
Abstract
Breast cancer, the most frequent female malignancy, is often curable when detected at an early stage. The treatment of metastatic breast cancer is more challenging and may be unresponsive to conventional therapy. Immunotherapy is crucial for treating metastatic breast cancer, but its resistance is a major limitation. The tumor microenvironment (TME) is vital in modulating the immunotherapy response. Various tumor microenvironmental components, such as cancer-associated fibroblasts (CAFs), tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs), are involved in TME modulation to cause immunotherapy resistance. This review highlights the role of stromal cells in modulating the breast tumor microenvironment, including the involvement of CAF-TAM interaction, alteration of tumor metabolism leading to immunotherapy failure, and other latest strategies, including high throughput genomic screening, single-cell and spatial omics techniques for identifying tumor immune genes regulating immunotherapy response. This review emphasizes the therapeutic approach to overcome breast cancer immune resistance through CAF reprogramming, modulation of TAM polarization, tumor metabolism, and genomic alterations.
Similar content being viewed by others

Introduction
Breast cancer is the most diagnosed malignancy in females, with about 2.3 million new cases globally in the year 2020, which accounted for 11.7% of all cancer incidences [1]. According to the International Agency for Research on Cancer (IARC), these numbers are estimated to increase to over 3 million by 2040 [1]. The cancer progression is a multistep integrated process controlled by several genetic and epigenetic factors. Some researchers stated that epigenetic alteration is another hallmark of most cancers due to its critical role in the initiation of carcinogenesis [2,3,4,5]. However, cancer develops because of a chaotic tumor microenvironment (TME), including various infiltrating immune cells like tumor-associated macrophages (TAMs), dendritic cells (DCs), lymphocytes, and other stromal cells like cancer-associated fibroblasts (CAFs), endothelial cells, pericytes, and extracellular matrix (ECM) [6]. All these components participate in a complex manner through cell-cell and cell-matrix interactions to shape the microenvironment conducive to tumor progression [3, 7, 8]. Cancer cells frequently educate stromal cells, such as fibroblasts, macrophages, vascular cells, adipocytes, and immune cells, to support their growth and spread to distant sites. The stromal component might dominate the tumor tissue in most solid cancers [9]. CAFs constitute a significant part of stroma, and various tumor-derived factors are known to induce the activation of fibroblasts to CAFs [10]. In addition to cancer cells, different infiltrating immune cells like tumor-associated neutrophils (TANs), TAMs, DCs, and mast cells (MCs) have subsequently appeared to enhance the activation of stromal cells, which, in turn, shape the immune suppressive TME. This interplay comprises immune-inhibitory circles to provide a favorable TME for tumor growth. Moreover, CAFs indirectly alter anticancer immunity and induce T cell dysfunction and immunologic tolerance by upregulating the expression of immune checkpoint molecules like PD1/PD-L1 [7]. The dynamic and mutual association between cancer cells and the TME has the potential to either curb or promote the spread of the disease. Immune cells that have invaded the tumors prevent their growth by destroying immunomodulatory neoplastic cells. However, they might also be responsible for developing tumor resistance to treatment by influencing tumor immunogenicity and selecting tumor clones that can cause immune exhaustion [11]. Moreover, the immune cells in the TME have a dual function in cancer development and metastasis. The type 1 helper T cells (Th1), cytotoxic T lymphocytes (CTLs), and natural killer cells (NK cells) are associated with an immune stimulant microenvironment. In contrast, the regulatory cells of the TME, including type 2 helper T cells (Th2), TAMs, regulatory T cells (Tregs), and myeloid-derived suppressor cells (MDSCs), are associated with immunosuppressive microenvironment and poor outcomes [12, 13]. These cells prevent tumor growth by eradicating immunogenic neoplastic cells or altering tumor immunogenicity, aiding tumor escape [14]. Besides these cells, chemokines and cytokines are essential members of the tumor immune microenvironment (TIME) and play a significant role in maintaining the equilibrium between protumor and antitumor immune responses [15]. The intricate interactions between the cancer cells and the immunological niche affect immunotherapy and many other anticancer therapies.
The development of immuno-based therapy in breast cancer has made significant progress over the past two decades. Though different immunotherapeutic strategies have been explored in breast cancer, the number of immunotherapy-based clinical trials increased after the advent of immune checkpoint inhibitors (ICIs) and antibody-drug conjugates (ADCs). As of January 2022, according to the data identified on clinicaltrials.gov, there were 745 immunotherapy-based trials enrolling patients with solid tumors of different cancers, out of which 450 trials (60.4%) were explicitly dedicated to breast cancer [16]. The ongoing development of immunotherapy has contributed to improved outcomes for many breast cancer patients. Nevertheless, insights from clinical landscapes highlight that TIME composition strongly influences the efficacy of immunotherapy [17]. Importantly, immune cells are now recognized as critical players in the emergence of resistance mechanisms to immunotherapy in breast cancer. These mechanisms hinder the establishment of long-lasting treatments and cause cancer growth [11].
This review article focuses on the complex and dynamic function of TME to elucidate the interplay between the stromal and immune cells. It aims to explore therapeutic strategies that may reverse immunotherapy resistance in breast cancer.
Recent clinical advances in breast cancer immunotherapy
Immunotherapy is a rapidly evolving field in the treatment of cancer. It involves harnessing the body’s immune system to recognize, target, and eliminate cancer cells. Several types of immunotherapy strategies have shown promising results in treating various cancers. These involve developing ICIs including monoclonal antibodies (mAbs) to block the immunosuppressive molecules and to improve the cytotoxicity of tumor-infiltrating lymphocytes such as CTLA4, PD1, and PD-L1. In addition, ADCs and cancer vaccines have also exhibited the potential to deliver cytotoxic drugs and boost the immune system. Although the FDA has approved various immunotherapeutic agents for treating many cancers, only a few are in clinical settings or undergoing clinical trials for the treatment of breast cancer [18].
Immune checkpoint inhibitors
The interaction of PD1 expressed in T cells with PD-L1 on cancer cells suppresses the proliferation and survival of T cells, which ultimately leads to immunosuppression. Pembrolizumab and nivolumab are ICIs that target PD1 to prevent PD1/PD-L1 interaction [19]. In contrast, atezolizumab and durvalumab act against PD-L1 to inhibit its interaction with PD1 [20, 21]. The FDA recently approved pembrolizumab for combinatorial application with chemotherapy to treat recurrent, unresectable and metastatic TNBCs [22]. FDA has approved atezolizumab and nab-paclitaxel combination therapy for treating locally advanced or metastatic TNBCs with PD-L1-positive tumors [23]. In contrast, atezolizumab was approved earlier for the treatment of TNBC along with paclitaxel in breast cancer but later used for other cancers but not for breast [23]. In the phase I clinical trial, another ICI, avelumab, which targets PD-L1, yielded an overall response rate of 3.0%, whereas in the case of TNBC patients, the overall response rate is 5.2% [24]. CTLA4, or CD152, is another immune checkpoint constitutively expressed on Treg and activated effector T cells [25, 26]. During an immune response, particularly in the priming phase of T cell activation, CTLA4 is highly upregulated. It induces negative feedback through the binding of CD80/CD86 to prevent CD28 co-stimulation and reduce T cell activation by competitive inhibition [27, 28]. Ipilimumab and tremelimumab, two anti-CTLA4 humanized monoclonal antibodies, attenuate negative signals on T cell co-stimulation [29, 30]. Ipilimumab, in combination with nivolumab and paclitaxel, is used to treat the resistance in the early stages of TNBC [31]. Tremelimumab and durvalumab are under clinical trial to treat metastatic TNBC, as the former alone did not exhibit promising results [32]. Although TNBC has excellent response rates to ICIs as compared to other sub-types of breast cancers, the efficacy as a single therapeutic agent is still poor. Moreover, these ICIs exhibit several adverse effects such as hypophysitis, colitis, thyroid dysfunction and pneumonitis etc [33, 34].
Monoclonal antibodies
mAbs have revolutionized cancer treatment and are widely used as immunotherapeutic agents. They target specific molecules or antigens on cancer cells and modulate the immune response to fight against cancer [35]. Trastuzumab is the first FDA-approved mAb for HER2+ breast cancer treatment [36]. It is employed along with other chemotherapeutic drugs to manage early-stage and metastatic HER2+ breast cancer [37, 38]. It inhibits the HER2 pathway to cause G1 phase arrest and inflicts apoptosis and angiogenesis in breast cancer cells by inhibiting the PI3K pathway [39, 40]. Trastuzumab also stimulates innate and adaptive immune responses through NK cells, activation of CTLs, and suppression of Treg cells [41]. However, this mAb is reported to be cardiotoxic in nature [42]. Pertuzumab is another mAb-approved drug used in combination with trastuzumab as the first-line treatment for HER2+ as well as non-hormonal metastatic breast cancer therapy [43]. It blocks the dimerization of HER2 with HER3 and EGFR to exhibit cytotoxic effects. This combination is also recommended for early treatment as well as trastuzumab-resistant breast cancers [44]. Margetuximab is also used in combination with chemotherapeutic agents for the management of HER2+ metastatic breast cancer. It enhances NK cells activity due to its high affinity for CD16A and poor binding to inhibitory CD32B [45]. This drug also activates macrophages, and neutrophils to elicit immune responses [46, 47]. Leronlimab, an anti-CCR5 antibody, is currently in a phase I clinical trial for TNBC treatment [48]. There is an ongoing clinical study to establish the efficacy of trastuzumab in combination with other chemotherapeutic drugs for treating breast cancer including TNBC [49]. Bispecific antibodies are also promising for breast cancer treatment. Zanidatamab (ZW25), targeting ECD II and ECD IV domains of HER2, is currently being clinically tested for HER2+ metastatic breast cancer cases (NCT04224272) [50] (Table 1). Zenocutuzumab (MCLA-128) and KN026 are also in clinical trials for the treatment of HER2+ metastatic breast cancer [51, 52]. However, these mAbs rarely cause severe allergic or inflammatory reactions [49].
Antibody-drug conjugates
Antibody-drug conjugates (ADCs) have been developed to deliver a high concentration of anticancer drugs in the cells that overexpress the targeted antigen recognized by its antibody. This targeted delivery approach allows more efficient and selective delivery of the cytotoxic payload to cancer cells, thereby minimizing damage to healthy cells. While ADCs are not traditionally classified as immunotherapy, they utilize the immune system’s mechanisms for targeted delivery and enhanced efficacy [53]. Trastuzumab-emtansine (T-DM1), an ADC, is produced by conjugating trastuzumab with emtansine, a microtubule inhibitor and blocks HER2 signalling [40]. This ADC is approved by the European Medicines Agency (EMA) and the FDA for treating HER2+ early invasive and metastatic breast cancer patients as a third-line therapy [54, 55]. Combining tucatinib with trastuzumab and capecitabine increases OS and reduces brain metastasis in patients with HER2+ breast cancer (NCT02614794) [56]. Trastuzumab-deruxtecan (T-DXd) is another EMA and FDA-approved ADC to treat metastatic HER2+ and HER2-low breast cancer as a second-line therapy when surgical removal is not recommended [57]. Deruxtecan, present in this ADC, is a Topo I inhibitor, causing inhibition of DNA replication, cell cycle arrest, and apoptosis [56]. However, T-DXd frequently exhibits several adverse effects including interstitial lung disease or pneumonitis. Depending on the severity of this adverse effect, the treatment may need to be discontinued. Proper optimization of the treatment and the adverse effect management are required for maximal benefit [58, 59]. Although most ADCs, such as trastuzumab-duocarmazine, MM-302, and RC48-ADC, are based on targeting HER2, researchers are also exploring other ADCs like ladiratuzumab-vedotin and cofetuzumab-pelidotin by selecting TNBC-expressing LIV1 and PI3K as molecular targets, respectively [26]. Ladiratuzumab-vedotin (SGN-LIV1A) is another ADC, consisting of an anti-LIV-1 monoclonal antibody linked to monomethyl auristatin E (MMAE), which is a potent microtubule-disrupting agent. LIV-1 is a membrane-type metalloprotease overexpressed in most TNBCs [60]. Clinical trials are conducted in patients with LIV1 positive, unresectable, locally advanced, or metastatic breast cancers with this ADC (NCT01969643) [61]. In this trial, SGN-LIV1A is tested in TNBC patients in one arm and the HER2+ patients in another arm. After completion of this trial, an overall response rate (ORR) was found to be 32% and a progression-free survival (PFS) of 11.3 weeks in patients with metastatic TNBC [61]. Sacituzumab-govitecan, also known as IMMU-132, is an antibody-ADC targeting TROP2, an antigen often overexpressed in TNBC. The antibody component of sacituzumab-govitecan binds to TROP2 on the surface of cancer cells, allowing the targeted delivery of SN-38 (Topo I inhibitor) to the tumor cells [62]. This ADC has been shown to improve the ORR and PFS of metastatic TNBC patients [63]. Datopotamab deruxtecan, another anti-TROP2 mAb, is being investigated in clinical trial for unresectable or metastatic TNBC as well as HER2+/ HER2− breast cancer cases (NCT05374512; NCT05104866) [64, 65]. It has exhibited better therapeutic efficacy with lesser adverse effects compared to sacituzumab-govitecan [64, 65]. Moreover, glycoprotein-NMB (gpNMB), significantly expressed in 40% TNBC, was explored to develop glembatumumab vedotin (CDX-011) for MMAE delivery and reported to achieve better ORR [66]. However, this ADC was less effective than capecitabine in the METRIC phase II trial (NCT01997333) [67]. Ongoing clinical trials, such as SGN-LIV1A and IMMU-132, are designed to further evaluate the potential benefits of these ADCs in treating TNBC (NCT04230109, NCT04468061, NCT03424005) [68]. Although all of these ADCs are found to be well-tolerated, these may cause cardiotoxicity, hematologic disorders, gastrointestinal problems, hepatoxicity and oral mucositis which require proper monitoring and therapeutic attention [68].
Vaccines
HER2 has been used as a target to develop breast cancer vaccines. Due to its large molecular weight, vaccines have been generated by targeting HER2 based on one or more HER2-derived peptides. E75 or nelipepimut-S is a peptide-based vaccine that targets HLA-A2-restricted nonapeptide derived from the extracellular domain of HER2 protein [69]. GP2 is another peptide vaccine that targets HLA-A2-restricted nonapeptide based on the transmembrane domain of the HER2 protein [70]. Moreover, the AE37 vaccine is a 12-mer peptide that targets the intracellular domain of modified HER2 [71]. Four amino acids containing peptides have been added to the intracellular domain of the HER2 protein to enhance immunogenicity. HLA-A2-restricted peptides (E75 and GP2) are the epitope of MHC class I molecules and primarily activate CD8+ cytotoxic T cells [72]. In contrast, the AE37 peptide is presented by MHC class II molecules and primarily stimulates CD4+ T cell activation to elicit an immune response [73]. These vaccines are reported to be effective against low HER2-expressing breast cancer and TNBC patients [74, 75]. Researchers are actively exploring the development of vaccines targeting non-HER2 tumor-associated antigens (TAAs). Cancer-testis antigens (CTAs) are often found to be overexpressed in cancer. NY-ESO-1 is an important CTA selected to generate breast cancer vaccine [76, 77]. Other CTAs chosen for vaccine development are Wilms tumor protein 1 (WT1), the melanoma-associated antigen-12 (MAGE-12), the folate receptor alpha (FRα), T-box transcription factor brachyury and the tumor suppressor transcription factor p53 [78,79,80]. MUC1 vaccines have been generated based on mucin1 TAA and are being evaluated in clinical trials for TNBC (NCT00986609) [81] (Table 1). Other vaccines developed based on TAA for TNBC patients are PVX-410 (peptide vaccine) and STEMVAC (DNA vaccine) [82]. Moreover, tumor-associated carbohydrate (TAC) antigens are also used for developing vaccines. P10s-PADRE is a TAC vaccine currently in a clinical trial for TNBC patients [83]. Recently, an α-lactalbumin-targeted vaccine is also in a phase I clinical trial [84]. However, vaccines are not equally active in all patients due to spatial mutational heterogeneity within individual tumors. Therefore, personalized neoantigen vaccines are generated and tested in clinical trials against TNBC [85, 86].
Recently, DC vaccines are being developed and investigated for different cancer therapies including breast cancer. Two types of DC vaccines: DC polypeptide vaccine and DC gene vaccine, are primarily studied for breast cancer immunotherapy. One DC vaccine containing MUC1 antigen along with two adjuvants has shown to stimulate cytokine release and CD4+ and CD8+ T cell-mediated immune response in TNBC mouse model [87]. However, MUC1-based vaccines are not found to be clinically effective in early breast cancer [88]. DC vaccine loaded with P32 peptide exerts immune response using in vivo breast cancer model [89]. Another DC vaccine with MHC-II binding HER3 peptide exhibits anti-HER3 CD4+ Th1 immune response to inhibit tumor growth in HER3 overexpressing in vivo breast cancer murine model [90]. Oxidized cell lysate-loaded spherical nucleic acids (SNAs) act as potent immunotherapeutic agent for TNBC [91]. Exosome-loaded DC vaccines are also being investigated using in vivo breast cancer mouse and ex vivo organoid models [92]. DC vaccines developed using CD133 mRNA and MUC1 mRNA along with CTLA4 blockade have shown enhanced immune response and inhibition of tumor growth using in vivo murine TNBC models [93, 94]. Several clinical trials are in progress to evaluate the therapeutic efficacy of DC vaccines in breast cancer (NCT03450044, NCT01431196, NCT01730118, NCT02061332, NCT001070211) [95,96,97,98,99].
Novel immunotherapy-based approaches
Recently, several novel immunotherapeutic strategies have been explored for solid cancers. Activation and expansion of DCs are promising immunotherapeutic approaches. TLR agonists, the potent activators of DCs have exhibited to stimulate immune response for better treatment in breast cancer [100]. Clinical trial is underway to investigate the efficacy of TLR agonist in breast cancer (NCT00821964) [101]. Moreover, these agonists are being clinically tested along with Flt3L, a stimulator of DC, for the treatment of advanced cases (NCT03789097) [102].
CAR-T, CAR-M, CAR-NK, and bispecific antibodies are reported to be very effective for breast cancer. Several CAR-T therapies are in clinical trials to investigate their safety and efficacy. The efficacy of MUC1-specific-CAR-T cells in TNBC has been established. Moreover, its safety is also being evaluated (NCT04020575) [103] (Table 1). However, c-Met-CAR-T cells are well tolerated in TNBC patients (NCT01837602) [103]. Mesothelin-targeted CAR-T cells are in phase I study in TNBC (NCT02580747) [104] (Table 1). CAR-M therapy, such as CAR-147, has shown reduced ECM deposition and enhanced T cell infiltration using HER2+in vivo breast cancer models [105]. CT-0508, an anti-HER2 CAR-M is in phase I trial for refractory HER2+ breast cancer patients with a parallel assignment intervention model (NCT04660929) [106] (Table 1). CD44v6-specific CAR-NK cells have been found to be effective in a mammosphere model of TNBC [107]. However, engineered immune cell application also involves several limitations. Unavailability of suitable target, low antigen density, loss of antitumor activity due to exhaustion and poor infiltration possess challenges in CAR-T therapy [108]. Moreover, this therapy exhibits several adverse effects due to targeting antigens present in the normal tissues [109]. Common adverse effects such as neurotoxicity, thrombocytopenia, cytokine release syndromes may be life threatening [110]. C-reactive protein along with various inflammatory cytokines are also elevated [111]. CAR-NK and CAR-M also exhibit several issues like limited proliferation capacity, insufficient infiltration, limited availibility, etc [108].
