
Abstract
Background
Familial hypercholesterolemia (FH) is the first molecularly and clinically characterized genetic disease of lipid metabolism. It is an autosomal dominant disorder with significantly elevated levels of total cholesterol and low density of lipoprotein cholesterol in serum, which would lead to extensive xanthomas and premature coronary heart disease. Mutations in low density lipoprotein receptor (LDLR), proprotein convertase subtilisin/kexin type 9 and Apo lipoprotein B-100 (APOB) have been identified to be the underlying cause of this disease.
Methods
Genetic testing and reports of the mutations in the Chinese population are still limited. In this study, 11 unrelated Chinese FH families were enrolled to detect the candidate gene variants by DNA direct sequencing.
Results and conclusion
We identified 12 mutations (11 in LDLR and one in APOB) in ten FH families. Three novel LDLR mutations (c.516C>A/p.D172E, c.1720C>A/p.R574S and c.760C>T/p.Q254X) were identified and co-segregated with the affected individuals in the families. Our discoveries not only further supports the significant role of LDLR in FH, but also expands the spectrum of LDLR mutations. These new insights will contribute to the genetic diagnosis and counseling of FH patients.
Similar content being viewed by others
Background
Dyslipidemia is a common disorder of lipid metabolism and major cardiovascular risk factor, accounting for 54% of population-attributable risk for myocardial infarction (Yusuf et al. 2004). Familial hypercholesterolemia (FH, OMIM#143890) is one of the most severe lipid dysfunctions, characterized by elevated total cholesterol and low density of lipoprotein cholesterol amounts in serum (Jannes et al. 2015). It is inherited in an autosomal dominant fashion, with frequencies of heterozygotes and homozygotes estimated at 1:200 and 1:300,000 worldwide (Foody and Vishwanath 2016). Total cholesterol and LDL-C concentrations in heterozygous patients often range between 9 and 14 mmol/L and 5–10 mmol/L, whereas homozygous patients show levels from 17 to 26 mmol/L and >10 mmol/L, respectively (European Association for Cardiovascular Prevention & Rehabilitation et al. 2011; Goldberg et al. 2011; Hovingh et al. 2013). Such high plasma TC and LDL-C levels may result in xanthelasmas and atherosclerotic plaques, the primary factors causing premature coronary heart disease (CHD) (Najam and Ray 2015). However, the levels of TC and LDL-C can be effectively reduced by statin (Vogt 2015).
To date, more than 1741 low density lipoprotein-receptor gene (LDLR) variants have been reported in the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) (Lahtinen et al. 2015). Meanwhile, two distinct disease-causing genes were identified in FH patients: proprotein convertase subtilisin/kexin type9 (PCSK9) (Al-Mashhadi et al. 2013) and Apo lipoprotein B-100 (APOB) (Alves et al. 2014). The clinical phenotypes resulting from these gene mutations vary. For example, APOB mutations may cause the least severe phenotype of the three (Soutar and Naoumova 2007). Besides LDLR, APOB and PCSK9 mutations, some copy number variants (CNVs) (Myocardial Infarction Genetics, Kathiresan et al. 2009; Costelloe et al. 2012) and rare mutations in associated genes, such as LDLRAP1 (Maglio et al. 2014), PNPLA5 (Lange et al. 2014) and APOC3 (Jorgensen et al. 2014) have also been reported in FH patients.
LDLR gene mutations represent 85–90% of disease-causing mutations in FH patients (Futema et al. 2014), however, most countries (including China) do not have valid nationwide registries for FH. Indeed, no more than 20 studies have assessed Chinese FH patients using genetic analysis, and novel variants identified remain scarce (Dai et al. 2011).
Therefore, in this study we investigated the possible causative gene in Chinese FH families. We identified three novel mutations (c.516C>A/p.D172E, c.1720C>A/p.R574S and c.760C>T/p.Q254X) in the affected members of their families. Based on the best of our knowledge, these mutations have not been reported in previous studies and were not presented in either our control cohorts, dbSNP or Exome Variant Server database (http://evs.gs.washington.edu/EVS/).
Methods
The Review Board of The Second Xiangya Hospital of the Central South University has approved this research. All related subjects have consented to this study.
