome-wide association study (not published), in the future, we will focus on several candidate genes/proteins that occured in KEGG enrichment analysis, such as jasmonic acid-amido synthetase, auxin transporter-like protein and so on.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author upon reasonable request.
Abbreviations
- Frby:
-
Friendly boy
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- PPIs:
-
Protein-protein interactions
- DAPs:
-
Differentially accumulated proteins
- BP:
-
Biological process
- MF:
-
Molecular function
- CC:
-
Cellular component
- Cdc5L:
-
Cell division cycle 5-like protein Chronic wrap restraint stress
- GBS:
-
Genotyping-by-sequencing
- FGF:
-
Fibroblast growth factor stimulus
References
Zheng HY, Li Y, Jin XH, Zhao Y, Luo GJ. The variation of plant morphology and secondary metabolite of male and female Actinidia arguta during growth. Agric Sci J Yanbian Univ. 2018;40:16–21.
Zhu DY, Yi GT, Qi WB. Difference in soluble sugar content of male and female plants of Actinidia chinensis. J Central South Forestry Univ. 1983;8:160–3.
Han GJ, Jia CZ, Deng X, Han XC, Meng CJ, Mao ZX, et al. Photosynthetic Characteristics and Chlorphyll Content Between Male and Female Plants with Differernt Ploidy in Actinidia arguta var. Purpurea. Guizhou Agric Sci. 2017;45:40–3.
Sun H, Li X, Cao WW, Jiang D, Piao YL. Differences in protective enzyme activity andcontents of Vc and pigments between female and male plants of Actinidia arguta (Seib.et Zucc.) Plangch.ex Miq. Agric Sci J Yanbian Univ. 2015;37:107–10.
Takashi A, Isabelle MH, Haruka O, Takuya M, Kenji B, Ikuo K, et al. A Y-encoded suppressor of feminization arose via lineage-specific duplication of a Cytokinin response regulator in kiwifruit. Plant Cell. 2018;30:780–95.
Takashi A, Sarah MP, Erika VG, Isabelle MH, Shigeo SS, Minori S, et al. Two Y chromosome-encoded genes determine sex in kiwifrui. bioRxiv. 2019; first posted online Apr. 22. https://doi.org/10.1101/615666.
Zhou WY, Qu D, Zhang XJ, Zhang WH, Liu YF, Zhang Y. Bioinformatics analysis and cloning of fasciclin gene family in kiwifruit. J Fruit Sci. 2020;37:1144–55.
Xiong EH. Proteomic identification of sex related proteins in Deocious plant Pistacia chinensis; 2013.
Chen YF, Shu SC, Bai Q, Ruan FN. Differential Proteomics Analysis on Flower Buds of Mono ecious Pistacia chinensis in Sex Differentiation Phases by Label-free LC-MS/MS. J Northeast Forestry Univ. 2019;47:34–43.
Zhang XW. Study on the differentially expressed genes in male and female flower buds of Ginkgo biloba L.[Ph. D. Dissertation]. Tai’an: Shandong Agricultural University; 2014.
Wisniewski JR, Alexandre Z, Nagarjuna N, Matthias M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6(5):359–62. https://doi.org/10.1038/nmeth.1322.
Luber CA, Cox J, Lauterbach H, Fancke B, Selbach M, Tschopp J, et al. Quantitative proteomics reveals subset-specific viral recognition in dendritic cells. Immunity. 2010;32(2):279–89. https://doi.org/10.1016/j.immuni.2010.01.013.
Gao X. Research progress of plant phenylalanine ammonia lyase. Modern Agric Sci Technol. 2009;01:30–3.
Jiang M, Cao JS. Chalcone synthase gene. J Cell Biol. 2007;29:525–9.
Cui JD, Li Y, Mou DH. Research progress of phenylalanine ammonia lyase (PAL). Sci Technol Food Ind. 2008;29:306–8.
Xu F. Cloning and expression of gbpal and gbans genes in Ginkgo biloba and the effect of ALA on flavonoid content. [Ph. D. Dissertation]. Tai’an: Shandong Agricultural University; 2008.
Dai LY, Li X, Luo K. Jasmonate and its functions. J Hunan Agric Univ. 2000;26:150–4.
Carey CC, Strahle JT, Selinger DA, Chandler VL. Mutations in the pale aleurone colorI regulatory gene of the Zea mays anthocyanin pathway have distinct phenotypes relative to the functionally similar TRANSPARENT TESTA GLABRal gene in Arabidopsis thaliana. Plant Cell. 2004;16(2):450–64. https://doi.org/10.1105/tpc.018796.
Mathews H, Clendennen SK, Caldwell CG, Liu XL, Wagner DR. Activation tagging in tomato identifies a transcriptional regulator of anthocyanin biosynthesis, modification, and transport. Plant Cell. 2003;15(8):1689–703. https://doi.org/10.1105/tpc.012963.
Kubo H, Peeters AJM, Aarts MGM, Pereira A, Koornneef M. ANTHOCY ANINLESS2, a homeobox gene affectinganthocyanin distribution and root development in Arabidopsis. Plant Cell. 1999;11(7):1217–26. https://doi.org/10.1105/tpc.11.7.1217.
Nesi N, Jond C, Debeaujon I, Caboche M, Lepiniec L. The Arabidopsis TT2 gene encodes an R2R3 MYB domain protein that acts as a key determinant for proanthocyanidin accumulation in developing seed. Plant Cell. 2001;13(9):2099–114. https://doi.org/10.1105/TPC.010098.
Nesi N, Debeaujon I, Jond C, Pelletier G, Caboche M, Lepiniec L. The TT8 gene encodes a basic helix-loop-helix domain protein required for expression of DFR and BAN genes in Arabidopsis siliques. Plant Cell. 2000;12(10):1863–78. https://doi.org/10.1105/tpc.12.10.1863.