Tumor microenvironment-mediated immune resistance: role of immune cells
CAF-TAM crosstalk-mediated immune resistance
CAFs and immune microenvironment regulation
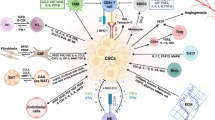
CAFs, primarily generated by the trans-differentiation of resident fibroblasts, have emerged as critical therapeutic targets [10, 112, 113]. Although recent studies have identified a subset of CAFs with tumor-restricting function, modulating CAFs in combination with immunotherapy improved outcomes in different preclinical models [114,115,116,117]. Different subsets of CAFs have been identified based on the expressions of various biomarkers. Characterization and exploring the immunomodulatory role of CAF populations will be beneficial in dealing with immune resistance [118, 119] (Fig. 1). Single-cell RNA sequencing (scRNA seq) of TNBC revealed the presence of two CAF subpopulations. One state is related to features of myofibroblasts, and the other is characterized by high expression of growth factors and immunomodulatory molecules [120]. This study has indicated the involvement of a diverse array of immunoregulatory molecules in the stromal-immune crosstalk in breast cancer. Exploring gene signatures from inflammatory CAFs (iCAFs) and differentiated‐perivascular cells revealed a strong association with cytotoxic T cell dysfunction and exclusion [120]. Costa et al. have identified four different subsets of CAFs (CAF-S1 to CAF-S4) with distinct properties and levels of activation in TNBC patients’ specimens by simultaneous analysis of six fibroblast markers (FAP, integrin β1, α-SMA, FSP1, PDGFRβ, and CAV1) [121]. Among these subpopulations, CAF-S1 exhibits the most prominent immunosuppression action. This CAF subset activates Treg cells and promotes Treg-mediated inhibition of T cell proliferation [122]. FAP+CAFs exhibit an immunosuppressive effect and diminish the efficacy of anti-PD-L1 therapy by secreting CXCL12 in murine pancreatic ductal adenocarcinoma (PDAC) in vivo model [115, 122]. FAP expressed by FAP+CAFs activates STAT3-CCL2 signaling and induces inflammatory characteristics in CAFs. This FAP+/CAF-S1 population boosts MDSCs recruitment, resulting in immunosuppressive TME [123, 124]. CAF-S1/iCAF subtype shapes the immunosuppressive TME by recruiting CD4+ CD25+ T cells and promoting their differentiation to Treg cells through the secretion of CXCL12 [121, 125]. Cremasco et al. have identified two distinct populations of FAP+ mesenchymal cells based on PDPN expression in breast cancer [126]. Myofibroblastic CAF-S1 and PDPN+ CAF subsets exhibit reduced IL2 activity and contribute to immunosupppression in breast cancer [127]. The FAP+PDPN+ population of CAFs is enriched at the outer edge of the tumor, in close contact with T cells, whereas the FAP+PDPN− population of cancer-associated pericytes (CAPs) is located around the vessels. Finally, FAP+PDPN+ CAFs diminish the proliferation of T cells in a NO-dependent manner, while FAP+PDPN− pericytes are not immunosuppressive [126].

Crosstalk between different cell types in the tumor microenvironment. TAMs, CAFs, NK cells, T cells, lymphocytes, and other cells present in the tumor microenvironment modulate each other by secreting different cytokines and chemokines. This crosstalk promotes ECM remodeling and angiogenesis and causes immune suppression in the breast cancer microenvironment
TGFβ-regulated CAFs can also contribute to immunosuppression by synthesizing ECM proteins in different types of cancers [128]. The CAFs-synthesized ECM may impact CD8+ T cell recruitment, thereby modulating the immunosuppressive environment [7]. A recent study has reported that CD16+ fibroblasts induce trastuzumab resistance in HER2+ breast cancer by causing matrix stiffness through VAV2 signaling [129]. Kieffer et al. have further identified eight CAF-S1 subclusters by analyzing CAF-S1 fibroblasts derived from breast cancer patient samples [122]. MyoCAFs from clusters 0 and 3 are characterized by ECM proteins and TGFβ signaling. The cluster 0/ECM-myoCAF enhances the expression of PD1 and CTLA4 in Treg cells, subsequently leading to increased TGFβ-myoCAF cellular content [122]. This study has highlighted a positive feedback loop between specific CAF-S1 clusters and Treg cells and discloses their role in immunotherapy resistance [122] (Fig. 2). The TGFβ driven expression of leucine-rich repeat-containing protein 15 (LRRC15) is associated with poor response to immune check point blockade in PDAC [130]. Targeting TGFβ signaling in myoCAFs might be beneficial for overcoming resistance to immunotherapy as TGFβ signaling plays a critical role in the formation of myoCAFs and restriction of T cell recruitment [131]. Grauel et al. have unbiasedly interrogated tumor mesenchymal cells, delineating the co-existence of distinct CAF subsets in the microenvironment of murine carcinomas [131]. This study has shown the neutralization of TGFβ signaling significantly reduces the myofibroblast subset under in vivo conditions. However, it promoted the formation of a distinct fibroblast population that displays a more robust response to interferon and enhanced immunomodulatory properties [131]. These changes correlate with improved antitumor immunity and greater efficacy of anti-PD1 immunotherapy. It has been reported that a subset of CAFs displays the expression of PD-L1 in TNBC patients, suggesting their involvement in immunomodulation and immunotherapy response [132].

Role of different CAF subsets in immune tolerance. CAF-S1/FAP+ subset induces immune suppression in tumors by secreting CXCL-12, increasing the recruitment and activation of CD8+ CD25+ T cells into Tregs. CAF-S1 subtype is further classified into TGFβ-CAFs and ECM-CAFs. ECM-CAFs enhance the expression of PD1 and CTLA4 in T cells, increasing the TGFβ-CAFs. This feedback loop induces resistance to immunotherapy. CAF subsets also exclude T cells from entering the cancer cell region by providing a nest around cancer cells, leading to immunotherapy resistance. However, NOX4 inhibition leads to the distribution of T cells into cancer cell region, thereby improving the efficacy of immunotherapy in cancer
However, CAF-rich tumors also show resistance to immunotherapy by the exclusion of CD8+ T cells through the secretion of chemokines. CAF inhibits TNF- and IFN-induced T cell-mediated necrosis and promotes immunosuppression by activating NF-κB signaling through the secretion of IL-6 and IL-8 in human intrahepatic biliary epithelial cells [133]. Upregulation of Hedgehog signaling in CAF population leads to higher iCAF production leading to activation of Treg cells to cause immunosuppression [134]. Biglycan (BGN), a prognostic biomarker for predicting immunotherapy response is highly upregulated under immune-resistant conditions in CAFs- derived from TNBC patient’s specimens [135]. CAF-derived BGN regulates ECM remodeling and immune response in breast cancer by facilitating the interaction of CAFs with immune cells, inhibiting NK cells, CD8+ cells, and MDSCs while stimulating tumor-favorable macrophage activation [136].
TAMs and immune microenvironment regulation
Macrophages play a significant role in cancer immune surveillance. These cells are associated with a poor prognosis, malignant phenotype, and negative hormone receptor status in breast cancer [137]. TAMs are the most abundant population of tumor-infiltrating immune cells and represent the major component of the innate immune system in TME [138]. Macrophages are considered immunoreactive cells due to their phagocytic and cytotoxic characteristics. They undergo polarization in response to microenvironmental signals into classically activated macrophages (M1) and alternatively activated macrophages (M2) [139]. M1 subtypes are activated by the Th1 cytokines, including tumor necrosis factor (TNF) and interferon-γ (IFN-γ). M1 macrophages exhibit their antitumor property by producing pro-inflammatory cytokines such as TNF, interleukin 2 (IL-2), and reactive oxygen and nitrogen intermediates [140, 141]. The M2 subtypes are stimulated by the Th2 cytokines such as IL-4, IL-10, and IL-13 and express CD206 (mannose receptor), arginase 1 (ARG1), and scavenger receptors [140]. TAMs are similar to the M2-macrophages that secret pro-tumor cytokines, facilitating tumor progression [142,143,144]. Additionally, TAMs influence angiogenesis and promote cell proliferation and metastasis by suppressing the activity of CD8+ T cells [145, 146].
Several cancer cell-derived factors induce the polarization of M2 macrophages. These cells, in turn, cause tumor progression by enhancing tumor angiogenesis, immune suppression, invasion and metastasis, and ECM remodeling [147]. The functional diversity of TAMs is greatly appreciated in cancer invasion, migration, tumorigenesis, angiogenesis, therapy resistance, and tumor suppression [148]. Several studies have explored targeting TAMs in various therapeutic approaches, including immune therapy and anti-angiogenic therapy. Ongoing clinical trials are underway to investigate the therapeutic efficacy of macrophage repolarization, antibodies targeting CFSRs (a receptor of GMCSF), and macrophage depletion for cancer therapy [149]. However, novel technologies like single-cell omics have explored the information about the molecular and functional diversity of TAMs in various cancers. A recent review has reported seven TAM subsets based on their molecular signatures in almost all cancer types [150]. Among seven subsets, angio-TAMs are pivotal in promoting multiple aspects of tumor progression. The expressions of VEGF-A and SPP1 (OPN) act as molecular signature of this particular subset of TAMs [151]. In addition, TAM-associated angiogenic factors like VCAN, FCN1, and THBS1 are also reported as molecular signatures of breast cancer progression [152].
Role of TAMs in immunotherapy resistance
TAMs mainly affect the tumor-killing ability of effector T cells to facilitate cancer progression [153]. They primarily target arginine metabolism for inhibiting T cell activity. TAMs are found to induce ARG1-mediated hydrolysis of L-arginine in early-stage breast cancer patients. The L-arginine is essential for the functioning of the effector T cells [154]. Further, nitric oxide synthase (NOS), a molecular marker of M1 macrophages, metabolizes L-arginine to produce NO, inhibiting the activity of effector T cells [155]. Since TAMs exhibit reduced expression of MHC II using in vivo 4T1 breast cancer mice model, they are less efficient in activating T cells and antigen presentation [156, 157]. TAMs also secrete various cytokines to regulate the expression of immune checkpoints and their ligands, including PD1/PD-L1 [158]. Moreover, in vivo studies have revealed that the genetic deficiency of macrophage common lymphatic endothelial and vascular endothelial receptor 1 (CLEVER1) suppresses tumor progression by activating the tumor-killing ability in effector T cells [159]. These TAM populations impede infiltrating T cells while upregulating Treg cells in TNBC [160]. TAMs express different ligands for PD1 and CTLA4 to inhibit T cell activation [161]. They also secrete various immunosuppressive factors, including CCL20, CCL22, TGFβ, IL-6, and IL-10, that can directly inhibit both CD8+ and CD4+ T cell effector function as well as recruitment of Tregs into the tumor lesion [153, 162,163,164,165]. TAM-secreted IL-10 inhibits antigen-presenting DCs, thereby hindering tumor immunity [166]. TAM-secreted prostaglandins (PGs) and cyclooxygenase-2 (COX-2) also contribute to immunosuppression [153]. PGE2, the primary product of COX-2, is crucial for breast cancer progression as it binds to EP1-EP4 prostanoid receptors on various immune cells [167]. COX-2 inhibitors, including aspirin, can decrease the production of PGE2, which is associated with a lower risk of breast cancer progression as COX-2 is constitutively expressed at high levels in breast cancer cells [168]. Both immune and cancer cells in the TME release PGE2, which stimulates bone marrow progenitors to differentiate into MDSCs and DCs and facilitates their recruitment and activation [169]. Moreover, PGE2 induces the M2 polarisation of macrophages and their production of PD-L1. Therefore, blocking PD-L1 by anti-PD1/PD-L1 immunotherapy impairs T cell-mediated immune response against cancer [170]. However, TAMs also stimulate IL-6 by modulating PD1 signaling in response to anti-PD1/PD-L1 treatment, resulting in an immunosuppressive environment in tumors [171]. Moreover, TAMs express Fc receptors that inhibit the binding of anti-PD1 antibodies to T cells, thereby preventing the suppression of PD1/PD-L1 signaling, leading to resistance to anti-PD1 therapy using in vivo tumor models [172]. Additionally, TAMs are found to inhibit NK cell-mediated anti-tumor activity, causing immunosuppression using in vivo murine breast cancer model [153, 173].
Interplay between TAM and CAF in remodelling the TIME
Tumor cells interact with stromal cells by secreting an array of cytokines, chemokines, and other tumor-promoting factors in the TME. The tumor-stromal cell interaction induces non-cancerous cells to acquire new tumor-promoting phenotypes, increasing tumor progression, multidrug resistance, distant metastasis, and immune suppression [3]. Studies using patient specimens have also shown a positive and reciprocal feedback responses among stromal cells. As discussed in the earlier section, CAFs are one of the most critical stromal cells in the TME, which is known to participate in various stages of tumor development through multiple mechanisms (Fig. 1). Among all immune cells, macrophages play a vital role in the TIME and are known to enhance several hallmarks of cancer by infiltrating into tumors [174]. Macrophages display a wide range of plasticity and various functional activities in TIME. TAMs are the most prominent immune cells near CAF-populated areas, suggesting strong interactions between these two cell types [175]. Several studies in spheroid/ in vivo models have reported that macrophage recruitment and differentiation are triggered by CAFs via several secretory factors and regulatory networks, thereby imparting pro-tumorigenic capabilities in TAMs [176,177,178]. For instance, in melanoma, CAF-secreted cytokines such as IL-10, IL-8, CCL2, and TGFβ stimulate macrophage recruitment and polarisation into the M2 phenotype with tumor-promoting functions [7, 179]. Similarly, CAFs trigger monocyte recruitment and provoke differentiation of monocytes to M2 macrophages by secreting SDF-1 (CXCL12), monocyte chemotactic protein 1 (MCP1), and CHI3L1 in breast cancer [180]. The CAF-induced TAMs exhibit an elevated expression of immune checkpoints such as PD1 and cause immunosuppression by reducing T cell activation and proliferation [181]. In breast cancer, the recruitment of monocytes to the tumor is triggered by the CAF-driven CXCL12/CXCR4 axis, which also supports the acquisition of an immunosuppressive lipid-associated macrophage (LAM) phenotype [182]. MSCs acquire CAF-like phenotype through macrophage-activated signaling, inducing TME remodeling and promoting oncogenic transformation [183]. The interaction of CAFs and TAMs can enhance EMT by IL-6 and SDF-1, leading to activation of CAFs [184]. TAMs can also differentiate MSCs into CAFs through various signaling cascades [185]. Single-cell and spatial transcriptomic analyses revealed that IL-1, chemerin, and TGFβ interact with FAP+ fibroblasts and SPP1+ macrophages, allowing immune escape and restricting T-cell invasion [186]. FAP+ CAFs induce scavenger receptor A (SR-A)+ TAM adhesion via cleaving type I collagen [187]. TAMs with the M2 phenotype also control the activation and generation of CAFs [188]. In addition to their stimulatory action on TAMs, CAFs may hinder specific functions of TAMs. ERα signaling in CAFs has been shown to decrease the expression of specific cytokines and chemokines, such as CCL5 (also known as RANTES) and IL-6, that disrupt macrophage infiltration and cancer cell invasion [189]. Furthermore, M-CSF-1, IL-6, and CCL2 play a vital role in the recruitment of monocytes and the elevation of the M2/M1 macrophage ratio [190]. Additionally, co-culture of cancer cells with CAF-like BM-MSCs does not have an invasive ability but supports the proliferation of cancer cells, whereas cancer cells co-cultured with TAM-like macrophages had the opposite effect [191]. Active CAFs produced by macrophage-induced signaling boost TAM activity and create a positive feedback loop to support cancer growth and inhibit the immune response in the TME [192]. Although several reports showed the role of CAFs on TAM regulation, further studies are needed to understand the impact of macrophages on the regulation of CAF phenotypes. The CAF-TAM interaction in shaping TIME in breast cancer is elucidated in Fig. 3.

Crosstalk between CAF and TAM in the tumor immune microenvironment. CAFs trigger macrophage recruitment and differentiation through various secretory factors and regulatory networks, inducing the pro-tumorigenic capabilities of TAMs. TAMs can also induce CAF generation and activation. The interaction of CAF and TAM causes immunosuppression to promote cancer growth via a feedback loop
MDSC-mediated immune tolerance
MDSCs are critical immunosuppressive components in the TME. An increased monocytic MDSC population is clinically correlated with more aggressive metastatic breast cancer [193]. These cells are activated and differentiated into atypical T cell suppressive neutrophils. Prolonged G-CSF exposure may encourage the tumor-promoting function of these immunosuppressive neutrophils [194]. MDSCs are also involved in regulating the function of B cells. They transform B cells into Breg cells to suppress T cell-mediated immune response [195]. Both monocytic and granulocytic MDSCs deplete ARG1 and induce PD-L1 expression to recruit Treg cells in the TME [196]. These cells also activate STAT3 signaling that causes T cell inhibition in response to IL6 using in vivo 4T1 breast cancer murine models [197]. MDSCs modulate MHC I to impede antigen presentation of cytotoxic T cells, resulting in immune tolerance in breast cancer [198].
DC-mediated immune tolerance
DC is crucial component of TIME due to its antigen presenting function, leading to T cell activation and immune response. However, maturation and activation of DCs are vital for their immunostimulatory action. Immature DCs fails to activate T lymphocytes due to their high endocytic action, causing immune tolerance [199]. Immature DCs can also increase the expressions of inhibitory receptor to impede T cell activation [200]. DCs with high PD-L1 expression may inhibit CD80, leading to T cell inactivation [200]. CCL4 is associated with DC activation and T cell-mediated anti-tumor immune response [201, 202]. In contrast, CAF-secreted TGF-β interferes in the maturation of DCs, inhibiting Treg differentiation [203]. The PGE2 secreted by tumor reduces the cytokine and their receptor expressions in both NK cells and DCs to facilitate immune suppression [204].