Patients and subjects
Eleven unrelated Chinese FH patients were enrolled after being diagnosed and treated at Department of Cardiology, The Second Xiangya Hospital of Central South University. Definition of FH was based on the standard (TC > 9 mmol/L and LDL-C > 5 mmol/L) formulated by European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS) (European Association for Cardiovascular Prevention & Rehabilitation et al. 2011; Goldberg et al. 2011; Hovingh et al. 2013). We have also taken CHD and xanthelasmas patients into account. Two hundred unrelated healthy Chinese subjects were recruited as control subjects to detect whether any sequence changes might be a common polymorphism (Xiang et al. 2014). Clinical data and detailed family history were collected for each subjects.
Methods
DNA extraction
Genomic DNA was extracted from peripheral blood of all the subjects by using a DNeasy Blood & Tissue Kit (Qiagen, Valencia, CA) as previously described (Xiang et al. 2014).
Mutation sequencing
The entire coding regions and flanking intronic sequences of LDLR (NM_000527) and PCSK9 (NM_174936) together with the p.R3527 mutation (part of exon 26) of APOB (NM_000384) were performed with polymerase chain reaction (PCR; primer sequences will be provided upon requests). Sanger sequencing was applied by the ABI 3100 Genetic Analyzer (ABI, Foster City, CA).
Multiple sequence alignments and bioinformatic prediction of mutation
The standard sequences of LDLR, PCSK9 and APOB refer to Ensemble database. The polyphen2 (polymorphism phenotyping, http://genetics.bwh.harvard.edu/pph2/) (Sunyaev et al. 2000), Sorting Intolerant From Tolerant (SIFT, http://provean.jcvi.org/) (Ng and Henikoff 2003) and MutationTaster (www.mutationtaster.org) programs (Schwarz et al. 2010) will be used for the prediction of pathogenicity of genetic mutations.
Results
Clinic data
A total of 11 unrelated FH probands were enrolled in this study, among whom four and seven showed homozygous and heterozygous phenotypes, respectively. Demographic details, clinical features, and lipid levels are shown in Table 1. In addition, the proband F3 had a history of xanthomas (Fig. 1), while proband F8 had a history of CHD.

Xanthomas of FH homozygous individual (proband F3). On elbow (a) and knee (b)
Mutation spectrum
Eleven mutations in LDLR and one mutation in APOB were found by DNA direct sequencing in ten probands and co-segregated with all the affected members (Table 2). No mutation of PCSK9 was found in any probands. Among these ten probands with variants, proband F1 carried the homozygous mutation, probands F3 and F8 carried compound heterozygous mutations. All three patients showed xanthomas, CHD or high TC and LDL-C levels. The mean serum TC was 18.57 mmol/l (min 17.05 mmol/l, max 20.15 mmol/l), and the mean serum LDL-C was 17.12 mmol/l (minimum 16.54 mmol/l, maximum 18.21 mmol/l). Other probands (F2, F4, F5, F6, F7 and F9) carried heterozygous mutations in LDLR. The mean serum TC was 9.12 mmol/l (min 7.52 mmol/l, max 11.23 mmol/l), and the mean serum LDL-C was 7.80 mmol/l (minimum 5.50 mmol/l, maximum 11.2 mmol/l). The proband F10 was detected a heterozygous mutation in APOB, whose serum TC was 7.8 mmol/l and serum LDL-C was 5.47 mmol/l. Currently none mutation of candidate genes was identified in proband F11. The serum TC was 18.91 mmol/l and serum LDL-C was 16.84 mmol/l.
Novel mutations
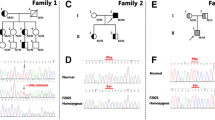
By sequencing analysis of LDLR, PCSK9 and APOB, three novel mutations in LDLR (c.516C>A/p.D172E, c.1720C>A/p.R574S and c.760C>T/p.Q254X) were detected and co-segregated with the affected FH family members in our study (Fig. 2). These newly identified mutations were not found in either our control cohort of 200 patients, dbSNP or the Exome Variant Server database (http://evs.gs.washington.edu/EVS/). Alignment of LDLR amino acid sequences from Human, Ptroglodytes, Mmulatta, Mmusculus, Trubripes, Drerio etc., revealed that the affected amino acids were evolutionarily conserved (Fig. 3). Three programs for analyzing protein functions, MutationTaster, polyphen2 and SIFT, predicted that these three variants are disease causing, probably damaging and deleterious, respectively (Table 2). All three different algorithm based bioinformatics programs showed a consistent result of detrimental effect of these variants, suggesting that these three sites (D172, Q254 and R574) play important roles in the function of LDLR.