Quattrocchio F, Wing JF, Woude K, van der Woude K, Mol JNM, Koes R. Analysis of bHLH and MYB domain proteins: species-specific regulatory differences are caused by divergent evolution of target anthocyanin genes. Plant J. 1998;13(4):475–88. https://doi.org/10.1046/j.1365-313X.1998.00046.x.
Wang J, Wu XH, Qi LMG, Te RGL, Qi Z, Kang Y. JA mediated signaling pathway regulate plant defense responses of root knot nematode. Acta Agriculturae Boreali-Sinica. 2014;29:234–8.
Cheng YF, Dai XH, Zhao YD. Anxin biosynthesis by the YUCCA flavin monooxygenases controls the formation of floral organs and vascular tissues in Arabidopsis. Genes Dev. 2006;20(13):1790–9. https://doi.org/10.1101/gad.1415106.
Gan ZY. Study on the regulatory effects of apple auxin transporter PIN on dwarfing of M9 interstem and pollen development[Ph. D. Dissertation]. Beijing: China Agricultural University; 2018.
Wang MX. Function Research of Flavonoid Biosynthesis in Style Regulating Pollen Tube Growth. Shandong Agricultural University, Tai’an:Shandong Agricultural University. 2015.
Yang HQ, Gao HJ. Physiological function of arginine and its metabolites in plants. J Plant Physiol Mol Biol. 2007;33(01):1–8.
Qiu HY, Zhang XB, Ni WK, Shi WD, Fan H, Xu J, et al. Expression and clinical role of Cdc5L as a novel cell cycle protein in hepatocellular carcinoma. Dig Dis Sci. 2016;61(3):p795–805. https://doi.org/10.1007/s10620-015-3937-9.
Zhang ZW, Mao WP, Wang LS, Liu MG, Zhang WT, Wu Y, et al. Depletion of CDC5L inhibits bladder cancer tumorigenesis. J Cancer. 2020;11(2):353–63. https://doi.org/10.7150/jca.32850.
Liu XM, Wang YK, Liu YH, Yu XX, Wang PC, Li X, et al. Single-cell transcriptome sequencing reveals that cell division cycle 5-like protein is essential for porcine oocyte maturation. J Biol Chem. 2018;293(5):1767–80. https://doi.org/10.1074/jbc.M117.809608.
Cai YX, Wu ZX, Wang Y,Wu YM. Expression of CDC5L in neuroblastoma and its clinical and prognostic significance. J Clin Pediatr Surg. 2019;18(6):472-9.
Agren J. Sexual differences in biomass and nutrient allocation in the dioecious rubus Chamaemorus. Ecology. 1988;69(4):962–73. https://doi.org/10.2307/1941251.
Popp JW, Reinartz JA. Sexual dimorphism in biomass fllocation and clonal growth of xanthoxylum americanum. Am J Bot. 1988;75(11):1732–41. https://doi.org/10.1002/j.1537-2197.1988.tb11249.x.
Xu X, Yang F, Yin CY, Li CY. Research advances in sex-specific responses in dioecious plants to environmental stresses. Chin J Appl Ecol. 2007;18(11):2626–31.
Chen XH, Zeng GW, Chen YP, Cao BS. Relationship of plant hormones and polyamine with sex reversion of cucumber (Cucumis sativus L.). J Zhejiang Univ (Agriculture and Life Sciences). 2001;27:639–42.
Xu LY, Jiang D, Guo MY, Huang YB, Piao YL. Determination of polyamine content in leaves of the male and female plants of Actinidia arguta by HPLC. Agric Sci J Yanbian Univ. 2016;38:220–5.
Hale I, Melo ATO, Gustafson H. Sex-linked molecular markers for two cold-hardy kiwifruit species, Actinidia arguta and A kolomikta. Eur J Horticultural Sci. 2018;83:236–46.
Gill GP, Harvey CF, Gardner RC, Fraser LG. Development of sex-linked PCR marker for gender identification in Actinidia. Theor Appl Genet. 1998;97(3):439–45. https://doi.org/10.1007/s001220050914.
Yao CC, Wang YJ, Liu XF, Long ZX. Obtainment and application of RAPD marker S1032—850 linked to male Genein Actinidia. J Agric Biotechnol. 2005;13:557–16.
Li J, Yu M, Geng LL. The fasciclin-like arabinogalactan protein gene, FLA3, is involved in microspore development of Arabidopsis. Plant J. 2010;64(3):482–97. https://doi.org/10.1111/j.1365-313X.2010.04344.x.
Chen LH, Zhang S, Zhao HX, Korpelainen H, Li CY. Sex-related adaptive responses to interaction of drought and salinity in Populus yunnanensis. Plant Cell Environ. 2010;3:1767–78.
Acknowledgements
We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Funding
This work was supported by the Sci-technological Project of Shaanxi Province “Research and development of genetic characteristics of main kiwifruit germplasm resources in Qinba area” (2021NY-085), the Department of Education Project of Shaanxi Province “Research of Genetic Basison and Construction of Kiwifruit Germplasm Resource Nursery in Southern Shaanxi”.
Author information
Authors and Affiliations
Contributions
Yu Z conceived and designed the experiments. Yue x W, Wan y Z and Shi m Z participated in the data analysis.Yu Z provided guidance for preparing the manuscript. Wen h Z provided samples. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, Y., Wang, Y., Zhou, W. et al. Comparative proteomics analysis reveals differentially accumulated proteins associated with male and female A. chinensis var. chinensis bud development. Proteome Sci 19, 8 (2021). https://doi.org/10.1186/s12953-021-00176-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12953-021-00176-w