Role of metabolic dysregulation and epigenetic alteration in tumor microenvironment-mediated immune tolerance
Metabolic dysregulation and immune tolerance
Due to metabolic reprogramming, cancer cells exhibit the Warburg effect, which describes the observation that cancer cells show increased glucose consumption and preferentially rely on glycolysis for energy production, even in the presence of oxygen. They use glycolytic products to promote their growth and proliferation [205]. The glycolytic glucose utilization by tumor cells limits the glucose availability in the TME, impeding T cell infiltration and IFN-γ production as T cells highly depend on glycolysis for their differentiation and effector function [206, 207]. Moreover, tumor glycolysis stabilizes Treg cells and inhibits T cell activation by modulating the glucose/lactate ratio in TME, thereby contributing to immune tolerance [208]. In low-glucose TME, Treg cells can depend on lactate for their survival and immunosuppressive action. Therefore, lactate-rich TME induces Treg polarization, leading to immune resistance [209,210,211]. HIF1α is also involved in metabolic reprogramming-mediated immune resistance in breast cancer. It induces PDK1 phosphorylation to inhibit pyruvate dehydrogenase, impeding pyruvate consumption in the TCA cycle [212]. Glycolytic end products such as pyruvate and lactate stabilize HIF1α to stimulate glycolysis further [213, 214]. Carbonic anhydrase IX (CAIX) is overexpressed in TNBC and promotes tumor growth, invasion, and migration. Lactate upregulates CAIX through HIF1α stabilization [215, 216]. High lactate and lactate dehydrogenase A (LDHA) expression levels stimulate HIF1α-mediated neovascularization and metastasis [217]. Therefore, lactate-induced HIF1α accumulation in TME contributes to immune tolerance. Pyrimidine metabolism, considered one of the hallmarks of cancer, is reported to be associated with immunotherapy response. Inhibition of pyrimidine synthesis results in lower CTLA4+ T cells in the TME [218]. Earlier studies have established the link between pyrimidine metabolism-related genes and immunotherapy efficacy [219].
Other immune tolerance mechanisms are also associated with immunotherapy failure (Fig. 4A). For example, high plasma IL-6 levels impair CD8+ CTL function through STAT3-mediated basic leucine zipper ATF-like transcription factor (BATF). This cytokine impedes CTL effector differentiation and gene expression, including IFNγ and perforin expression, leading to anti-PD-L1 therapy resistance in preclinical tumor model [220]. Placenta-specific 8 (PLAC8) protein also plays a pivotal role in modulating the immune response, cancer growth, and progression in TNBC [221,222,223,224]. The role of PLAC8 protein has been established using both breast cancer cell lines and patients’ specimens. This protein is overexpressed in TNBC, and its stability is regulated by ubiquitin-fold modifier 1 (UFM1), a ubiquitination modifier [225, 226]. PLAC8 has been reported to modify PD-L1 expression in TNBC via its ubiquitination, leading to immune tolerance [227, 228]. Moreover, cuproptosis, non-apoptotic programmed cell death due to high intracellular copper accumulation, is involved in immune tolerance [229]. Cuproptosis-related genes (CRGs) regulate TME and immune cell infiltration, leading to tolerance of immunotherapy. RAD23B is a vital CRG that affects immunotherapy efficacy of ICIs in breast cancer [230]. The tumors also exhibit endogenous immune response-mediated acquired immune resistance (AIR). Endogenous immune response-mediated AIR in tumors hinders the antitumor immune response and impacts overall survival rates by CD8+ cell-induced expression of CD163 and/or FoxP3 [231]. These data suggested a direct correlation between densities of CD8+ cells with CD163+ and FoxP3+ cells in the breast cancer patients’ specimens.

Tumor microenvironment-mediated immune tolerance in breast cancer. (A) Tumor microenvironment-mediated immunotherapy resistance mechanisms. Different cell types of the tumor microenvironment, such as CAF, TAM, and MDSC, contribute to immune tolerance by inducing differentiation of Treg cells, antigen uptake and maturation of dendritic cells, and M2 polarization. These cells also suppress CD8+ and NK cell activation and recruitment. Tumor metabolism, particularly glycolysis and TCA cycle, are also associated with immune tolerance in breast cancer. (B) Therapeutic strategies to overcome immunotherapy resistance. Nanoparticles, photoimmunotherapy, and several drugs and inhibitors may target CAF, TAM, MDSC, and tumor metabolism for sensitizing breast cancer to immunotherapy
Tumor mutation burden and immune tolerance
Immune suppression within the tumor niche majorly contributes to the failure of immunotherapy and its resistance. Differential expression of neo-antigens presented by APC due to mutations provokes poor T-lymphocyte recognition and activation, which results from tumor mutation burden (TMB) [232]. The T cells must be able to distinguish between tumor and normal cells after being reactivated. It is easier to identify cancer cells if their surface displays immunogenic neo-antigens. Since neo-antigens result from genomic alteration in cancer, with increases in TMB, there is an increase in immunogenicity, allowing T cells to recognize them and eradicate cancer cells [232]. However, the nature and type of mutations in TMB as a predictor in response to PD1/PD-L1 immune therapy failure in melanoma can be partially explained by the different turnover rates between genomic events and the last stages of MHC presentation [233]. In addition, it is critical to emphasize that TMB has significant limitations as a predictive biomarker, particularly when used alone, from the perspective of immune therapy response [234]. First, T cells only recognize a limited percentage of non-synonymous mutations as neo-antigens. Second, the distinct tumor molecular fingerprints and the clonality associated with these neo-antigens enhance the capacity to produce a successful anticancer therapy response [235]. Finally, the ability of T cells to penetrate the tumor site, the balance of TIME components and the equilibrium between suppressive and activating cytokines within the TIME, modulation of metabolic pathways in immune and cancer cells, and the type of checkpoint presentation by tumor influence the T cell-mediated tumor elimination [236]. Thus, while TMB is correlated with improved outcomes following ICI administration, TMB must be considered in combination with several other parameters to optimize ICI response due to the intricacy of the immune response.
Epigenetic modification and immune tolerance
Changes in epigenetic alterations can impact the growth and development of healthy cells, potentially resulting in oncogenic transformation. Additionally, they are shown to affect immune cells’ aberrant function, normal stimulation, and activation in the TIME. These alterations modulate the activation of various signaling pathways in immune cells, affecting tumor growth [237]. Histone modification enzymes including DNA methyltransferases (DNMTs), DNA demethylases, histone methyltransferases (HMTs), histone demethylases (HDMs), histone deacetylases (HDACs), histone acetyltransferases (HATs) contribute to carcinogenesis [238]. In addition, chromatin remodeling, RNA modification, and noncoding RNAs regulate various biological processes crucial to cancer development. Zeste homology 2 (EZH2), the catalytic subunit of PRC2 provokes the release of LOXL4 in tumor cells to modulate the activation of macrophage into TAMs via miR-29b/miR-30d-LOXL4 axis in TNBC [239]. DNMT1 and EZH2 are linked with low tumor-infiltrating CD8+ T cells and poor patient outcomes [240]. EZH2 has also been demonstrated to play a key role in the development of Treg cells, which dampen immunological responses. Furthermore, DNMT1 attenuates the tumor cell-derived Th1-type CXCL9 and CXCL10 expression, which influences effector T cell trafficking into the TME in ovarian cancer [241].
Similarly, in TNBC, another methyltransferase known as coactivator-associated arginine methyltransferase 1 (CARM1) primarily targets BAF155 by blocking the interferon pathway, which reduces the host immune response [242]. Lysine-specific demethylase 1 (LSD1), a histone demethylase involved in epigenetic EMT regulation, the acquisition of cancer stem cell markers (CSCs), and treatment resistance in breast cancer, could be a promising target for overcoming anti-PD-L1 therapy resistance. LSD1 is inversely correlated with CD8+ T cells in breast cancer, non-small-cell lung cancer, and melanoma [243]. By suppressing the MHC-I-producing genes, H2-D1, H2-K2 and LSD1 significantly impact the normal expression of MHC-I protein antigen in tumor cells. This enhances the exclusion of MHC-I identification by CD8+ T lymphocytes, which could result in immunological escape [244]. The K acetyltransferase 6 A (KAT6A) acetylation of SMAD3 controls macrophage recruitment, metastasis, and immunosuppression. Combining anti-PD-L1 therapy and KAT6A inhibitor reduces metastases and improves survival in TNBC xenograft-bearing mice [245]. TNBC is caused by hypermethylation of the DNA methyltransferase 1 (DNMT1) gene [246]. In addition, the histone demethylase, KDM5B has been shown to promote the migration, proliferation, and modulation of cellular physiology of tumor cells. Therefore, KDM5B silencing in breast cancer reprograms lipid metabolism to stimulate the migration and proliferation of breast cancer cells via activation of AMPK [247]. BRD4 inhibition has been found to induce macrophage reprogramming from the M2 to M1 phenotype and proinflammatory cytokine production, resulting in T cell activation. Similarly, this suppression was correlated with increased expression of MHC 1 genes by tumor cells and an increase in the CD8+ T cells/Tregs ratio [248].
Combination of immunotherapy and epi-drugs in immunotherapy
Extensive heterogeneity has impeded the management of TNBC by causing therapy resistance. Combination drug therapy (or CDT) has gained an improved pathological clinical response (PCR), progression-free survival (PFS), and overall survival (OS) in various malignancies [249]. Epigenetic drugs or epi-drugs are shown to have promising results in various solid cancers, including breast. Goswami et al. have uncovered an additional benefit of combining immunotherapy with EZH2 inhibitors [249]. They have shown that combination treatment might boost the therapeutic efficacy of antibodies that target CTLA-4 and decline the number of immune-suppressive cells [249]. In recent days, bromodomain and extra-terminal (BET) inhibition have gained significant attraction in the treatment of breast cancer. Several BET inhibitors, such as birabresib, molibresib, and mivebresib act by modulating the interaction between the enhancer and promoter for transcriptional repression [250]. Additionally, new strategies that target chimeric chemicals (BET-PROTACs) by BET-proteolysis have been explored in TNBC with encouraging results, even in BET-resistant cancers, and they bind a ubiquitin ligase while allosterically inhibiting BET bromodomains [251]. BET inhibitor resistance in TNBC is correlated with TAM infiltration in the TME [252]. Entinostat, a HDAC inhibitor augments the anti-tumor effects of IL-15 agonist and vaccine in 4T1 TNBC mouse models [253]. In addition, clinical trials investigating this epi-drug in combination with atezolizumab showed overall response rate of 10% (NCT02708680) [254]. A recent phase Ib trial with entinostat, nivolumab along with ipilimumab in hormone receptor positive and advanced TNBC suggested the overall response rate of 25%, with 10% in hormone receptor positive and 40% in TNBC and recommended a further study of these combinations in phase II trial (NCT02453620) [255].
Furthermore, additional investigation is needed to determine whether ICI, along with different epi-drugs specifically targeting each type of immunosuppressive cells may be more beneficial for breast cancer therapy. These cutting-edge therapeutic strategies may be enhanced by developing the precisely targeted drug delivery systems for the treatment of breast cancer.
Therapeutic strategies to overcome immunotherapy resistance
Several groups have explored developing new therapeutic strategies for immunotherapy tolerance. CMTM7, a PD-L1 regulator, is frequently deleted or downregulated in TNBC to cause therapeutic resistance. TNBC with higher CMTM7 expression is more sensitive to chemotherapy and immunotherapy [256]. This protein exhibits a positive correlation with immune cell infiltration and immune checkpoints. High CMTM7 protein level leads to a better therapeutic response of anti-PD1 or anti-PD-L1 therapy. Hence, CMTM7 can be considered a predictive biomarker for immunotherapy response, and its expressions can be modulated to deal with immune tolerance in breast cancer [256]. PSME2, overexpressed in breast cancer, is involved in the proteasomal degradation of several proteins. It positively correlates with immune response and good prognosis in HER2+ breast cancer patients [257]. It promotes immune cell infiltration and checkpoint functions, improving immunotherapy outcomes. This novel biomarker in breast cancer may also be modulated to overcome immune resistance [257]. Earlier reports have revealed three TAAs: CD74, IRF1, and PSME2, associated with immune cell infiltration in breast cancer. These three TAAs are found to be overexpressed, amplified, or mutated in breast cancer and directed for developing mRNA vaccines in this cancer [258]. Moreover, HSP90 inhibitors reduce PD-L1 and PD-L2 surface expressions and increase CD8+ T cell infiltration in the tumor [259]. NDNB1182, an HSP90β inhibitor, blocks CDK4 and stimulates the expressions of IFN-mediated genes. HSP90 inhibitor with immune checkpoint blockade is employed for treating immunotherapy-resistant murine breast cancer [260]. Another natural product-based HSP90 inhibitor, 17-AAG, is found to be effective along with trastuzumab in treating trastuzumab-refractory HER2+ breast cancer [261]. However, it exhibited dose-limiting hepatotoxicity and gallbladder toxicity in preclinical study [262]. TNFα is also involved in trastuzumab resistance in HER2+ breast cancer. It upregulates the expression of MUC4, which interacts with HER2 through its MUC4β subunit and promotes tumor metastasis [263, 264]. MUC4 shelters the trastuzumab epitope in HER2 protein, blocking trastuzumab interaction [265]. Soluble TNFα-mediated inhibition of MUC4 downregulation modulates macrophages and NK cells to reverse immunosuppressive environment and trastuzumab resistance [266]. MSA2, a stimulator of interferon genes, boosts dendritic cell maturation and its antigen-presenting ability. It also promotes macrophage activation along with the release of chemokines and cytokines [267]. Improved T cell migration with chemotaxis leads to a better innate and adaptive immune responses against breast cancer. NSA2 is used with YM101, an anti-TGFβ/PD-L1 antibody, to deal with immune resistance in non-inflamed tumors [267]. Direct Akt activation stimulates the immune system in PD1 checkpoint blockade-resistant tumors to suppress their growth. The Akt downregulates Treg cells while upregulates CD4+ and CD8+ tumor-infiltrating lymphocytes (TILs), leading to IFNγ expression and thereby inducing an anti-tumor immune response [268]. A combination of anti-PD-L1 monoclonal antibodies and PARP inhibitors are also effective in treating breast cancer patients [269]. Combinatorial treatment with albumin-paclitaxel and pembrolizumab also exhibits efficacy in TNBC cases exhibiting higher PD1 on T cells [270]. Moreover, determining the drug exposure of tumor tissue is important to understand the mechanism of immune tolerance. [68Ga]Ga-DK223-PET has been developed using Gallium-68-labeled peptide and investigated for monitoring tumor status with anti-PD-L1 therapy. This strategy helps to optimize immunotherapy for effective treatment [271]. A non-invasive method has been developed for analyzing the blood-based TMB and copy number profiling to predict outcomes in breast cancer patients undergoing treatment with the combination of endocrine therapy along with CDK4/6 inhibitor, a standard treatment for HR+/HER2− metastatic breast cancer [272, 273]. Along with these approaches, there are several reports employing various therapeutic strategies to overcome immunotherapy tolerance.
Targeting CAFs
CAFs have been shown to contribute to immunotherapy tolerance significantly. Researchers have developed various therapeutic interventions to improve immunotherapy response by targeting the CAF population in the TME. Ford et al. have assessed the potential of CAF targeting the NADPH oxidase 4 (NOX4) inhibition in several cancers [274]. NOX4 is an enzyme involved in the differentiation of myoCAFs, and it’s inhibition reverts the myoCAFs into a quiescent phenotype and promotes intra-tumoral infiltration of CD8+ T cells. NOX4 inhibition has shown to overcome the CD8+ T cell exclusion and potentiate anti-PD1 immunotherapy response using in vivo murine breast cancer model [274] (Fig. 2). GKT137831 is a NOX4/1 inhibitor reported to repress CAF-mediated immune tolerance [275, 276]. TGFβ1-induced ECM remodelling in CAFs stimulate hepatocellular cancer cell invasion [277]. Upregulation of IL2 in TGFβ+ and PDPN+ CAF populations sensitizes the trastuzumab-resistant HER2+ breast tumor [127, 278]. TGFβ inhibitors may also address immunotherapy resistance by targeting the CAF population [127]. The depletion of CAF is alternative strategy for improved immunotherapy efficacy. Depleting FAP+ CAF has been reported to boost the effectiveness of vaccines against cancer [117]. AMD3100 blocks the action of CAF-mediated CXCL12 signaling and immunosuppression by targeting CXCL12-CXCR4 interaction [115, 279]. However, targeting CAF has certain limitations due to the existence and insufficient understanding of various CAF subpopulations; therefore, precisely targeting CAF is more challenging. Selective CAF subpopulations possess anti-tumorigenic properties, leading to ineffective therapy [280]. Most CAF-targeted therapy could be combined with other immunotherapy for successful breast cancer treatment [281]. Lack of preclinical and clinical data is a limitation affecting CAF-targeted therapy [282].
Targeting TAMs
TAMs are reported to involve in immune resistance through various mechanisms. Several groups have explored many therapeutic options along with anti-PD1/PD-L1 therapy [283, 284]. However, a detailed understanding of the heterogeneity of TAM needs to be explored further [285, 286]. Single-cell RNA seq has established the existence of both immunosuppressive and immunostimulatory TAM subpopulations at the tumor site. CD8+ T cell enrichment reduces memory T cells and inhibitory macrophages among various TAM populations [287]. Folate receptor 2 expressing macrophages (FOLR2+) is involved in the anticancer immune response. FOLR2+ macrophages induce higher CD8+ T cells and dendritic cell infiltration into the breast tumor niche [288]. In contrast, CX3CR1+CCR2−/low TAMs induce tumor-promoting TME [286]. TREM2+ macrophage subpopulation contributes to immune resistance via exhausting T cells [283]. The preclinical result of emactuzumab, a CSF1R antibody, established its efficacy in targeting the CD163+ TAM population in breast cancer [289]. BTH1677, an agonist of Dectin receptor and pembrolizumab treatment, exhibits repolarization of M2 macrophages, and a phase II trial is under process to study the efficacy of this combination therapy in metastatic TNBC (NCT02981303) [290]. PLX3397 (Pexidarnitib), another anti-CSF1R antibody, is also being investigated along with eribulin for the treatment of brain metastatic cases of breast cancer (NCT01596751) [291]. Although several reports are available on repolarization, reprogramming, and depletion of TAM in cancer, these strategies are not sufficient enough to overcome breast cancer immune resistance.
Targeting MDSC
A cryo-thermal treatment strategy has been developed to target metastatic tumors by activating innate and adaptive immune responses [292]. This therapy inhibits MDSCs by inducing their differentiation into antigen-presenting cells. However, to improve the in vivo efficacy of this therapy, all-trans retinoid acid (ATRA) is employed to stimulate the maturation of functional MDSCs and inhibit immunosuppressive molecules [293]. This combination treatment inhibits Th2 and Treg subpopulations while stimulating cytotoxic CD8+ T cells and NK cells to address MDSC-mediated immune tolerance [293]. Moreover, MDSC biogenesis may be targeted to reduce MDSC load and enhance immunotherapy response. Dihydroorotate dehydrogenase inhibitors downregulate MDSC generation and maturation, improving immunotherapy efficacy in an in vivo TNBC model [294] (Fig. 4B). Brequinar, an inhibitor of dihydroorotate dehydrogenase, has been reported to enhance the effectiveness of immune checkpoint inhibition in refractory HER2+ breast cancer [295]. ADAM12 is a metalloproteinase that is highly expressed in TNBC [296]. This protein is negatively correlated with the expression of MDSC genes. ADAM12 inhibition suppresses MDSCs and improves T and B cell infiltration at the tumor site using an in vivo TNBC model. Combination of anti-PD1 and anti-CTLA4 therapy has enhanced the efficacy of immunotherapy upon abrogation of ADAM12 using in vivo murine breast cancer model [297].