Pedigrees and sequencing results of the LDLR mutations of the families affected with FH. The hypercholesterolemic patient is indicated by a black symbol. The normal cholesterolemic individuals are indicated by open symbols. N normal, M mutant, arrow the proband

Analysis of the mutations of LDLR. Alignment of multiple LDLR protein sequences across species (from Ensemble). Red columns show conserved regions in site D172 (a), R574 (b) and Q264 (c) respectively
Discussion and future perspective
According to EAS data, the estimated percentage of individuals diagnosed with FH in 2013 was less than 1% in approximately 180 countries/territories, including China. Moreover, China is a multi-racial nation, and such heterogeneous population is expected to harbor a number of novel gene mutations (Nordestgaard et al. 2013). In the present study, we employed direct sequencing to explore mutations of possible causative genes for FH. Twelve LDLR and APOB variants were detected, including three unique mutations (c.516C>A/p.D172E, c.1720C>A/p.R574S and c.760C>T/p.Q254X). The incidence rates of LDLR and APOB mutations were 82 and 9% in these Chinese FH families, respectively. These data corroborated previous reports demonstrating that over 85% of FH cases are due to hereditary mutations in LDLR, with the APOB variant (p.Arg3527) accounting for 5% of FH cases (Futema et al. 2014).
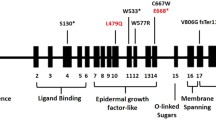
The novel mutations (c.516C>A/p.D172E, c.1720C>A/p.R574S and c.760C>T/p.Q254X) were detected in Families F1, F2 and F3, respectively. In Family F1, one homozygous and four heterozygous (c.516C>A/p.D172E) patients were identified. This mutation is found in the highly conserved ligand binding domain of LDLR, and may affect LDL binding (Gent and Braakman 2004). In Family F2, four patients (c.1720C>A/p.R574S) were diagnosed as FH. The substitution of the alkaline amino acid (Arg) by the polar but not charged amino acid (Ser) at position 574 of LDLR may be the genetic basis for FH. Proband F3 was a compound heterozygous mutation (c.760C>T/p.Q254X/c.1216C>A) carrier. The disease-causing SNP (c.1216C>A) is a splicing site that was used to exclude the natural splicing site, and causes a deletion of 31 bp from the mRNA, probably introducing premature termination of four codons after R406 (Bourbon et al. 2007). If the mRNA carries a nonsense mutation (c.760C>T/p.Q254X), it will be degraded by nonsense mediated mRNA decay. The LDLR protein without the C-terminal domain will not be found in the cell membrane. Therefore, serum TC and LDL levels were consistent with homozygous mutation carriers, such as proband F1.
Furthermore, APOB mutation (c.10579C>T/p.R3527W) was detected in Family F10. This mutation could influence the conformation and structure of APOB in the binding domain. This may decrease LDL degradation and increase TC and LDL-C levels (Gaffney et al. 1995). Besides, APOB mutations often show a lighter phenotype than LDLR and PCSK9 mutations in patients. Our clinical and molecular data also confirmed this viewpoint.
Among all LDLR mutations, 27% (three out of eleven) of variants are found in exon 4. According to previous studies assessing Chinese FH patients, 24% of variants are found in exon 4 of LDLR, and our data are consistent with this percentage (Austin et al. 2004). Such a high frequency may be caused by the large exon size, but could be also related to selection bias.
In addition, no disease causing mutations in candidate genes were detected in proband F11, despite high TC and LDL-C levels in the patient. This might be caused by variations in other genes such as APOC3 and PNPLA5 (Jorgensen et al. 2014, Lange et al. 2014). Furthermore, CNVs also play a crucial role in FH for unique cases (Myocardial Infarction Genetics, Kathiresan, et al. 2009, Costelloe et al. 2012). Considering the serious phenotype of proband F11, we believe that genetic factors may have had a dominant effect. This will be identified through whole-exome sequencing in the future.
In conclusion, we detected mutations of LDLR, APOB and PCSK9 in 11 Chinese FH families, among which ten were found to be deleterious mutations. Meanwhile, three novel LDLR mutations (c.516C>A/p.D172E, c.1720C>A/p.R574S and c.760C>T/p.Q254X) were identified. More patients were not available for statistical analyses, and no percentage of Chinese FH patients with positive genetic diagnosis could be revealed in this study. However, the present identification of three novel mutations and other mutations not only further supports the significant role of LDLR in FH, but also expands the spectrum of LDLR mutations. These new insights will contribute to the genetic diagnosis and counseling of FH patients.