Modulation of metabolism
Tumor metabolism plays a crucial role in regulating tumor immunity and immunotherapy response. The high nutrient demand of cancer cells for their rapid growth and proliferation limits the availability of metabolic nutrients for immune cells, leading to diminished immune activity [298]. Cancer cell metabolism suppresses T cell metabolism and its immune function. Tumor cells with high glycolytic activity affect the growth of T cells by depleting glucose in TME to impede T cell-mediated cytokine secretion. Targeting CTLA4 and PD-1 improve T cell metabolism and restore its immune function [28]. Amino acid metabolism is also crucial for the function of T cells [299]. TAM, especially M2 macrophages, are associated with metabolism-dependent immunosuppression. M2 macrophages reduce the glycolytic flux by inducing PD-L1 expression to avoid the competition for oxidative phosphorylation. High PD-L1 expression in TAMs leads to immunosuppression [207, 300]. CSF1R inhibitors have the potential to block the transcriptional activation of genes involved in TAM-mediated immunosuppression [146]. M1 repolarization may also effectively suppress M2-dependent metabolic modulation of TIME [158]. Lipid metabolism also significantly affects ICI therapy by hampering T cell proliferation and activation. Metabolites may also act as a signaling agent to modulate immune activity [301]. Several reports are available on metabolic reprogramming for enhanced immunotherapy response. CD28 and CTLA4 are involved in competitive regulation of metabolism by binding with common receptors, CD80 and CD86. Highly abundant CD28 exhibits a lower affinity to these receptors, while less abundant CTLA4 shows a higher affinity of it [302]. CD28 promotes glucose utilization through glycolysis in T cells, which is required for their activation [303]. CTLA4 inhibits CD28-mediated glucose metabolism in T cells through Akt inhibition, impeding T cell activation [304]. Anti-CTLA4 therapy promotes CD28 stimulation in antigen-presenting T cells and hampers glucose-dependent Treg cell stabilization [206]. Hence, CTLA4 inhibitors can be usefull in dealing with immune tolerance in breast cancer [305]. Tumor glycolysis can also be targeted to inhibit MDSCs infiltration-mediated immunosuppression in TME by LDHA knockdown in TNBC [306]. LDHA knockdown also destabilizes HIF1α to promote immune cell infiltration, improving immunotherapy outcomes in murine breast cancer [217, 307]. FX-11, an LDH inhibitor, is combined with anti-PD1 therapy to induce cytotoxic CD8+ T cells and NK cells, resulting in improved antitumor immune response in TNBC [308]. It has been reported that CAIX inhibition can boost immune checkpoint inhibitor’s efficacy in TNBC. Preclinical data have shown that SLC-0111, a CAIX inhibitor, acts synergistically with immune checkpoint inhibitors such as anti-PD1 or anti-CTLA4 to suppress tumor vascularization and metastasis in a TNBC xenograft model [309, 310].
Photodynamic therapy and photoimmunotherapy
Photodynamic therapy (PDT), which employs a photosensitizer to generate light-induced ROS, along with immunotherapy overcome the immune tolerance [311]. PDT-induced oxidative stress leads to calreticulin-mediated necrosis of tumor cells and secretes damage-associated molecular patterns (DAMPs) to cause antigen-presenting T cell activation [312]. PDT also selectively activates macrophages in a dose-dependent manner [313]. It stimulates macrophages to secrete lysophosphatidylcholine (LPC), which forms macrophage activating factor (MAF) via T and B cell signaling to induce anticancer effects. PDT-induced phagocytosis in macrophages results in CD8+ T-cell activation [314]. PDT inhibits immunosuppressive TME along with its anti-tumor immune response [311]. It is used with immunomodulatory agents to prevent metastasis through improved CD8+ T cell stimulation [315].
Phototherapy combined with immunotherapy (photoimmunotherapy; PIT) is applied to escape the immunosuppressive TME. PIT activates the system’s immune response to achieve long-term antitumor immunity. It also targets metastasized tumors and prevents breast cancer recurrence [316]. Two-dimensional black phosphorus (BP) nanostructures have gained popularity for their PIT applications. CpG oligodeoxynucleotide encapsulating NIR and ROS-sensitive BP nanovesicles (BPNVs) stimulates cytokine release by antigen-presenting cells (APCs) [317]. It generates NIR laser irradiation-responsive ROS to release CpG at the tumor site. APCs take up the released CpG to activate the cytokine-mediated immune response against the tumor. The in vivo efficacy of this nanosystem is also established using in vivo 4T1 tumor-bearing BALB/c mice models [318].
Photothermal therapy (PTT) can also be efficiently employed for potential tumor immunotherapy. A biomimetic NIR-responsive black phosphorus quantum dots (BPQDs) formulation is developed by coating with erythrocyte membrane to achieve better tumor accumulation and prolonged circulation [319]. It induces PTT for immune system activation in breast cancer to target metastatic and residual tumors. NIR irradiation leads to dendritic cell recruitment and elicits CD8+ T cell response at the tumor site. A combination of PD1 therapy and BPQDs exhibits more potent activity against primary and secondary cancers [319]. In addition, tumor-specific PTT is developed using cancer cell membrane coating for BPQDs [320]. PD-L1 combination with BPQDs improves dendritic cell maturation, and T cell-mediated anticancer immunity leads to better tumor cell recognition and tumor-specific lethal efficiency. This immunotherapy combination exhibits an immunological memory effect, resulting in more efficient action for recurrence and metastatic TNBC [320]. Another BPNP is fabricated using PEGylated hyaluronic acid (HABPs) and applied with PTT, PDT, and PIT. This formulation downregulates CD206 expression and upregulates CD86, leading to the repolarisation of TAMs into M1 macrophages. In vitro and in vivo studies demonstrated that combining PDT, PTT, and HABPs immunotherapy causes immunogenic cell death. This combination therapy secretes DAMPs for robust anticancer immunity through improved dendritic cell maturation and effector cell stimulation [321]. All these potential therapeutic strategies to overcome immune tolerance in breast cancer have been depicted in Fig. 4B.
Regulation of tumor immune genes and immunotherapy response
Recent advances using in vivo CRISPR screens have identified various genes that make cancer cells evade anti-tumor immunity or regulate response to ICI therapy. Deletion of these tumor immune genes improves the immunotherapy outcome. In vivo CRISPR knockout screening using a syngeneic TNBC mouse model has revealed the role of E3 ubiquitin ligase, Cop1, in regulating immunotherapy response. The first screen utilized a customized lentiviral guide RNA library called MuSCK, targeting 4500 genes involved in cancer progression and immune evasion. The library was transduced into 4T1 cells and then injected into mice. A subsequent secondary screening with the MuSCK 2.0 library was performed to validate 79 hits. Cop1 deletion also improves the anti-tumor immune response by inhibiting chemokine secretion and macrophage infiltration in the TME [322]. Another in vivo CRISPR screen employed a guide RNA library, DrIM, targeting 2796 human disease-associated immune genes transduced into 4T1-Cas9 cells. Validation in immunocompetent and immunodeficient mice identified Ido1 and Lgals2 as immunotherapy targets in TNBC. These genes also promote TAMs and M2 polarization [323]. Dong et al. conducted an unbiased genome-wide CRISPR screening using a single guide RNA library called mouse knockout (MKO) library [324]. E0771 murine TNBC cells expressing the ovalbumin tumor antigen were employed, and Cas9-expressing CD8+ T cells, transduced with the MKO library, were injected into mice with E0771-ovalbumin transplants. The screening has identified Dhx37 as a regulator of T cell function in the TME. The study established that Dhx37 dampens the CD8+ T cell activity by physically interacting with the NF-κB pathway in TNBC [324]. A pooled in vivo CRISPR screen approach has identified defective IFNγ signaling responsible for immunotherapy resistance. The protein tyrosine phosphatase PTPN2 deletion improves IFNγ-mediated antigen presentation, leading to better immunotherapy response [325].
Combination therapy and nanoparticle-mediated immunotherapy
Immunostimulatory and immunomodulatory molecules must be precisely and efficiently delivered to the right target immune cells to employ cutting-edge therapeutic strategies in reshaping TIME without off-target effects. Moreover, nanocarrier-mediated combinatorial approaches are very promising to achieve better immunotherapy response. Kim et al. have shown the efficacy of SGT-53 to potentiate the action of anti-PD1 therapy using in vivo 4T1 breast cancer model [326]. SGT-53, a nanocarrier containing a plasmid encoding p53 gene, stimulates immune response and sensitize the resistant tumor to anti-PD1 antibody. Moreover, this combinatorial treatment also limits the immune-related adverse effect [326]. Combination of chemotherapy and immunotherapy is also investigated to improve the treatment outcome. Phase III trial was conducted with the atezolizumab and nanoparticle albumin-bound paclitaxel (nab-paclitaxel) for the treatment of metastatic TNBC. The results have shown the improved efficacy of atezolizumab in TNBC particularly in PD-L1+ tumors [327]. pH responsive micellar nanosystem has been developed for co-delivery of anti-PD-L1 siRNA and photosensitizer. This nanocarrier elicits PDT-induced antitumor immune response and overcomes the immune tolerance in melanoma [328]. Kang et al. have investigated the potential of chemoimmunotherapy using nanoparticulate system [329]. Paclitaxel and imiquimod have been co-assembled for improved immunotherapy response as well as antitumor efficacy [329]. Nanomaterials have also been shown to possess an intrinsic ability to influence immune cells directly, including macrophage polarisation, thus becoming an appealing strategy to reprogram TAMs [330]. Various metallic nanoparticles are reported to activate immune response. Iron oxide nanoparticles have shown to encourage immunosuppressive M2 TAMs to repolarize into the pro-inflammatory M1 phenotype using in vivo murine breast cancer model [330]. In addition, a study has shown that gold and silver nanoparticles can provoke an immune response in TAMs. Gold and silver nanoparticles contribute to TAM reprogramming into M1-like phenotype through downregulating TNF-α and IL-10 and upregulating IL-12 [331]. Treg depletion or suppression of their immunosuppressive actions may potentially restore effector T cell antitumor activity, inhibiting tumor growth. Restoration of antitumor immune response within the TIME is a challenging task. Recently, a nanoparticle-based MUC1 mRNA vaccine (NP) in combination with anti-CTLA-4 has shown to induce cytotoxic T lymphocyte response, leading to enhancement of antitumor activity as compared with vaccine or anti-CTLA4 alone in 4T1 mice models [94]. Understanding the drug targets and off-targets in immune and stromal cells requires robust drug delivery systems within the TIME. Therefore, selecting suitable delivery systems, dose optimization, drug combinations, and their pharmacokinetics and pharmacodynamics in TIME will open new avenues to overcome immunotherapy resistance in breast cancer.
Conclusion and future perspective
Immunotherapy has therapeutic potential for treating breast cancer, resistance to conventional therapies. Several lines of investigation are in progress for an in-depth understanding of the immune tolerance mechanisms to develop novel therapeutic approaches for combating immune resistance. The study of intricate biological phenomena has been revolutionized by single-cell omics tools, such as assay for transposase accessible chromatin (ATAC) sequencing and single-cell RNA, which enable detailed monitoring of the TME during immunotherapy. Incorporating single-cell CRISPR screens improves the resolution of biological event analysis, while concurrent spatial omics studies offer simultaneous spatial information and protein expression. Researchers have carried out a parallel single-cell RNA-seq and T cell receptor (TCR) profiling using breast cancer patient samples to understand the tumor microenvironmental changes following anti-PD1 therapy [287]. Single-cell analysis revealed specific immune cell subsets linked with anti-PD-L1 therapy in TNBC [332]. This study identified responders with an increased pool of CXCL13+ T cells by mapping alterations in immune cells after anti-PD-L1 and paclitaxel therapy using paired single-cell transcriptome, ATAC, and TCR sequencing [332]. Additionally, single-cell RNA-seq has clarified the intricate biology of T cell exhaustion in the TME [333]. Algorithms facilitating co-registration of single-cell and spatial NGS data offer unprecedented insights into the co-evolution of immune cell clones. They may aid in identifying targets for combination therapy in cancer immunotherapy regimens. Therefore, a detailed understanding of the tumor microenvironmental changes during immunotherapy through single-cell omics, multi-omics, or multiplexed in situ spatial protein profiling may aid in developing novel therapeutic strategies to overcome immune tolerance in breast cancer.
The intricate interplay between CAF and TAM is crucial in determining immune therapy efficacy with the TIME. For instance, a pan cancer analysis revealed iCAFs stimulate cancer cell proliferation, epithelial-mesenchymal transition (EMT), and the create an immunosuppressive TIME in breast cancer patients receiving anti-PD1 immunotherapy [334]. Similarly, Li et al. have characterized two CAFs clusters A and B where cluster B is abundant with immunosuppressive macrophages and resulted in poor overall survival than cluster A [335]. TAM depletion by sequential administration of TAM targeting T cells followed by cancer targeting T cells, resulting in reduction in tumor size and longer survival in mouse ovarian cancer model [336]. CAF-derived IL-6 contributes to immune therapy resistance and inhibition of the IL-6-STAT3/Akt-PD-L1 axis resensitizes to immune therapy in breast cancer patients [337]. In addition, MDSC is an essential component of the TME to be considered in immunotherapy outcomes. Tumor metabolism, gaining attention in cancer research, is also linked to immunotherapy resistance. Thus, developing strategies targeting CAF and TAM regulated core transcriptional networks and its heterogeneity could provide better immune therapy-based strategy for the treatment of breast cancer. Immune therapy resistance is also conferred by epigenetic modifications in various cells within the TIME. Particularly immune cells are regulated epigenetically in modulating immune cell function and developing immune therapy resistance. Combining epi-drugs along with various immune therapy-based approaches could provide better disease-free survival in breast cancer patients.
Nanotechnology is also being used to reprogram TME for immune stimulation. Nanoparticles can contribute to better drug uptake, improved CAF reprogramming, and better T cell infiltration, leading to more effective immunotherapy. PDT, PIT and PTT improves the immunotherapy efficacy thereby providing better therapeutic outcomes in breast cancer. Combining nanoparticles with photodynamic immunotherapy ablates TAM metabolism in TNBC [338]. Further addressing PDT related adverse effects in terms of structural and biological aspect while combining with immunotherapy could improve clinical manifestations in breast cancer treatment [339].
Moreover, non-specific immunostimulation by immunotherapy mimics autoimmune disorders and causes several immune-related adverse effects. Minor toxicity may be managed by temporary withdrawal of immunotherapy while it must be discontinued in case of severe toxicity. Different immunotherapies have their specific immune-related adverse effects. Along with immunotherapy resistance, addressing immune-related other adverse effects is also crucial for successful immunotherapy outcome [340]. All these factors should be considered to control the TIME for a better immunotherapeutic outcome.
Additionally, organ-specific differences in TME may cause different immune-resistant mechanisms and immune therapy responses [341]. Hence, we must understand the organ-specific TME to establish more precise therapy depending on the immune tolerance mechanism. Immuno-subtyping may be a successful approach to deal with immunotherapy failure. It will aid in understanding tumor heterogeneity among breast cancer patients [342]. Moreover, single-cell and spatial transcriptomics studies may detangle the complexity of TME-mediated immunomodulation. These will be beneficial to explore biomarkers for immunotherapy response prediction and targets for improved immunotherapy outcomes [343]. Advanced therapeutic strategies, including personalized immunotherapy and gene editing based therapeutics, may be helpful to modulate different TME components for enhanced immunotherapy efficacy.
In conclusion, various components of TME such as CAFs, TAMs, DCs and MDSCs are involved in TIME modulation, leading to immunotherapy resistance. In this review, we have highlighted various immunotherapy-based approaches and stromal-immune interplay such as CAF, TAM and MDSC-mediated immune tolerance in breast cancer. Moreover, alteration of tumor metabolism leads to immunotherapy failure. Furthermore, targeting these and their metabolic regulation with combination therapy could overcome immune resistance and enhance the efficacy of immunotherapy in breast cancer. This review emphasizes the therapeutic approaches to overcome breast cancer immune resistance in combination with immunotherapy such as photodynamic, photoimmunotherapy, epi-drug and nanoparticle mediated drug delivery. The comprehensive strategies emphasize that the resistance to immunotherapy needs to be further studied to develop therapeutic regimens for the successful overcome of immunotherapy resistant breast cancers.
Data availability
Not applicable.
Abbreviations
- CAF:
-
Cancer-associated fibroblast
- TAM:
-
Tumor-associated macrophage
- GLOBOCAN:
-
Global cancer observatory
- TME:
-
Tumor microenvironment
- DC:
-
Dendritic cell
- MC:
-
Mast cell
- ECM:
-
Extracellular matrix
- TAN:
-
Tumor-associated neutrophil
- PD1:
-
Programmed cell death protein 1
- PDL1:
-
Programmed cell death protein ligand 1
- Th:
-
T helper type
- NK:
-
Natural killer
- CTL:
-
Cytotoxic T lymphocyte
- Treg:
-
Regulatory T cell
- MDSC:
-
Myeloid-derived suppressor cell
- ICIs:
-
Immune checkpoint inhibitors
- TIME:
-
Tumor immune microenvironment
- mAB:
-
Monoclonal antibody
- ADC:
-
Antibody-drug conjugate
- EGFR:
-
Epidermal growth factor receptor
- CTLA4:
-
Cytotoxic T-lymphocyte–associated antigen 4
- FDA:
-
Food & drug administration
- T-DM1:
-
Trastuzumab-Emtansine
- TNBC:
-
Triple-negative breast cancer
- HER:
-
Human epidermal growth factor receptor
- EMA:
-
European Medicines Agency
- gpNMB:
-
Glycoprotein-NMB
- IMMU-132:
-
Sacituzumab govitecan
- SGN-LIV1A:
-
Ladiratuzumab vedotin
- MMAE:
-
Monomethyl-auristatin-E
- ORR:
-
Overall response rate
- OS:
-
Overall survival
- PFS:
-
Progression-free survival
- CDX-011:
-
Glembatumumab vedotin
- CTA:
-
Cancer-testis antigen
- WT1:
-
Wilms tumor protein 1
- MAGE-12:
-
Melanoma antigen gene-12
- FRα:
-
Folate receptor alpha
- TAC:
-
Tumor-associated carbohydrate
- MSC:
-
Mesenchymal stem cell
- ILC:
-
Invasive lobular carcinoma
- IDC:
-
Invasive ductal carcinoma
- P4H:
-
Prolyl 4-hydroxylase
- NG2:
-
Chondroitin sulfate proteoglycan
- PDGFR:
-
Platelet-derived growth factor receptor
- FAP:
-
Fibroblast activation protein
- PDPN:
-
Podoplanin
- FSP:
-
Fibroblast specific protein
- ER:
-
Estrogen receptor
- myoCAF:
-
Myofibroblastic CAF
- iCAF:
-
Inflammatory CAF
- apCAF:
-
Antigen-presenting CAF
- α-SMA:
-
α-Smooth muscle actin
- CAV1:
-
Caveolin 1
- STAT3:
-
Signal transducer and activator of transcription 3
- CAP:
-
Cancer-associated pericyte
- NO:
-
Nitric oxide
- TGF:
-
Transforming growth factor
- LRRC15:
-
Leucine-rich repeat-containing protein 15
- PDAC:
-
Pancreatic ductal adenocarcinoma
- CXC:
-
Cysteine X cysteine
- TNF:
-
Tumor necrosis factor
- IFN:
-
Interferon
- IL:
-
Interleukin
- FASL:
-
Fas ligand
- BGN:
-
Biglycan
- ARG:
-
Arginase
- iNOS:
-
Inducible Nitric oxide synthase
- MHC:
-
Major histocompatibility complex
- JAK:
-
Janus kinase
- PI3K:
-
Phosphatidylinositol-3 kinase
- AKT:
-
Protein kinase B
- PG:
-
Prostaglandin
- COX2:
-
Cyclooxygenase-2
- PEG2:
-
Prostaglandin E2
- VEGF:
-
Vascular endothelial growth factor
- SPP1:
-
Secreted phosphoprotein 1
- BM-MSC:
-
Bone marrow-derived mesenchymal stem cell
- MZF1:
-
Myeloid zinc finger 1
- ADF:
-
Adipocyte-derived fibroblast
- MCP1:
-
Monocyte chemotactic protein 1
- CHI3L1:
-
Chitinase-3-like protein 1
- LAM:
-
Lipid-associated macrophages
- EMT:
-
Epithelial-to-mesenchymal transition
- SDF1:
-
Stromal cell-derived factor 1
- G-CSF:
-
Granulocyte colony-stimulating factor
- Breg:
-
Regulatory B cell
- HIF1α:
-
Hypoxia-induced factor 1α
- PDK1:
-
3-Phosphoinositide-dependent kinase 1
- TCA:
-
Tricarboxylic acid
- LDHA:
-
Lactate dehydrogenase A
- PLAC8:
-
Placenta-specific 8
- UFM1:
-
Ubiquitin-fold modifier 1
- BATF:
-
Basic leucine zipper ATF-like transcription factor
- CRG:
-
Cuproptosis-related gene
- AIR:
-
Acquired immune resistance
- GEMM:
-
Genetically engineered mouse model
- DrIM:
-
Disease related immune gene
- NOD:
-
Non-obese diabetic
- MKO:
-
Mouse knockout
- ATAC:
-
Assay for transposase accessible chromatin
- TCR:
-
T cell receptor
- TAA:
-
Tumor-associated antigen
- HSP:
-
Heat shock protein
- CDK:
-
Cyclin-dependent kinase
- MUC4β:
-
Mucin 4β
- MSA2:
-
Mammary serum antigen 2
- TIL:
-
Tumor-infiltrating lymphocyte
- PET:
-
Positron emission tomography
- HR:
-
Hormone receptor
- NOX4:
-
NADPH oxidase 4
- FOLR:
-
Folate receptor
- TREM:
-
Triggering receptor expressed on myeloid cell
- ATRA:
-
All-trans retinoid acid
- ADAM12:
-
A disintegrin and metalloproteinase-12
- MCT1:
-
Monocarboxylate transporter-1
- CIAX:
-
Carbonic anhydrase IX
- PDT:
-
Photodynamic therapy
- ROS:
-
Reactive oxygen species
- DAMP:
-
Damage-associated molecular pattern
- MAF:
-
Macrophage activating factor
- PIT:
-
Photoimmunotherapy
- BP:
-
Black phosphorus
- BPNV:
-
BP nanovesicles
- APC:
-
Antigen-presenting cell
- NIR:
-
Near-infrared
- PTT:
-
Photothermal therapy
- BPQD:
-
Black phosphorus quantum dots
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a Cancer Journal for Clinicians. 2021;:caac.21660.
Bai R, Chen N, Li L, Du N, Bai L, Lv Z, et al. Mechanisms of Cancer Resistance to Immunotherapy. Front Oncol. 2020;10:1290.
Baghban R, Roshangar L, Jahanban-Esfahlan R, Seidi K, Ebrahimi-Kalan A, Jaymand M, et al. Tumor microenvironment complexity and therapeutic implications at a glance. Cell Commun Signal. 2020;18:59.
Sarkar S, Horn G, Moulton K, Oza A, Byler S, Kokolus S, et al. Cancer Development, Progression, and Therapy: an epigenetic overview. IJMS. 2013;14:21087–113.
Hanahan D. Hallmarks of Cancer: New dimensions. Cancer Discov. 2022;12:31–46.
Chew V, Toh HC, Abastado J-P. Immune Microenvironment in Tumor Progression: characteristics and challenges for Therapy. J Oncol. 2012;2012:1–10.
Mao X, Xu J, Wang W, Liang C, Hua J, Liu J, et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer. 2021;20:131.
Butti R, Kumar TVS, Nimma R, Banerjee P, Kundu IG, Kundu GC. Osteopontin Signaling in shaping Tumor Microenvironment Conducive to Malignant Progression. In: Birbrair A, editor. Tumor Microenvironment. Cham: Springer International Publishing; 2021. pp. 419–41.
Andersson P, Yang Y, Hosaka K, Zhang Y, Fischer C, Braun H, et al. Molecular mechanisms of IL-33–mediated stromal interactions in cancer metastasis. JCI Insight. 2018;3:e122375.
Butti R, Nimma R, Kundu G, Bulbule A, Kumar TVS, Gunasekaran VP, et al. Tumor-derived osteopontin drives the resident fibroblast to myofibroblast differentiation through Twist1 to promote breast cancer progression. Oncogene. 2021;40:2002–17.
Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 2018;32:1267–84.
Sadeghalvad M, Mohammadi-Motlagh H-R, Rezaei N. Immune microenvironment in different molecular subtypes of ductal breast carcinoma. Breast Cancer Res Treat. 2021;185:261–79.
Del Gil CR, Huh SJ, Ekram MB, Trinh A, Liu LL, Beca F, et al. Immune escape in breast Cancer during in situ to Invasive Carcinoma Transition. Cancer Discov. 2017;7:1098–115.
Salemme V, Centonze G, Cavallo F, Defilippi P, Conti L. The Crosstalk between Tumor Cells and the Immune Microenvironment in breast Cancer: implications for Immunotherapy. Front Oncol. 2021;11:610303.
Balkwill F. Cancer and the chemokine network. Nat Rev Cancer. 2004;4:540–50.
Debien V, De Caluwé A, Wang X, Piccart-Gebhart M, Tuohy VK, Romano E, et al. Immunotherapy in breast cancer: an overview of current strategies and perspectives. npj Breast Cancer. 2023;9:7.
Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018;24:541–50.
Garrido-Castro AC, Lin NU, Polyak K. Insights into Molecular classifications of Triple-negative breast Cancer: improving patient selection for treatment. Cancer Discov. 2019;9:176–98.
Stark MC, Joubert AM, Visagie MH. Molecular Farming of Pembrolizumab and Nivolumab. IJMS. 2023;24:10045.
Krasniqi E, Barchiesi G, Pizzuti L, Mazzotta M, Venuti A, Maugeri-Saccà M, et al. Immunotherapy in HER2-positive breast cancer: state of the art and future perspectives. J Hematol Oncol. 2019;12:111.
Schmid P, Cortes J, Pusztai L, McArthur H, Kümmel S, Bergh J, et al. Pembrolizumab for early triple-negative breast Cancer. N Engl J Med. 2020;382:810–21.
Adel NG. Current treatment landscape and emerging therapies for metastatic triple-negative breast cancer. Am J Manag Care. 2021;27(Suppl 5):S87–96.
Franzoi MA, Romano E, Piccart M. Immunotherapy for early breast cancer: too soon, too superficial, or just right? Ann Oncol. 2021;32:323–36.
Dirix LY, Takacs I, Jerusalem G, Nikolinakos P, Arkenau H-T, Forero-Torres A, et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: a phase 1b JAVELIN solid tumor study. Breast Cancer Res Treat. 2018;167:671–86.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64.
Henriques B, Mendes F, Martins D. Immunotherapy in breast Cancer: when, how, and what challenges? Biomedicines. 2021;9:1687.
Rowshanravan B, Halliday N, Sansom DM. CTLA-4: a moving target in immunotherapy. Blood. 2018;131:58–67.
Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. 2016;39:98–106.
Shimomura A, Fujiwara Y, Kondo S, Kodaira M, Iwasa S, Kitano S, et al. Tremelimumab-associated tumor regression following after initial progression: two case reports. Immunotherapy. 2016;8:9–15.
Tarhini A. CTLA-4 blockade: therapeutic potential in cancer treatments. OTT. 2010;3:15–25.
Loi S, Francis PA, Zdenkowski N, Gebski V, Fox SB, White M, et al. Neoadjuvant ipilimumab and nivolumab in combination with paclitaxel following anthracycline-based chemotherapy in patients with treatment resistant early-stage triple-negative breast cancer (TNBC): a single-arm phase 2 trial. JCO. 2022;40 16suppl:602–602.
Santa-Maria CA, Kato T, Park J-H, Kiyotani K, Rademaker A, Shah AN, et al. A pilot study of durvalumab and tremelimumab and immunogenomic dynamics in metastatic breast cancer. Oncotarget. 2018;9:18985–96.
Hwang WL, Pike LRG, Royce TJ, Mahal BA, Loeffler JS. Safety of combining radiotherapy with immune-checkpoint inhibition. Nat Rev Clin Oncol. 2018;15:477–94.
Barroso-Sousa R, Barry WT, Garrido-Castro AC, Hodi FS, Min L, Krop IE, et al. Incidence of endocrine dysfunction following the use of different Immune checkpoint inhibitor regimens: a systematic review and Meta-analysis. JAMA Oncol. 2018;4:173.
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of Chemotherapy plus a monoclonal antibody against HER2 for metastatic breast Cancer that overexpresses HER2. N Engl J Med. 2001;344:783–92.
Muntasell A, Cabo M, Servitja S, Tusquets I, Martínez-García M, Rovira A, et al. Interplay between natural killer cells and Anti-HER2 antibodies: perspectives for breast Cancer immunotherapy. Front Immunol. 2017;8:1544.
Early Breast Cancer Trialists’ Collaborative group (EBCTCG). Trastuzumab for early-stage, HER2-positive breast cancer: a meta-analysis of 13 864 women in seven randomised trials. Lancet Oncol. 2021;22:1139–50.
Kreutzfeldt J, Rozeboom B, Dey N, De P. The trastuzumab era: current and upcoming targeted HER2 + breast cancer therapies. Am J Cancer Res. 2020;10:1045–67.
García-Aranda M, Redondo M, Immunotherapy. A challenge of breast Cancer Treatment. Cancers. 2019;11:1822.
Hunter FW, Barker HR, Lipert B, Rothé F, Gebhart G, Piccart-Gebhart MJ, et al. Mechanisms of resistance to trastuzumab emtansine (T-DM1) in HER2-positive breast cancer. Br J Cancer. 2020;122:603–12.
Nuti M, Bellati F, Visconti V, Napoletano C, Domenici L, Caccetta J, et al. Immune effects of Trastuzumab. J Cancer. 2011;2:317–23.
Klein P. Trastuzumab and cardiac dysfunction: update on preclinical studies. Semin Oncol. 2003;30:49–53.
Swain SM, Baselga J, Kim S-B, Ro J, Semiglazov V, Campone M, et al. Pertuzumab, Trastuzumab, and Docetaxel in HER2-Positive metastatic breast Cancer. N Engl J Med. 2015;372:724–34.
Loi S, Giobbie-Hurder A, Gombos A, Bachelot T, Hui R, Curigliano G, et al. Pembrolizumab plus Trastuzumab in trastuzumab-resistant, advanced, HER2-positive breast cancer (PANACEA): a single-arm, multicentre, phase 1b–2 trial. Lancet Oncol. 2019;20:371–82.
Royce M, Osgood CL, Amatya AK, Fiero MH, Chang CJG, Ricks TK, et al. FDA approval Summary: Margetuximab plus Chemotherapy for Advanced or metastatic HER2-Positive breast Cancer. Clin Cancer Res. 2022;28:1487–92.
Rugo HS, Im S-A, Cardoso F, Cortés J, Curigliano G, Musolino A, et al. Efficacy of Margetuximab vs Trastuzumab in patients with pretreated ERBB2-Positive advanced breast Cancer: a phase 3 Randomized Clinical Trial. JAMA Oncol. 2021;7:573.
Tarantino P, Morganti S, Uliano J, Giugliano F, Crimini E, Curigliano G. Margetuximab for the treatment of HER2-positive metastatic breast cancer. Expert Opin Biol Ther. 2021;21:127–33.
Jiao X, Wang M, Zhang Z, Li Z, Ni D, Ashton AW, et al. Leronlimab, a humanized monoclonal antibody to CCR5, blocks breast cancer cellular metastasis and enhances cell death induced by DNA damaging chemotherapy. Breast Cancer Res. 2021;23:11.
Behl A, Wani ZA, Das NN, Parmar VS, Len C, Malhotra S, et al. Monoclonal antibodies in breast cancer: a critical appraisal. Crit Rev Oncol/Hematol. 2023;183:103915.
Swain SM, Shastry M, Hamilton E. Targeting HER2-positive breast cancer: advances and future directions. Nat Rev Drug Discov. 2023;22:101–26.
Hamilton EP, Petit T, Pistilli B, Goncalves A, Ferreira AA, Dalenc F, et al. Clinical activity of MCLA-128 (zenocutuzumab), trastuzumab, and vinorelbine in HER2 amplified metastatic breast cancer (MBC) patients (pts) who had progressed on anti-HER2 ADCs. JCO. 2020;38 15suppl:3093–3093.
Zhang J, Ji D, Cai L, Yao H, Yan M, Wang X, et al. First-in-human HER2-targeted bispecific antibody KN026 for the treatment of patients with HER2-positive metastatic breast Cancer: results from a phase I study. Clin Cancer Res. 2022;28:618–28.
Peters C, Brown S. Antibody–drug conjugates as novel anti-cancer chemotherapeutics. Biosci Rep. 2015;35:e00225.
Arab A, Yazdian-Robati R, Behravan J. HER2-Positive breast Cancer Immunotherapy: a focus on Vaccine Development. Arch Immunol Ther Exp. 2020;68:2.
Galván Morales MA, Barrera Rodríguez R, Santiago Cruz JR, Teran LM. Overview of new treatments with immunotherapy for breast Cancer and a proposal of a combination therapy. Molecules. 2020;25:5686.
Keam SJ. Trastuzumab Deruxtecan: first approval. Drugs. 2020;80:501–8.
Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, et al. The genomic Landscape of Endocrine-Resistant Advanced breast cancers. Cancer Cell. 2018;34:427–e4386.
Rugo HS, Bianchini G, Cortes J, Henning J-W, Untch M. Optimizing treatment management of trastuzumab deruxtecan in clinical practice of breast cancer. ESMO Open. 2022;7:100553.
Chiu JWY, Lee SC, Ho JC, Park YH, Chao T-C, Kim S-B, et al. Clinical Guidance on the monitoring and management of Trastuzumab Deruxtecan (T-DXd)-Related adverse events: insights from an Asia-Pacific Multidisciplinary Panel. Drug Saf. 2023;46:927–49.
Sussman D, Smith LM, Anderson ME, Duniho S, Hunter JH, Kostner H, et al. SGN–LIV1A: a novel antibody–drug Conjugate Targeting LIV-1 for the treatment of metastatic breast Cancer. Mol Cancer Ther. 2014;13:2991–3000.
Modi S, Pusztai L, Forero A, Mita M, Miller K, Weise A, et al. Abstract PD3-14: phase 1 study of the antibody-drug conjugate SGN-LIV1A in patients with heavily pretreated triple-negative metastatic breast cancer. Cancer Res. 2018;78(4Supplement):PD3–14.
Cardillo TM, Govindan SV, Sharkey RM, Trisal P, Arrojo R, Liu D, et al. Sacituzumab Govitecan (IMMU-132), an Anti-TROP2/SN-38 antibody–drug conjugate: characterization and efficacy in pancreatic, gastric, and other cancers. Bioconjug Chem. 2015;26:919–31.
Bardia A, Mayer IA, Diamond JR, Moroose RL, Isakoff SJ, Starodub AN, et al. Efficacy and safety of Anti-TROP2 antibody drug Conjugate Sacituzumab Govitecan (IMMU-132) in heavily pretreated patients with metastatic triple-negative breast Cancer. J Clin Oncol. 2017;35:2141–8.
Shastry M, Jacob S, Rugo HS, Hamilton E. Antibody-drug conjugates targeting TROP2: clinical development in metastatic breast cancer. Breast. 2022;66:169–77.
Dent RA, Cescon DW, Bachelot T, Jung KH, Shao Z-M, Saji S, et al. TROPION-Breast02: Datopotamab deruxtecan for locally recurrent inoperable or metastatic triple-negative breast cancer. Future Oncol. 2023;19:2349–59.
Yardley DA, Weaver R, Melisko ME, Saleh MN, Arena FP, Forero A, et al. EMERGE: a randomized phase II study of the antibody-drug Conjugate Glembatumumab Vedotin in Advanced Glycoprotein NMB–Expressing breast Cancer. JCO. 2015;33:1609–19.
Vahdat LT, Schmid P, Forero-Torres A, Blackwell K, Telli ML, Melisko M, et al. Glembatumumab vedotin for patients with metastatic, gpNMB overexpressing, triple-negative breast cancer (METRIC): a randomized multicenter study. npj Breast Cancer. 2021;7:57.
D’Arienzo A, Verrazzo A, Pagliuca M, Napolitano F, Parola S, Viggiani M, et al. Toxicity profile of antibody-drug conjugates in breast cancer: practical considerations. eClinicalMedicine. 2023;62:102113.
Clifton GT, Peoples GE, Mittendorf EA. The development and use of the E75 (HER2 369–377) peptide vaccine. Future Oncol. 2016;12:1321–9.
Clifton GT, Gall V, Peoples GE, Mittendorf EA. Clinical Development of the E75 vaccine in breast Cancer. Breast Care (Basel). 2016;11:116–21.
Brown TA, Mittendorf EA, Hale DF, Myers JW, Peace KM, Jackson DO, et al. Prospective, randomized, single-blinded, multi-center phase II trial of two HER2 peptide vaccines, GP2 and AE37, in breast cancer patients to prevent recurrence. Breast Cancer Res Treat. 2020;181:391–401.
Zhao L, Zhang M, Cong H. Advances in the study of HLA-restricted epitope vaccines. Hum Vaccin Immunother. 2013;9:2566–77.
Clifton GT, Mittendorf EA, Peoples GE. Adjuvant HER2/neu peptide cancer vaccines in breast cancer. Immunotherapy. 2015;7:1159–68.
Mittendorf EA, Clifton GT, Holmes JP, Schneble E, Van Echo D, Ponniah S, et al. Final report of the phase I/II clinical trial of the E75 (nelipepimut-S) vaccine with booster inoculations to prevent disease recurrence in high-risk breast cancer patients. Ann Oncol. 2014;25:1735–42.
Mittendorf EA, Ardavanis A, Symanowski J, Murray JL, Shumway NM, Litton JK, et al. Primary analysis of a prospective, randomized, single-blinded phase II trial evaluating the HER2 peptide AE37 vaccine in breast cancer patients to prevent recurrence. Ann Oncol. 2016;27:1241–8.