References
Al-Mashhadi RH, Sorensen CB, Kragh PM, Christoffersen C, Mortensen MB, Tolbod LP, Thim T, Du Y, Li J, Liu Y, Moldt B, Schmidt M, Vajta G, Larsen T, Purup S, Bolund L, Nielsen LB, Callesen H, Falk E, Mikkelsen JG, Bentzon JF (2013) Familial hypercholesterolemia and atherosclerosis in cloned minipigs created by DNA transposition of a human PCSK9 gain-of-function mutant. Sci Transl Med 5(166):161–166
Alves AC, Etxebarria A, Soutar AK, Martin C, Bourbon M (2014) Novel functional APOB mutations outside LDL-binding region causing familial hypercholesterolaemia. Hum Mol Genet 23(7):1817–1828
Austin MA, Hutter CM, Zimmern RL, Humphries SE (2004) Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol 160(5):407–420
Bourbon M, Sun XM, Soutar AK (2007) A rare polymorphism in the low density lipoprotein (LDL) gene that affects mRNA splicing. Atherosclerosis 195(1):e17–e20
Costelloe SJ, El-Sayed Moustafa JS, Drenos F, Palmen J, Li Q, Whiting S, Thomas M, Kivimaki M, Kumari M, Hingorani AD, Tzoulaki I, Jarvelin MR, Ruokonen A, Hartikainen AL, Pouta A, Walters RG, Blakemore AI, Humphries SE, Coin LJ, Talmud PJ (2012) Gene-targeted analysis of copy number variants identifies 3 novel associations with coronary heart disease traits. Circ Cardiovasc Genet 5(5):555–560
Dai YF, Sun LY, Zhang XB, Wang LY (2011) Research progression of LDLR mutations in Chinese familial hypercholesterolemia. Yi Chuan 33(1):1–8
European Association for Cardiovascular Prevention & Rehabilitation, Reiner Z, Catapano AL, De Backer G, Graham I, Taskinen MR, Wiklund O, Agewall S, Alegria E, Chapman MJ, Durrington P, Erdine S, Halcox J, Hobbs R, Kjekshus J, Filardi PP, Riccardi G, Storey RF, Wood D, ESC Committee for Practice Guidelines (CPG) 2008–2010 and 2010–2012 Committees (2011) ESC/EAS guidelines for the management of dyslipidaemias: the task force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur Heart J 32(14):1769–1818
Foody JM, Vishwanath R (2016) Familial hypercholesterolemia/autosomal dominant hypercholesterolemia: molecular defects, the LDL-C continuum, and gradients of phenotypic severity. J Clin Lipidol 10(4):970–986
Futema M, Plagnol V, Li K, Whittall RA, Neil HA, Seed M, Simon Broome C, Bertolini S, Calandra S, Descamps OS, Graham CA, Hegele RA, Karpe F, Durst R, Leitersdorf E, Lench N, Nair DR, Soran H, Van Bockxmeer FM, U. K. Consortium, Humphries SE (2014) Whole exome sequencing of familial hypercholesterolaemia patients negative for LDLR/APOB/PCSK9 mutations. J Med Genet 51(8):537–544
Gaffney D, Reid JM, Cameron IM, Vass K, Caslake MJ, Shepherd J, Packard CJ (1995) Independent mutations at codon 3500 of the apolipoprotein B gene are associated with hyperlipidemia. Arterioscler Thromb Vasc Biol 15(8):1025–1029
Gent J, Braakman I (2004) Low-density lipoprotein receptor structure and folding. Cell Mol Life Sci 61(19–20):2461–2470
Goldberg AC, Hopkins PN, Toth PP, Ballantyne CM, Rader DJ, Robinson JG, Daniels SR, Gidding SS, de Ferranti SD, Ito MK, McGowan MP, Moriarty PM, Cromwell WC, Ross JL, Ziajka PE, National Lipid Association Expert Panel on Familial Hypercholesterolemia (2011) Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol 5(3 Suppl):S1–S8
Hovingh GK, Davidson MH, Kastelein JJ, O’Connor AM (2013) Diagnosis and treatment of familial hypercholesterolaemia. Eur Heart J 34(13):962–971
Jannes CE, Santos RD, de Souza Silva PR, Turolla L, Gagliardi AC, Marsiglia JD, Chacra AP, Miname MH, Rocha VZ, Filho WS, Krieger JE, Pereira AC (2015) Familial hypercholesterolemia in Brazil: cascade screening program, clinical and genetic aspects. Atherosclerosis 238(1):101–107
Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjaerg-Hansen A (2014) Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med 371(1):32–41
Lahtinen AM, Havulinna AS, Jula A, Salomaa V, Kontula K (2015) Prevalence and clinical correlates of familial hypercholesterolemia founder mutations in the general population. Atherosclerosis 238(1):64–69
Lange LA, Hu Y, Zhang H, Xue C, Schmidt EM, Tang ZZ, Bizon C, Lange EM, Smith JD, Turner EH, Jun G, Kang HM, Peloso G, Auer P, Li KP, Flannick J, Zhang J, Fuchsberger C, Gaulton K, Lindgren C, Locke A, Manning A, Sim X, Rivas MA, Holmen OL, Gottesman O, Lu Y, Ruderfer D, Stahl EA, Duan Q, Li Y, Durda P, Jiao S, Isaacs A, Hofman A, Bis JC, Correa A, Griswold ME, Jakobsdottir J, Smith AV, Schreiner PJ, Feitosa MF, Zhang Q, Huffman JE, Crosby J, Wassel CL, Do R, Franceschini N, Martin LW, Robinson JG, Assimes TL, Crosslin DR, Rosenthal EA, Tsai M, Rieder MJ, Farlow DN, Folsom AR, Lumley T, Fox ER, Carlson CS, Peters U, Jackson RD, van Duijn CM, Uitterlinden AG, Levy D, Rotter JI, Taylor HA, Gudnason V Jr, Siscovick DS, Fornage M, Borecki IB, Hayward C, Rudan I, Chen YE, Bottinger EP, Loos RJ, Saetrom P, Hveem K, Boehnke M, Groop L, McCarthy M, Meitinger T, Ballantyne CM, Gabriel SB, O’Donnell CJ, Post WS, North KE, Reiner AP, Boerwinkle E, Psaty BM, Altshuler D, Kathiresan S, Lin DY, Jarvik GP, Cupples LA, Kooperberg C, Wilson JG, Nickerson DA, Abecasis GR, Rich SS, Tracy RP, Willer CJ, NHLBI Grand Opportunity Exome Sequencing Project (2014) Whole-exome sequencing identifies rare and low-frequency coding variants associated with LDL cholesterol. Am J Hum Genet 94(2):233–245
Maglio C, Mancina RM, Motta BM, Stef M, Pirazzi C, Palacios L, Askaryar N, Boren J, Wiklund O, Romeo S (2014) Genetic diagnosis of familial hypercholesterolaemia by targeted next-generation sequencing. J Intern Med 276(4):396–403
Myocardial Infarction Genetics Consortium, Kathiresan S, Voight BF, Purcell S, Musunuru K, Ardissino D, Mannucci PM, Anand S, Engert JC, Samani NJ, Schunkert H, Erdmann J, Reilly MP, Rader DJ, Morgan T, Spertus JA, Stoll M, Girelli D, McKeown PP, Patterson CC, Siscovick DS, O’Donnell CJ, Elosua R, Peltonen L, Salomaa V, Schwartz SM, Melander O, Altshuler D, Ardissino D, Merlini PA, Berzuini C, Bernardinelli L, Peyvandi F, Tubaro M, Celli P, Ferrario M, Fetiveau R, Marziliano N, Casari G, Galli M, Ribichini F, Rossi M, Bernardi F, Zonzin P, Piazza A, Mannucci PM, Schwartz SM, Siscovick DS, Yee J, Friedlander Y, Elosua R, Marrugat J, Lucas G, Subirana I, Sala J, Ramos R, Kathiresan S, Meigs JB, Williams G, Nathan DM, MacRae CA, O’Donnell CJ, Salomaa V, Havulinna AS, Peltonen L, Melander O, Berglund G, Voight BF, Kathiresan S, Hirschhorn JN, Asselta R, Duga S, Spreafico M, Musunuru K, Daly MJ, Purcell S, Voight BF, Purcell S, Nemesh J, Korn JM, McCarroll SA, Schwartz SM, Yee J, Kathiresan S, Lucas G, Subirana I, Elosua R, Surti A, Guiducci C, Gianniny L, Mirel D, Parkin M, Burtt N, Gabriel SB, Samani NJ, Thompson JR, Braund PS, Wright BJ, Balmforth AJ, Ball SG, Hall A, Wellcome Trust Case Control Consortium, Schunkert H, Erdmann J, Linsel-Nitschke P, Lieb W, Ziegler A, Konig I, Hengstenberg C, Fischer M, Stark K, Grosshennig A, Preuss M, Wichmann HE, Schreiber S, Schunkert H, Samani NJ, Erdmann J, Ouwehand W, Hengstenberg C, Deloukas P, Scholz M, Cambien F, Reilly MP, Li M, Chen Z, Wilensky R, Matthai W, Qasim A, Hakonarson HH, Devaney