Thomas R, Al-Khadairi G, Roelands J, Hendrickx W, Dermime S, Bedognetti D, et al. NY-ESO-1 based immunotherapy of Cancer: current perspectives. Front Immunol. 2018;9:947.
Wang C, Gu Y, Zhang K, Xie K, Zhu M, Dai N, et al. Systematic identification of genes with a cancer-testis expression pattern in 19 cancer types. Nat Commun. 2016;7:10499.
O’Shaughnessy J, Roberts LK, Smith JL, Levin MK, Timis R, Finholt JP, et al. Safety and initial clinical efficacy of a dendritic cell (DC) vaccine in locally advanced, triple-negative breast cancer (TNBC) patients (pts). JCO. 2016;34 15suppl:1086–1086.
Higgins M, Curigliano G, Dieras V, Kuemmel S, Kunz G, Fasching PA, et al. Safety and immunogenicity of neoadjuvant treatment using WT1-immunotherapeutic in combination with standard therapy in patients with WT1-positive stage II/III breast cancer: a randomized phase I study. Breast Cancer Res Treat. 2017;162:479–88.
Chung VM, Kos F, Hardwick N, Yuan Y, Chao J, Li M, et al. A phase 1 study of p53MVA vaccine in combination with pembrolizumab. JCO. 2018;36 5suppl:206–206.
Kimura T, Finn OJ. MUC1 immunotherapy is here to stay. Expert Opin Biol Ther. 2013;13:35–49.
Hosseini M, Seyedpour S, Khodaei B, Loghman A-H, Seyedpour N, Yazdi M-H, et al. Cancer vaccines for Triple-negative breast Cancer: a systematic review. Vaccines. 2023;11:146.
Rugo HS, Cortes J, Xu B, Huang C-S, Kim S-B, Melisko ME, et al. A phase 3, randomized, open-label study of the anti-globo H vaccine adagloxad simolenin/obi-821 in the adjuvant treatment of high-risk, early-stage, Globo H-positive triple-negative breast cancer. JCO. 2022;40 16suppl:TPS611–611.
Stevens KN, Vachon CM, Couch FJ. Genetic susceptibility to Triple-negative breast Cancer. Cancer Res. 2013;73:2025–30.
Gray A, Yan L, Kast WM. Prevention is Better Than cure: the case for clinical trials of Therapeutic Cancer vaccines in the prophylactic setting. Mol Interv. 2010;10:197–203.
Brito Baleeiro R, Liu P, Chard Dunmall LS, Di Gioia C, Nagano A, Cutmore L, et al. Personalized neoantigen viro-immunotherapy platform for triple-negative breast cancer. J Immunother Cancer. 2023;11:e007336.
Zhang S, Liu Y, Zhou J, Wang J, Jin G, Wang X. Breast Cancer Vaccine containing a Novel toll-like receptor 7 agonist and an aluminum adjuvant exerts Antitumor effects. IJMS. 2022;23:15130.
Singer CF, Pfeiler G, Hubalek M, Bartsch R, Stöger H, Pichler A, et al. Efficacy and safety of the therapeutic cancer vaccine tecemotide (L-BLP25) in early breast cancer: results from a prospective, randomised, neoadjuvant phase II study (ABCSG 34). Eur J Cancer. 2020;132:43–52.
Dehghan-Manshadi M, Nikpoor AR, Hadinedoushan H, Zare F, Sankian M, Fesahat F, et al. Protective immune response against P32 oncogenic peptide-pulsed PBMCs in mouse models of breast cancer. Int Immunopharmacol. 2021;93:107414.
Basu A, Albert GK, Awshah S, Datta J, Kodumudi KN, Gallen C, et al. Identification of immunogenic MHC class II Human HER3 peptides that mediate Anti-HER3 CD4 + Th1 responses and potential use as a Cancer Vaccine. Cancer Immunol Res. 2022;10:108–25.
Callmann CE, Cole LE, Kusmierz CD, Huang Z, Horiuchi D, Mirkin CA. Tumor cell lysate-loaded immunostimulatory spherical nucleic acids as therapeutics for triple-negative breast cancer. Proc Natl Acad Sci USA. 2020;117:17543–50.
Huang L, Rong Y, Tang X, Yi K, Qi P, Hou J, et al. Engineered exosomes as an in situ DC-primed vaccine to boost antitumor immunity in breast cancer. Mol Cancer. 2022;21:45.
Tay AS-MS, Amano T, Edwards LA, Yu JS. CD133 mRNA-transfected dendritic cells induce coordinated cytotoxic and helper T cell responses against breast cancer stem cells. Mol Therapy - Oncolytics. 2021;22:64–71.
Liu L, Wang Y, Miao L, Liu Q, Musetti S, Li J, et al. Combination immunotherapy of MUC1 mRNA Nano-vaccine and CTLA-4 Blockade effectively inhibits growth of Triple negative breast Cancer. Mol Ther. 2018;26:45–55.
Santisteban M, Solans BP, Hato L, Urrizola A, Mejías LD, Salgado E, et al. Final results regarding the addition of dendritic cell vaccines to neoadjuvant chemotherapy in early HER2-negative breast cancer patients: clinical and translational analysis. Ther Adv Med Oncol. 2021;13:175883592110646.
Maeng HM, Moore BN, Bagheri H, Steinberg SM, Inglefield J, Dunham K, et al. Phase I clinical trial of an autologous dendritic cell vaccine against HER2 shows Safety and preliminary clinical efficacy. Front Oncol. 2021;11:789078.
Lowenfeld L, Mick R, Datta J, Xu S, Fitzpatrick E, Fisher CS, et al. Dendritic cell vaccination enhances Immune responses and induces regression of HER2pos DCIS Independent of Route: results of Randomized Selection Design Trial. Clin Cancer Res. 2017;23:2961–71.
Lowenfeld L, Zaheer S, Oechsle C, Fracol M, Datta J, Xu S, et al. Addition of anti-estrogen therapy to anti-HER2 dendritic cell vaccination improves regional nodal immune response and pathologic complete response rate in patients with ER pos /HER2 pos early breast cancer. OncoImmunology. 2017;6:e1207032.
Bernal-Estévez DA, Ortíz Barbosa MA, Ortíz-Montero P, Cifuentes C, Sánchez R, Parra-López CA. Autologous dendritic cells in Combination with Chemotherapy restore responsiveness of T cells in breast Cancer patients: a single-arm phase I/II trial. Front Immunol. 2021;12:669965.
Adams S, Kozhaya L, Martiniuk F, Meng T-C, Chiriboga L, Liebes L, et al. Topical TLR7 agonist Imiquimod can induce Immune-mediated rejection of skin metastases in patients with breast Cancer. Clin Cancer Res. 2012;18:6748–57.
Salazar LG, Lu H, Reichow JL, Childs JS, Coveler AL, Higgins DM, et al. Topical Imiquimod Plus Nab-paclitaxel for breast Cancer cutaneous metastases: a phase 2 clinical trial. JAMA Oncol. 2017;3:969.
Hammerich L, Marron TU, Upadhyay R, Svensson-Arvelund J, Dhainaut M, Hussein S, et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat Med. 2019;25:814–24.
Dees S, Ganesan R, Singh S, Grewal IS. Emerging CAR-T cell therapy for the treatment of Triple-negative breast Cancer. Mol Cancer Ther. 2020;19:2409–21.
Dey A, Ghosh S, Jha S, Hazra S, Srivastava N, Chakraborty U, et al. Recent advancement in breast cancer treatment using CAR T cell therapy:- a review. Adv Cancer Biology - Metastasis. 2023;7:100090.
Zhang W, Liu L, Su H, Liu Q, Shen J, Dai H, et al. Chimeric antigen receptor macrophage therapy for breast tumours mediated by targeting the tumour extracellular matrix. Br J Cancer. 2019;121:837–45.
Schepisi G, Gianni C, Palleschi M, Bleve S, Casadei C, Lolli C, et al. The New Frontier of Immunotherapy: Chimeric Antigen Receptor T (CAR-T) cell and macrophage (CAR-M) therapy against breast Cancer. Cancers. 2023;15:1597.
Raftery MJ, Franzén AS, Radecke C, Boulifa A, Schönrich G, Stintzing S, et al. Next Generation CD44v6-Specific CAR-NK cells effective against Triple negative breast Cancer. IJMS. 2023;24:9038.
Zhang P, Zhang G, Wan X. Challenges and new technologies in adoptive cell therapy. J Hematol Oncol. 2023;16:97.
Long B, Brem E, Koyfman A. Oncologic emergencies: Immune-Based Cancer therapies and complications. WestJEM. 2020;21:566–80.
Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–95.
Fajgenbaum DC, June CH. Cytokine storm. N Engl J Med. 2020;383:2255–73.
Wu F, Yang J, Liu J, Wang Y, Mu J, Zeng Q, et al. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Sig Transduct Target Ther. 2021;6:218.
Simon T, Salhia B. Cancer-Associated fibroblast subpopulations with diverse and dynamic roles in the Tumor Microenvironment. Mol Cancer Res. 2022;20:183–92.
Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. IL1-Induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 2019;9:282–301.
Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci USA. 2013;110:20212–7.
Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu C-C, Simpson TR, et al. Depletion of Carcinoma-Associated fibroblasts and fibrosis induces immunosuppression and accelerates Pancreas Cancer with reduced survival. Cancer Cell. 2014;25:719–34.
Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of Antitumor immunity by stromal cells expressing fibroblast activation Protein–α. Science. 2010;330:827–30.
Hu D, Li Z, Zheng B, Lin X, Pan Y, Gong P, et al. Cancer-associated fibroblasts in breast cancer: challenges and opportunities. Cancer Commun. 2022;42:401–34.
Chen Y, McAndrews KM, Kalluri R. Clinical and therapeutic relevance of cancer-associated fibroblasts. Nat Rev Clin Oncol. 2021;18:792–804.
Wu SZ, Roden DL, Wang C, Holliday H, Harvey K, Cazet AS et al. Stromal cell diversity associated with immune evasion in human triple-negative breast cancer. EMBO J. 2020;39:e104063.
Costa A, Kieffer Y, Scholer-Dahirel A, Pelon F, Bourachot B, Cardon M, et al. Fibroblast heterogeneity and immunosuppressive environment in human breast Cancer. Cancer Cell. 2018;33:463–e47910.
Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B, et al. Single-cell analysis reveals fibroblast clusters linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020;10:1330–51.
Chen L, Qiu X, Wang X, He J. FAP positive fibroblasts induce immune checkpoint blockade resistance in colorectal cancer via promoting immunosuppression. Biochem Biophys Res Commun. 2017;487:8–14.
Yang X, Lin Y, Shi Y, Li B, Liu W, Yin W, et al. FAP promotes immunosuppression by Cancer-Associated fibroblasts in the Tumor Microenvironment via STAT3–CCL2 signaling. Cancer Res. 2016;76:4124–35.
Cords L, Tietscher S, Anzeneder T, Langwieder C, Rees M, De Souza N, et al. Cancer-associated fibroblast classification in single-cell and spatial proteomics data. Nat Commun. 2023;14:4294.
Cremasco V, Astarita JL, Grauel AL, Keerthivasan S, MacIsaac K, Woodruff MC, et al. FAP delineates heterogeneous and functionally divergent stromal cells in Immune-excluded breast tumors. Cancer Immunol Res. 2018;6:1472–85.
Rivas EI, Linares J, Zwick M, Gómez-Llonin A, Guiu M, Labernadie A, et al. Targeted immunotherapy against distinct cancer-associated fibroblasts overcomes treatment resistance in refractory HER2 + breast tumors. Nat Commun. 2022;13:5310.
Chakravarthy A, Khan L, Bensler NP, Bose P, De Carvalho DD. TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat Commun. 2018;9:4692.
Liu X, Lu Y, Huang J, Xing Y, Dai H, Zhu L, et al. CD16 + fibroblasts foster a trastuzumab-refractory microenvironment that is reversed by VAV2 inhibition. Cancer Cell. 2022;40:1341–e135713.
Dominguez CX, Müller S, Keerthivasan S, Koeppen H, Hung J, Gierke S, et al. Single-cell RNA sequencing reveals stromal evolution into LRRC15 + myofibroblasts as a determinant of patient response to Cancer Immunotherapy. Cancer Discov. 2020;10:232–53.
Grauel AL, Nguyen B, Ruddy D, Laszewski T, Schwartz S, Chang J, et al. TGFβ-blockade uncovers stromal plasticity in tumors by revealing the existence of a subset of interferon-licensed fibroblasts. Nat Commun. 2020;11:6315.
Yoshikawa K, Ishida M, Yanai H, Tsuta K, Sekimoto M, Sugie T. Prognostic significance of PD-L1-positive cancer-associated fibroblasts in patients with triple-negative breast cancer. BMC Cancer. 2021;21:239.
Yokoyama T, Komori A, Nakamura M, Takii Y, Kamihira T, Shimoda S, et al. Human intrahepatic biliary epithelial cells function in innate immunity by producing IL-6 and IL‐8 via the TLR4‐NF‐κB and ‐MAPK signaling pathways. Liver Int. 2006;26:467–76.
Steele NG, Biffi G, Kemp SB, Zhang Y, Drouillard D, Syu L, et al. Inhibition of hedgehog signaling alters fibroblast composition in pancreatic Cancer. Clin Cancer Res. 2021;27:2023–37.
Zheng S, Zou Y, Tang Y, Yang A, Liang J-Y, Wu L, et al. Landscape of cancer-associated fibroblasts identifies the secreted biglycan as a protumor and immunosuppressive factor in triple-negative breast cancer. OncoImmunology. 2022;11:2020984.
Zheng S, Liang J, Tang Y, Xie J, Zou Y, Yang A et al. Dissecting the role of cancer-associated fibroblast‐derived biglycan as a potential therapeutic target in immunotherapy resistance: a tumor bulk and single‐cell transcriptomic study. Clin Translational Med. 2023;13:e1189.
Deligne C, Midwood KS. Macrophages and extracellular matrix in breast Cancer: partners in crime or protective allies? Front Oncol. 2021;11:620773.
Qiu S-Q, Waaijer SJH, Zwager MC, de Vries EGE, van der Vegt B, Schröder CP. Tumor-associated macrophages in breast cancer: innocent bystander or important player? Cancer Treat Rev. 2018;70:178–89.
Arora S, Dev K, Agarwal B, Das P, Syed MA. Macrophages: their role, activation and polarization in pulmonary diseases. Immunobiology. 2018;223:383–96.
Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13.
Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889–96.
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 2002;23:549–55.
Su S, Liu Q, Chen J, Chen J, Chen F, He C, et al. A positive Feedback Loop between Mesenchymal-Like Cancer cells and macrophages is essential to breast Cancer metastasis. Cancer Cell. 2014;25:605–20.
Sousa S, Brion R, Lintunen M, Kronqvist P, Sandholm J, Mönkkönen J, et al. Human breast cancer cells educate macrophages toward the M2 activation status. Breast Cancer Res. 2015;17:101.
Xu M, Liu M, Du X, Li S, Li H, Li X, et al. Intratumoral Delivery of IL-21 overcomes Anti-Her2/Neu resistance through shifting Tumor-Associated macrophages from M2 to M1 phenotype. J Immunol. 2015;194:4997–5006.
Ruffell B, Coussens LM. Macrophages and therapeutic resistance in Cancer. Cancer Cell. 2015;27:462–72.
Fu L-Q, Du W-L, Cai M-H, Yao J-Y, Zhao Y-Y, Mou X-Z. The roles of tumor-associated macrophages in tumor angiogenesis and metastasis. Cell Immunol. 2020;353:104119.
Dong F, Ruan S, Wang J, Xia Y, Le K, Xiao X, et al. M2 macrophage-induced lncRNA PCAT6 facilitates tumorigenesis and angiogenesis of triple-negative breast cancer through modulation of VEGFR2. Cell Death Dis. 2020;11:728.
Cendrowicz E, Sas Z, Bremer E, Rygiel TP. The role of macrophages in Cancer Development and Therapy. Cancers. 2021;13:1946.
Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017;14:399–416.
Ma R-Y, Black A, Qian B-Z. Macrophage diversity in cancer revisited in the era of single-cell omics. Trends Immunol. 2022;43:546–63.
Cheng S, Li Z, Gao R, Xing B, Gao Y, Yang Y, et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell. 2021;184:792–e80923.
Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor-associated macrophages: an accomplice in solid tumor progression. J Biomed Sci. 2019;26:78.
de Boniface J, Mao Y, Schmidt-Mende J, Kiessling R, Poschke I. Expression patterns of the immunomodulatory enzyme arginase 1 in blood, lymph nodes and tumor tissue of early-stage breast cancer patients. OncoImmunology. 2012;1:1305–12.
Rath M, Müller I, Kropf P, Closs EI, Munder M. Metabolism via Arginase or nitric oxide synthase: two competing arginine pathways in macrophages. Front Immunol. 2014;5:532.
Movahedi K, Laoui D, Gysemans C, Baeten M, Stangé G, Van den Bossche J, et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010;70:5728–39.
Reeves E, James E. Antigen processing and immune regulation in the response to tumours. Immunology. 2017;150:16–24.
Vitale I, Manic G, Coussens LM, Kroemer G, Galluzzi L. Macrophages and metabolism in the Tumor Microenvironment. Cell Metabol. 2019;30:36–50.
Viitala M, Virtakoivu R, Tadayon S, Rannikko J, Jalkanen S, Hollmén M. Immunotherapeutic blockade of Macrophage Clever-1 reactivates the CD8 + T-cell response against immunosuppressive tumors. Clin Cancer Res. 2019;25:3289–303.
Santoni M, Romagnoli E, Saladino T, Foghini L, Guarino S, Capponi M et al. Triple negative breast cancer: key role of Tumor-Associated macrophages in regulating the activity of anti-PD-1/PD-L1 agents. Biochimica et Biophysica Acta (BBA) - reviews on Cancer. 2018;1869:78–84.
Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227–42.
Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196:254–65.
Annacker O, Asseman C, Read S, Powrie F. Interleukin-10 in the regulation of T cell-induced colitis. J Autoimmun. 2003;20:277–9.
Thomas DA, Massagué J. TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8:369–80.
Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CMT, Pryer N, et al. Macrophage IL-10 blocks CD8 + T cell-dependent responses to Chemotherapy by suppressing IL-12 expression in Intratumoral dendritic cells. Cancer Cell. 2014;26:623–37.
Mittal SK, Roche PA. Suppression of antigen presentation by IL-10. Curr Opin Immunol. 2015;34:22–7.
Finetti F, Travelli C, Ercoli J, Colombo G, Buoso E, Trabalzini L. Prostaglandin E2 and Cancer: insight into Tumor Progression and Immunity. Biology. 2020;9:434.
Half E, Tang XM, Gwyn K, Sahin A, Wathen K, Sinicrope FA. Cyclooxygenase-2 expression in human breast cancers and adjacent ductal carcinoma in situ. Cancer Res. 2002;62:1676–81.
Li K, Shi H, Zhang B, Ou X, Ma Q, Chen Y, et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Sig Transduct Target Ther. 2021;6:362.
DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. 2019;19:369–82.
Tsukamoto H, Fujieda K, Miyashita A, Fukushima S, Ikeda T, Kubo Y, et al. Combined blockade of IL6 and PD-1/PD-L1 signaling abrogates mutual regulation of their immunosuppressive effects in the Tumor Microenvironment. Cancer Res. 2018;78:5011–22.
Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS et al. In vivo imaging reveals a tumor-associated macrophage–mediated resistance pathway in anti–PD-1 therapy. Sci Transl Med. 2017;9:eaal3604.
Krneta T, Gillgrass A, Poznanski S, Chew M, Lee AJ, Kolb M, et al. M2-polarized and tumor-associated macrophages alter NK cell phenotype and function in a contact-dependent manner. J Leukoc Biol. 2017;101:285–95.
Dandekar RC, Kingaonkar AV, Dhabekar GS. Role of macrophages in malignancy. Ann Maxillofac Surg. 2011;1:150–4.
Herrera M, Herrera A, Domínguez G, Silva J, García V, García JM, et al. Cancer-associated fibroblast and M2 macrophage markers together predict outcome in colorectal cancer patients. Cancer Sci. 2013;104:437–44.
Mizutani Y, Kobayashi H, Iida T, Asai N, Masamune A, Hara A, et al. Meflin-positive Cancer-Associated fibroblasts inhibit pancreatic carcinogenesis. Cancer Res. 2019;79:5367–81.
Ksiazkiewicz M, Gottfried E, Kreutz M, Mack M, Hofstaedter F, Kunz-Schughart LA. Importance of CCL2-CCR2A/2B signaling for monocyte migration into spheroids of breast cancer-derived fibroblasts. Immunobiology. 2010;215:737–47.
Cohen N, Shani O, Raz Y, Sharon Y, Hoffman D, Abramovitz L, et al. Fibroblasts drive an immunosuppressive and growth-promoting microenvironment in breast cancer via secretion of chitinase 3-like 1. Oncogene. 2017;36:4457–68.
Martinez-Outschoorn UE, Lisanti MP, Sotgia F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Sem Cancer Biol. 2014;25:47–60.
Fiori ME, Di Franco S, Villanova L, Bianca P, Stassi G, De Maria R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol Cancer. 2019;18:70.
Farhood B, Najafi M, Mortezaee K. Cancer-associated fibroblasts: secretions, interactions, and therapy. J Cell Biochem. 2019;120:2791–800.
Timperi E, Gueguen P, Molgora M, Magagna I, Kieffer Y, Lopez-Lastra S, et al. Lipid-Associated macrophages Are Induced by Cancer-Associated fibroblasts and mediate Immune suppression in breast Cancer. Cancer Res. 2022;82:3291–306.
Lei X, Lei Y, Li J-K, Du W-X, Li R-G, Yang J, et al. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020;470:126–33.
Chen Z, Zhou L, Liu L, Hou Y, Xiong M, Yang Y, et al. Single-cell RNA sequencing highlights the role of inflammatory cancer-associated fibroblasts in bladder urothelial carcinoma. Nat Commun. 2020;11:5077.
Zhang Y, Liu Q, Liao Q. Long noncoding RNA: a dazzling dancer in tumor immune microenvironment. J Exp Clin Cancer Res. 2020;39:231.
Qi J, Sun H, Zhang Y, Wang Z, Xun Z, Li Z, et al. Single-cell and spatial analysis reveal interaction of FAP + fibroblasts and SPP1 + macrophages in colorectal cancer. Nat Commun. 2022;13:1742.
Mazur A, Holthoff E, Vadali S, Kelly T, Post SR. Cleavage of type I collagen by fibroblast activation Protein-α enhances class A scavenger receptor mediated macrophage adhesion. PLoS ONE. 2016;11:e0150287.
Ueshima E, Fujimori M, Kodama H, Felsen D, Chen J, Durack JC, et al. Macrophage-secreted TGF-β 1 contributes to fibroblast activation and ureteral stricture after ablation injury. Am J Physiology-Renal Physiol. 2019;317:F52–64.
Yeh C-R, Slavin S, Da J, Hsu I, Luo J, Xiao G-Q, et al. Estrogen receptor α in cancer associated fibroblasts suppresses prostate cancer invasion via reducing CCL5, IL6 and macrophage infiltration in the tumor microenvironment. Mol Cancer. 2016;15:7.
Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, et al. Pancreatic Cancer-Associated Stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-Dependent manner. Cancer Res. 2013;73:3007–18.
Hashimoto O, Yoshida M, Koma Y, Yanai T, Hasegawa D, Kosaka Y, et al. Collaboration of cancer-associated fibroblasts and tumour‐associated macrophages for neuroblastoma development. J Pathol. 2016;240:211–23.
Gunaydin G. CAFs interacting with TAMs in Tumor Microenvironment to Enhance Tumorigenesis and Immune Evasion. Front Oncol. 2021;11:668349.
Bergenfelz C, Roxå A, Mehmeti M, Leandersson K, Larsson A-M. Clinical relevance of systemic monocytic-MDSCs in patients with metastatic breast cancer. Cancer Immunol Immunother. 2020;69:435–48.
Casbon A-J, Reynaud D, Park C, Khuc E, Gan DD, Schepers K et al. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc Natl Acad Sci USA. 2015;112:E566–75.
Shen M, Wang J, Yu W, Zhang C, Liu M, Wang K, et al. A novel MDSC-induced PD-1 – PD-L1 + B-cell subset in breast tumor microenvironment possesses immuno-suppressive properties. OncoImmunology. 2018;7:e1413520.
Christmas BJ, Rafie CI, Hopkins AC, Scott BA, Ma HS, Cruz KA, et al. Entinostat converts Immune-resistant breast and pancreatic cancers into checkpoint-responsive tumors by reprogramming tumor-infiltrating MDSCs. Cancer Immunol Res. 2018;6:1561–77.
Oh K, Lee O-Y, Shon SY, Nam O, Ryu PM, Seo MW, et al. A mutual activation loop between breast cancer cells and myeloid-derived suppressor cells facilitates spontaneous metastasis through IL-6 trans-signaling in a murine model. Breast Cancer Res. 2013;15:R79.
Segovia-Mendoza M, Morales-Montor J. Immune Tumor Microenvironment in breast Cancer and the participation of Estrogen and its receptors in Cancer Physiopathology. Front Immunol. 2019;10:348.
Gelao L, Criscitiello C, Esposito A, Laurentiis MD, Fumagalli L, Locatelli MA, et al. Dendritic cell-based vaccines: clinical applications in breast cancer. Immunotherapy. 2014;6:349–60.
Zheng Y, Li S, Tang H, Meng X, Zheng Q. Molecular mechanisms of immunotherapy resistance in triple-negative breast cancer. Front Immunol. 2023;14:1153990.
Liang X, Fu C, Cui W, Ober-Blöbaum JL, Zahner SP, Shrikant PA, et al. β-Catenin mediates tumor-induced immunosuppression by inhibiting cross-priming of CD8 + T cells. J Leukoc Biol. 2013;95:179–90.
Vu SH, Vetrivel P, Kim J, Lee M-S. Cancer Resistance to Immunotherapy: Molecular mechanisms and tackling strategies. IJMS. 2022;23:10906.
Lin Y, Cai Q, Chen Y, Shi T, Liu W, Mao L, et al. CAFs shape myeloid-derived suppressor cells to promote stemness of intrahepatic cholangiocarcinoma through 5‐lipoxygenase. Hepatology. 2022;75:28–42.
Zelenay S, van der Veen AG, Böttcher JP, Snelgrove KJ, Rogers N, Acton SE, et al. Cyclooxygenase-dependent Tumor Growth through Evasion of Immunity. Cell. 2015;162:1257–70.
Liberti MV, Locasale JW. The Warburg Effect: how does it Benefit Cancer cells? Trends Biochem Sci. 2016;41:211–8.
Zappasodi R, Serganova I, Cohen IJ, Maeda M, Shindo M, Senbabaoglu Y, et al. CTLA-4 blockade drives loss of Treg stability in glycolysis-low tumours. Nature. 2021;591:652–8.
Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the Tumor Microenvironment is a driver of Cancer Progression. Cell. 2015;162:1229–41.
Gu J, Zhou J, Chen Q, Xu X, Gao J, Li X, et al. Tumor metabolite lactate promotes tumorigenesis by modulating MOESIN lactylation and enhancing TGF-β signaling in regulatory T cells. Cell Rep. 2022;39:110986.
Comito G, Iscaro A, Bacci M, Morandi A, Ippolito L, Parri M, et al. Lactate modulates CD4 + T-cell polarization and induces an immunosuppressive environment, which sustains prostate carcinoma progression via TLR8/miR21 axis. Oncogene. 2019;38:3681–95.
Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting Edge: distinct glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4 + T cell subsets. J Immunol. 2011;186:3299–303.
Wang H, Franco F, Tsui Y-C, Xie X, Trefny MP, Zappasodi R, et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat Immunol. 2020;21:298–308.
Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metabol. 2006;3:187–97.
Lu H, Forbes RA, Verma A. Hypoxia-inducible factor 1 activation by aerobic glycolysis implicates the Warburg Effect in Carcinogenesis. J Biol Chem. 2002;277:23111–5.
Kozlov AM, Lone A, Betts DH, Cumming RC. Lactate preconditioning promotes a HIF-1α-mediated metabolic shift from OXPHOS to glycolysis in normal human diploid fibroblasts. Sci Rep. 2020;10:8388.
Neumeister VM, Sullivan CA, Lindner R, Lezon-Geyda K, Li J, Zavada J, et al. Hypoxia-induced protein CAIX is associated with somatic loss of BRCA1 protein and pathway activity in triple negative breast cancer. Breast Cancer Res Treat. 2012;136:67–75.
Panisova E, Kery M, Sedlakova O, Brisson L, Debreova M, Sboarina M, et al. Lactate stimulates CA IX expression in normoxic cancer cells. Oncotarget. 2017;8:77819–35.
Serganova I, Cohen IJ, Vemuri K, Shindo M, Maeda M, Mane M, et al. LDH-A regulates the tumor microenvironment via HIF-signaling and modulates the immune response. PLoS ONE. 2018;13:e0203965.
Siddiqui A, Ceppi P. A non-proliferative role of pyrimidine metabolism in cancer. Mol Metabolism. 2020;35:100962.
Luo Y, Tian W, Lu X, Zhang C, Xie J, Deng X, et al. Prognosis stratification in breast cancer and characterization of immunosuppressive microenvironment through a pyrimidine metabolism-related signature. Front Immunol. 2022;13:1–19.
Huseni MA, Wang L, Klementowicz JE, Yuen K, Breart B, Orr C, et al. CD8 + T cell-intrinsic IL-6 signaling promotes resistance to anti-PD-L1 immunotherapy. Cell Rep Med. 2023;4:100878.
Mao M, Chen Y, Jia Y, Yang J, Wei Q, Li Z, et al. PLCA8 suppresses breast cancer apoptosis by activating the PI3k/AKT/NF-κB pathway. J Cell Mol Med. 2019;23:6930–41.
Mao M, Hu D, Yang J, Chen Y, Zhang X, Shen J, et al. Regulation of tamoxifen sensitivity by the PLAC8/MAPK pathway axis is antagonized by curcumin-induced protein stability change. J Mol Med. 2021;99:845–58.
Chen Y, Jia Y, Mao M, Gu Y, Xu C, Yang J, et al. PLAC8 promotes adriamycin resistance via blocking autophagy in breast cancer. J Cell Mol Med. 2021;25:6948–62.
Mao M, Cheng Y, Yang J, Chen Y, Xu L, Zhang X, et al. Multifaced roles of PLAC8 in cancer. Biomark Res. 2021;9:73.
Komatsu M, Chiba T, Tatsumi K, Iemura S, Tanida I, Okazaki N, et al. A novel protein-conjugating system for Ufm1, a ubiquitin-fold modifier. EMBO J. 2004;23:1977–86.
Yoo HM, Park JH, Jeon YJ, Chung CH. Ubiquitin-fold modifier 1 acts as a positive Regulator of breast Cancer. Front Endocrinol. 2015;6:36.
Mao M, Chen Y, Yang J, Cheng Y, Xu L, Ji F, et al. Modification of PLAC8 by UFM1 affects tumorous proliferation and immune response by impacting PD-L1 levels in triple-negative breast cancer. J Immunother Cancer. 2022;10:e005668.
Qin G, Wang X, Ye S, Li Y, Chen M, Wang S, et al. NPM1 upregulates the transcription of PD-L1 and suppresses T cell activity in triple-negative breast cancer. Nat Commun. 2020;11:1669.
Wang Y, Chen Y, Zhang J, Yang Y, Fleishman JS, Wang Y, et al. Cuproptosis: a novel therapeutic target for overcoming cancer drug resistance. Drug Resist Updates. 2024;72:101018.
Song S, Zhang M, Xie P, Wang S, Wang Y. Comprehensive analysis of cuproptosis-related genes and tumor microenvironment infiltration characterization in breast cancer. Front Immunol. 2022;13:1–18.
Fortis SP, Sofopoulos M, Goulielmaki M, Arnogiannaki N, Ardavanis A, Perez SA, et al. Association between Intratumoral CD8 + T cells with FoxP3 + and CD163 + cells: a potential Immune intrinsic negative feedback mechanism for Acquired Immune Resistance. Cancers. 2022;14:6208.
Chabanon RM, Pedrero M, Lefebvre C, Marabelle A, Soria J-C, Postel-Vinay S. Mutational Landscape and Sensitivity to Immune Checkpoint blockers. Clin Cancer Res. 2016;22:4309–21.
Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor Mutational Burden as an independent predictor of response to Immunotherapy in Diverse Cancers. Mol Cancer Ther. 2017;16:2598–608.
Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74.
Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature. 2012;482:400–4.
Blank CU, Haanen JB, Ribas A, Schumacher TN. CANCER IMMUNOLOGY. The cancer immunogram. Science. 2016;352:658–60.
Yoo CB, Jones PA. Epigenetic therapy of cancer: past, present and future. Nat Rev Drug Discov. 2006;5:37–50.
Cheng CJ, Bahal R, Babar IA, Pincus Z, Barrera F, Liu C, et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature. 2015;518:107–10.
Zhang Y, Chen J, Liu H, Mi R, Huang R, Li X, et al. The role of histone methylase and demethylase in antitumor immunity: a new direction for immunotherapy. Front Immunol. 2023;13:1–16.
Dunn J, Rao S. Epigenetics and immunotherapy: the current state of play. Mol Immunol. 2017;87:227–39.
Khodayari S, Khodayari H, Saeedi E, Mahmoodzadeh H, Sadrkhah A, Nayernia K. Single-cell transcriptomics for unlocking Personalized Cancer Immunotherapy: toward targeting the origin of Tumor Development Immunogenicity. Cancers. 2023;15:3615.
Kim E-J, Liu P, Zhang S, Donahue K, Wang Y, Schehr JL, et al. BAF155 methylation drives metastasis by hijacking super-enhancers and subverting anti-tumor immunity. Nucleic Acids Res. 2021;49:12211–33.
Lee DY, Salahuddin T, Iqbal J. Lysine-specific demethylase 1 (LSD1)-Mediated Epigenetic Modification of Immunogenicity and Immunomodulatory effects in breast cancers. Curr Oncol. 2023;30:2127–43.
Nguyen EM, Taniguchi H, Chan JM, Zhan YA, Chen X, Qiu J, et al. Targeting lysine-specific demethylase 1 rescues major histocompatibility Complex Class I Antigen Presentation and overcomes programmed death-ligand 1 Blockade Resistance in SCLC. J Thorac Oncol. 2022;17:1014–31.
Yu B, Luo F, Sun B, Liu W, Shi Q, Cheng S-Y, et al. KAT6A acetylation of SMAD3 regulates myeloid-derived suppressor cell recruitment, metastasis, and Immunotherapy in Triple-negative breast Cancer. Adv Sci (Weinh). 2022;9:e2105793.
Wong KK. DNMT1: a key drug target in triple-negative breast cancer. Sem Cancer Biol. 2021;72:198–213.
Zhang Z-G, Zhang H-S, Sun H-L, Liu H-Y, Liu M-Y, Zhou Z. KDM5B promotes breast cancer cell proliferation and migration via AMPK-mediated lipid metabolism reprogramming. Exp Cell Res. 2019;379:182–90.
Perrier A, Didelot A, Laurent-Puig P, Blons H, Garinet S. Epigenetic Mechanisms of Resistance to Immune Checkpoint Inhibitors. Biomolecules. 2020;10:1061
Goswami S, Apostolou I, Zhang J, Skepner J, Anandhan S, Zhang X, et al. Modulation of EZH2 expression in T cells improves efficacy of anti–CTLA-4 therapy. J Clin Invest. 2018;128:3813–8.
Sun H-Y, Du S-T, Li Y-Y, Deng G-T, Zeng F-R. Bromodomain and extra-terminal inhibitors emerge as potential therapeutic avenues for gastrointestinal cancers. World J Gastrointest Oncol. 2022;14:75–89.
Noblejas-López MDM, Nieto-Jimenez C, Burgos M, Gómez-Juárez M, Montero JC, Esparís-Ogando A, et al. Activity of BET-proteolysis targeting chimeric (PROTAC) compounds in triple negative breast cancer. J Exp Clin Cancer Res. 2019;38:383.
Qiao J, Chen Y, Mi Y, Jin H, Wang L, Huang T, et al. Macrophages confer resistance to BET inhibition in triple-negative breast cancer by upregulating IKBKE. Biochem Pharmacol. 2020;180:114126.
Hicks KC, Knudson KM, Lee KL, Hamilton DH, Hodge JW, Figg WD, et al. Cooperative Immune-mediated mechanisms of the HDAC inhibitor Entinostat, an IL15 superagonist, and a Cancer Vaccine effectively synergize as a Novel Cancer Therapy. Clin Cancer Res. 2020;26(3):704–16.
Joyce OS, et al. Results of ENCORE 602 (TRIO025), a phase II, randomized, placebo-controlled, double-blinded, multicenter study of atezolizumab with or without entinostat in patients with advanced triple-negative breast cancer (aTNBC). JCO. 2020;38:1014–1014.
Roussos Torres ET, Ho WJ, Danilova L, Tandurella JA, Leatherman J, Rafie C et al. Entinostat, Nivolumab and Ipilimumab for women with advanced HER2-negative breast cancer: a phase ib trial. Nat Cancer. 2024 Feb 14. Epub ahead of print.
Jiang X, Qian Z, Chen Y, Zhou T, Zhao C, Yin Y. CMTM7 recognizes an immune-hot tumor microenvironment and predicts therapeutic response of immunotherapy in breast cancer well. Front Genet. 2022;13:1–15.
Wu C, Zhong R, Sun X, Shi J. PSME2 identifies immune-hot tumors in breast cancer and associates with well therapeutic response to immunotherapy. Front Genet. 2022;13:1–12.
Li RQ, Wang W, Yan L, Song LY, Guan X, Zhang W, et al. Identification of tumor antigens and immune subtypes in breast cancer for mRNA vaccine development. Front Oncol. 2022;12:1–17.