J, Burnett MS, Pichard AD, Kent KM, Satler L, Lindsay JM, Waksman R, Knouff CW, Waterworth DM, Walker MC, Mooser V, Epstein SE, Rader DJ, Scheffold T, Berger K, Stoll M, Huge A, Girelli D, Martinelli N, Olivieri O, Corrocher R, Morgan T, Spertus JA, McKeown P, Patterson CC, Schunkert H, Erdmann E, Linsel-Nitschke P, Lieb W, Ziegler A, Konig IR, Hengstenberg C, Fischer M, Stark K, Grosshennig A, Preuss M, Wichmann HE, Schreiber S, Holm H, Thorleifsson G, Thorsteinsdottir U, Stefansson K, Engert JC, Do R, Xie C, Anand S, Kathiresan S, Ardissino D, Mannucci PM, Siscovick D, O’Donnell CJ, Samani NJ, Melander O, Elosua R, Peltonen L, Salomaa V, Schwartz SM, Altshuler D (2009) Genome-wide association of early-onset myocardial infarction with single nucleotide polymorphisms and copy number variants. Nat Genet 41(3):334–341
Najam O, Ray KK (2015) Familial hypercholesterolemia: a review of the natural history, diagnosis, and management. Cardiol Ther 4(1):25–38
Ng PC, Henikoff S (2003) SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 31(13):3812–3814
Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, Wiegman A, Santos RD, Watts GF, Parhofer KG, Hovingh GK, Kovanen PT, Boileau C, Averna M, Boren J, Bruckert E, Catapano AL, Kuivenhoven JA, Pajukanta P, Ray K, Stalenhoef AF, Stroes E, Taskinen MR, Tybjaerg-Hansen A, European Atherosclerosis Society Consensus Panel (2013) Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J 34(45):3478–3490
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D (2010) Mutation taster evaluates disease-causing potential of sequence alterations. Nat Methods 7(8):575–576
Soutar AK, Naoumova RP (2007) Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat Clin Pract Cardiovasc Med 4(4):214–225
Sunyaev S, Ramensky V, Bork P (2000) Towards a structural basis of human non-synonymous single nucleotide polymorphisms. Trends Genet 16(5):198–200
Vogt A (2015) The genetics of familial hypercholesterolemia and emerging therapies. Appl Clin Genet 8:27–36
Xiang R, Fan LL, Huang H, Cao BB, Li XP, Peng DQ, Xia K (2014) A novel mutation of GATA4 (K319E) is responsible for familial atrial septal defect and pulmonary valve stenosis. Gene 534(2):320–323
Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, Lanas F, McQueen M, Budaj A, Pais P, Varigos J, Lisheng L, Investigators IS (2004) Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case–control study. Lancet 364(9438):937–952
Authors’ contributions
RD, L-LF, J-JL and Z-JH carried out the genetic studies, participated in the sequence alignment and drafted the manuscript. M-JL, Y-QC and S-PZ participated in the sample collecting. RX, HH and KX participated in the design of the study and performed the statistical analysis. All authors read and approved the final manuscript.
Acknowledgements
We thank the patients and their families for participating in this study. We thank the State Key Laboratory of Medical Genetics of China for technical assistance. This study was supported by the National Natural Science Foundation of China (81370394), the National Basic Research Program of China (973 Program) (2012CB517900), the Fundamental Research Funds for Central Universities of Central South University (2015zzts274).
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Du, R., Fan, LL., Lin, MJ. et al. Mutation detection in Chinese patients with familial hypercholesterolemia. SpringerPlus 5, 2095 (2016). https://doi.org/10.1186/s40064-016-3763-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40064-016-3763-3