Zavareh RB, Spangenberg SH, Woods A, Martínez-Peña F, Lairson LL. HSP90 inhibition enhances Cancer Immunotherapy by modulating the Surface expression of multiple Immune Checkpoint proteins. Cell Chem Biology. 2021;28:158–e1685.
Rahmy S, Mishra SJ, Murphy S, Blagg BSJ, Lu X. Hsp90β inhibition upregulates interferon response and enhances immune checkpoint blockade therapy in murine tumors. Front Immunol. 2022;13:1005045.
Modi S, Stopeck A, Linden H, Solit D, Chandarlapaty S, Rosen N, et al. HSP90 inhibition is effective in breast Cancer: a phase II trial of Tanespimycin (17-AAG) plus trastuzumab in patients with HER2-Positive metastatic breast Cancer progressing on Trastuzumab. Clin Cancer Res. 2011;17:5132–9.
Ramanathan RK, Trump DL, Eiseman JL, Belani CP, Agarwala SS, Zuhowski EG, et al. Phase I pharmacokinetic-pharmacodynamic study of 17-(Allylamino)-17-Demethoxygeldanamycin (17AAG, NSC 330507), a Novel inhibitor of heat shock protein 90, in patients with Refractory Advanced cancers. Clin Cancer Res. 2005;11:3385–91.
Mercogliano MF, De Martino M, Venturutti L, Rivas MA, Proietti CJ, Inurrigarro G, et al. TNFα-Induced Mucin 4 expression elicits Trastuzumab Resistance in HER2-Positive breast Cancer. Clin Cancer Res. 2017;23:636–48.
Carraway KL, Rossi EA, Komatsu M, Price-Schiavi SA, Huang D, Guy PM, et al. An intramembrane modulator of the ErbB2 receptor tyrosine kinase that potentiates Neuregulin Signaling. J Biol Chem. 1999;274:5263–6.
Price-Schiavi SA, Jepson S, Li P, Arango M, Rudland PS, Yee L, et al. Rat Muc4 (sialomucin complex) reduces binding of anti-ErbB2 antibodies to tumor cell surfaces, a potential mechanism for herceptin resistance. Int J Cancer. 2002;99:783–91.
Bruni S, Mauro FL, Proietti CJ, Cordo-Russo RI, Rivas MA, Inurrigarro G, et al. Blocking soluble TNFα sensitizes HER2-positive breast cancer to trastuzumab through MUC4 downregulation and subverts immunosuppression. J Immunother Cancer. 2023;11:e005325.
Yi M, Niu M, Wu Y, Ge H, Jiao D, Zhu S, et al. Combination of oral STING agonist MSA-2 and anti-TGF-β/PD-L1 bispecific antibody YM101: a novel immune cocktail therapy for non-inflamed tumors. J Hematol Oncol. 2022;15:142.
Santinon F, Ezzahra BF, Bachais M, Sarabia Pacis A, Rudd CE. Direct AKT activation in tumor-infiltrating lymphocytes markedly increases interferon-γ (IFN-γ) for the regression of tumors resistant to PD-1 checkpoint blockade. Sci Rep. 2022;12:18509.
Jiao S, Xia W, Yamaguchi H, Wei Y, Chen M-K, Hsu J-M, et al. PARP inhibitor upregulates PD-L1 expression and enhances Cancer-Associated Immunosuppression. Clin Cancer Res. 2017;23:3711–20.
Li B, Tao W, Shao-hua Z, Ze-rui Q, Fu-quan J, Fan L, et al. Remarkable response with pembrolizumab plus albumin-bound paclitaxel in 2 cases of HER2-positive metastatic breast cancer who have failed to multi-anti-HER2 targeted therapy. Cancer Biol Ther. 2018;19:292–5.
Mishra A, Kumar D, Gupta K, Lofland G, Sharma AK, Banka DS, et al. Gallium-68–labeled peptide PET quantifies Tumor exposure of PD-L1 therapeutics. Clin Cancer Res. 2023;29:581–91.
Main SC, Cescon DW, Bratman SV. Liquid biopsies to predict CDK4/6 inhibitor efficacy and resistance in breast cancer. Cancer Drug Resist. 2022;5:727–48.
Chin YM, Shibayama T, Chan HT, Otaki M, Hara F, Kobayashi T, et al. Serial circulating tumor DNA monitoring of CDK4/6 inhibitors response in metastatic breast cancer. Cancer Sci. 2022;113:1808–20.
Ford K, Hanley CJ, Mellone M, Szyndralewiez C, Heitz F, Wiesel P, et al. NOX4 inhibition potentiates immunotherapy by overcoming Cancer-Associated fibroblast-mediated CD8 T-cell exclusion from tumors. Cancer Res. 2020;80:1846–60.
Hanley CJ, Mellone M, Ford K, Thirdborough SM, Mellows T, Frampton SJ, et al. Targeting the Myofibroblastic Cancer-Associated Fibroblast phenotype through inhibition of NOX4. JNCI: J Natl Cancer Inst. 2018;110:109–20.
Hanley CJ, Thomas GJ. T-cell tumour exclusion and immunotherapy resistance: a role for CAF targeting. Br J Cancer. 2020;123:1353–5.
Chakravarthy A, Khan L, Bensler NP, Bose P, De Carvalho DD. TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat Commun. 2018;9(1):4692.
Mhaidly R, Mechta-Grigoriou F. Role of cancer‐associated fibroblast subpopulations in immune infiltration, as a new means of treatment in cancer. Immunol Rev. 2021;302:259–72.
Chen IX, Chauhan VP, Posada J, Ng MR, Wu MW, Adstamongkonkul P, et al. Blocking CXCR4 alleviates desmoplasia, increases T-lymphocyte infiltration, and improves immunotherapy in metastatic breast cancer. Proc Natl Acad Sci USA. 2019;116:4558–66.
Glabman RA, Choyke PL, Sato N. Cancer-Associated fibroblasts: Tumorigenicity and Targeting for Cancer Therapy. Cancers. 2022;14:3906.
Rizzolio S, Giordano S, Corso S. The importance of being CAFs (in cancer resistance to targeted therapies). J Exp Clin Cancer Res. 2022;41:319.
Monteran L, Erez N. The Dark side of fibroblasts: Cancer-Associated fibroblasts as mediators of Immunosuppression in the Tumor Microenvironment. Front Immunol. 2019;10:1835.
Binnewies M, Pollack JL, Rudolph J, Dash S, Abushawish M, Lee T, et al. Targeting TREM2 on tumor-associated macrophages enhances immunotherapy. Cell Rep. 2021;37:109844.
Flores-Toro JA, Luo D, Gopinath A, Sarkisian MR, Campbell JJ, Charo IF et al. CCR2 inhibition reduces tumor myeloid cells and unmasks a checkpoint inhibitor effect to slow progression of resistant murine gliomas. Proceedings of the National Academy of Sciences. 2020;117:1129–38.
Zhu Y, Herndon JM, Sojka DK, Kim K-W, Knolhoff BL, Zuo C, et al. Tissue-Resident macrophages in Pancreatic Ductal Adenocarcinoma Originate from embryonic hematopoiesis and promote Tumor Progression. Immunity. 2017;47:323–e3386.
Soncin I, Sheng J, Chen Q, Foo S, Duan K, Lum J, et al. The tumour microenvironment creates a niche for the self-renewal of tumour-promoting macrophages in colon adenoma. Nat Commun. 2018;9:582.
Bassez A, Vos H, Van Dyck L, Floris G, Arijs I, Desmedt C, et al. A single-cell map of intratumoral changes during anti-PD1 treatment of patients with breast cancer. Nat Med. 2021;27:820–32.
Nalio Ramos R, Missolo-Koussou Y, Gerber-Ferder Y, Bromley CP, Bugatti M, Núñez NG, et al. Tissue-resident FOLR2 + macrophages associate with CD8 + T cell infiltration in human breast cancer. Cell. 2022;185:1189–e120725.
Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting Tumor-Associated macrophages with Anti-CSF-1R antibody reveals a strategy for Cancer Therapy. Cancer Cell. 2014;25:846–59.
Uhlik MT, Harrison B, Gorden K, Leonardo S, Walsh R, Ertelt K, et al. Abstract LB-129: Imprime PGG, a soluble yeast β-glucan PAMP, in combination with Pembrolizumab induces infiltration and activation of both innate and adaptive immune cells within tumor sites in melanoma and triple-negative breast cancer (TNBC) patients. Cancer Res. 2018;78(13Supplement):LB–129.
Anfray U. Andón, Allavena. Current strategies to Target Tumor-Associated-macrophages to improve Anti-tumor Immune responses. Cells. 2019;9:46.
Zhu J, Zhang Y, Zhang A, He K, Liu P, Xu LX. Cryo-thermal therapy elicits potent anti-tumor immunity by inducing extracellular Hsp70-dependent MDSC differentiation. Sci Rep. 2016;6:27136.
Lou Y, Peng P, Wang S, Wang J, Du P, Zhang Z, et al. Combining all-trans retinoid acid treatment targeting myeloid-derived suppressive cells with cryo-thermal therapy enhances antitumor immunity in breast cancer. Front Immunol. 2022;13:1–14.
Colligan SH, Amitrano AM, Zollo RA, Peresie J, Kramer ED, Morreale B, et al. Inhibiting the biogenesis of myeloid-derived suppressor cells enhances immunotherapy efficacy against mammary tumor progression. J Clin Invest. 2022;132:1–17.
Horvat NK, Lesinski GB. Bring on the brequinar: an approach to enforce the differentiation of myeloid-derived suppressor cells. J Clin Invest. 2022;132:1–3.
Mendaza S, Ulazia-Garmendia A, Monreal-Santesteban I, Córdoba A, de Azúa YR, Aguiar B, et al. ADAM12 is a potential therapeutic target regulated by Hypomethylation in Triple-negative breast Cancer. IJMS. 2020;21:903.
Wang G, Romero Y, Thevarajan I, Zolkiewska A. ADAM12 abrogation alters immune cell infiltration and improves response to checkpoint blockade therapy in the T11 murine model of triple-negative breast cancer. OncoImmunology. 2023;12:2158006.
Leone RD, Powell JD. Metabolism of immune cells in cancer. Nat Rev Cancer. 2020;20:516–31.
Kishton RJ, Sukumar M, Restifo NP. Metabolic regulation of T cell longevity and function in Tumor Immunotherapy. Cell Metabol. 2017;26:94–109.
Hartley GP, Chow L, Ammons DT, Wheat WH, Dow SW. Programmed cell death Ligand 1 (PD-L1) signaling regulates macrophage proliferation and activation. Cancer Immunol Res. 2018;6:1260–73.
Wang Y, Wang Y, Ren Y, Zhang Q, Yi P, Cheng C. Metabolic modulation of immune checkpoints and novel therapeutic strategies in cancer. Sem Cancer Biol. 2022;86:542–65.
Sansom DM. CD28, CTLA-4 and their ligands: who does what and to whom? The effects of CD28 and CTLA-4 ligands. Immunology. 2000;101:169–77.
Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 Signaling Pathway regulates glucose metabolism. Immunity. 2002;16:769–77.
Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-Cell activation by distinct mechanisms. Mol Cell Biol. 2005;25:9543–53.
Plitas G, Konopacki C, Wu K, Bos PD, Morrow M, Putintseva EV, et al. Regulatory T cells exhibit distinct features in human breast Cancer. Immunity. 2016;45:1122–34.
Li W, Tanikawa T, Kryczek I, Xia H, Li G, Wu K, et al. Aerobic glycolysis controls myeloid-derived suppressor cells and Tumor Immunity via a specific CEBPB isoform in Triple-negative breast Cancer. Cell Metabol. 2018;28:87–e1036.
Rizwan A, Serganova I, Khanin R, Karabeber H, Ni X, Thakur S, et al. Relationships between LDH-A, Lactate, and metastases in 4T1 breast tumors. Clin Cancer Res. 2013;19:5158–69.
Gong Y, Ji P, Yang Y-S, Xie S, Yu T-J, Xiao Y, et al. Metabolic-pathway-based subtyping of Triple-negative breast Cancer reveals potential therapeutic targets. Cell Metabol. 2021;33:51–e649.
Chafe SC, McDonald PC, Saberi S, Nemirovsky O, Venkateswaran G, Burugu S, et al. Targeting Hypoxia-Induced Carbonic anhydrase IX enhances Immune-Checkpoint Blockade locally and systemically. Cancer Immunol Res. 2019;7:1064–78.
Hedlund MD. Nemirovsky, Awrey, Jensen, Dedhar. Harnessing Induced Essentiality: Targeting Carbonic anhydrase IX and Angiogenesis reduces Lung Metastasis of Triple negative breast Cancer xenografts. Cancers. 2019;11:1002.
Jin H, Liao S, Yao F, Li J, Xu Z, Zhao K, et al. Insight into the crosstalk between photodynamic therapy and immunotherapy in breast Cancer. Cancers. 2023;15:1532.
Taber SW, Fingar VH, Coots CT, Wieman TJ. Photodynamic therapy using mono-L-aspartyl chlorin e6 (Npe6) for the treatment of cutaneous disease: a phase I clinical study. Clin Cancer Res. 1998;4:2741–6.
Vrouenraets MB, Visser GWM, Snow GB, van Dongen GAMS. Basic principles, applications in oncology and improved selectivity of photodynamic therapy. Anticancer Res. 2003;23:505–22.
Anzengruber F, Avci P, De Freitas LF, Hamblin MR. T-cell mediated anti-tumor immunity after photodynamic therapy: why does it not always work and how can we improve it? Photochem Photobiol Sci. 2015;14:1492–509.
Wachowska M, Gabrysiak M, Muchowicz A, Bednarek W, Barankiewicz J, Rygiel T, et al. 5-Aza-2′-deoxycytidine potentiates antitumour immune response induced by photodynamic therapy. Eur J Cancer. 2014;50:1370–81.
Soman S, Kulkarni S, Pandey A, Dhas N, Subramanian S, Mukherjee A, et al. 2D hetero-nanoconstructs of black phosphorus for breast Cancer theragnosis: Technological advancements. Biosensors. 2022;12:1009.
Gogoi M, Sarma HD, Bahadur D, Banerjee R. Biphasic magnetic nanoparticles–nanovesicle hybrids for chemotherapy and self-controlled hyperthermia. Nanomedicine. 2014;9:955–70.
Li Z, Hu Y, Fu Q, Liu Y, Wang J, Song J, et al. NIR/ROS-Responsive Black Phosphorus QD Vesicles as immunoadjuvant carrier for specific Cancer photodynamic immunotherapy. Adv Funct Mater. 2020;30:1905758.
Liang X, Ye X, Wang C, Xing C, Miao Q, Xie Z, et al. Photothermal cancer immunotherapy by erythrocyte membrane-coated black phosphorus formulation. J Controlled Release. 2019;296:150–61.
Zhao P, Xu Y, Ji W, Zhou S, Li L, Qiu L, et al. Biomimetic black phosphorus quantum dots-based photothermal therapy combined with anti-PD-L1 treatment inhibits recurrence and metastasis in triple-negative breast cancer. J Nanobiotechnol. 2021;19:181.
Zhang X, Tang J, Li C, Lu Y, Cheng L, Liu J. A targeting black phosphorus nanoparticle based immune cells nano-regulator for photodynamic/photothermal and photo-immunotherapy. Bioactive Mater. 2021;6:472–89.
Wang X, Tokheim C, Gu SS, Wang B, Tang Q, Li Y, et al. In vivo CRISPR screens identify the E3 ligase Cop1 as a modulator of macrophage infiltration and cancer immunotherapy target. Cell. 2021;184:5357–e537422.
Ji P, Gong Y, Jin ML, Wu HL, Guo LW, Pei YC et al. In Vivo multidimensional CRISPR screens identify Lgals2 as an immunotherapy target in triple-negative breast cancer. Sci Adv. 2022;8:eabl8247.
Dong MB, Wang G, Chow RD, Ye L, Zhu L, Dai X, et al. Systematic Immunotherapy Target Discovery using genome-scale in vivo CRISPR screens in CD8 T cells. Cell. 2019;178:1189–e120423.
Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature. 2017;547:413–8.
Kim S-S, Harford JB, Moghe M, Rait A, Chang EH. Combination with SGT-53 overcomes tumor resistance to a checkpoint inhibitor. OncoImmunology. 2018;7:e1484982.
Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-negative breast Cancer. N Engl J Med. 2018;379:2108–21.
Dai L, Li K, Li M, Zhao X, Luo Z, Lu L, et al. Size/Charge changeable acidity-responsive micelleplex for photodynamic‐improved PD‐L1 immunotherapy with enhanced tumor penetration. Adv Funct Mater. 2018;28:1707249.
Kang T, Li Y, Wang Y, Zhu J, Yang L, Huang Y, et al. Modular Engineering of targeted dual-drug nanoassemblies for Cancer Chemoimmunotherapy. ACS Appl Mater Interfaces. 2019;11:36371–82.
Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol. 2016;11:986–94.
Pal R, Chakraborty B, Nath A, Singh LM, Ali M, Rahman DS, et al. Noble metal nanoparticle-induced oxidative stress modulates tumor associated macrophages (TAMs) from an M2 to M1 phenotype: an in vitro approach. Int Immunopharmacol. 2016;38:332–41.
Zhang Y, Chen H, Mo H, Hu X, Gao R, Zhao Y, et al. Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell. 2021;39:1578–e15938.
Tietscher S, Wagner J, Anzeneder T, Langwieder C, Rees M, Sobottka B, et al. A comprehensive single-cell map of T cell exhaustion-associated immune environments in human breast cancer. Nat Commun. 2023;14:98.
Ma C, Yang C, Peng A, Sun T, Ji X, Mi J, et al. Pan-cancer spatially resolved single-cell analysis reveals the crosstalk between cancer-associated fibroblasts and tumor microenvironment. Mol Cancer. 2023;22(1):170.
Li C, Yang L, Zhang Y, Hou Q, Wang S, et al. Integrating single-cell and bulk transcriptomic analyses to develop a cancer-associated fibroblast-derived biomarker for predicting prognosis and therapeutic response in breast cancer. Front Immunol. 2024;14:1307588.
Rodriguez-Garcia A, Lynn RC, Poussin M, Eiva MA, Shaw LC, O’Connor RS, et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat Commun. 2021;12(1):877.
Chuangchot N, Jamjuntra P, Yangngam S, Luangwattananun P, Thongchot S, Junking M, et al. Enhancement of PD-L1-attenuated CAR-T cell function through breast cancer-associated fibroblasts-derived IL-6 signaling via STAT3/AKT pathways. Breast Cancer Res. 2023;25(1):86.
Chen Y, Shu X, Guo JY, Xiang Y, Liang SY, Lai JM, et al. Nanodrugs mediate TAMs-related arginine metabolism interference to boost photodynamic immunotherapy. J Control Release. 2024;367:248–64.
Marta Warszyńska JM, Dąbrowski. Photodynamic therapy combined with immunotherapy: Recent advances and future research directions. Coordination Chemistry Reviews, 495, 2023, 215350.
Johnson DB, Reynolds KL, Sullivan RJ, Balko JM, Patrinely JR, et al. Immune checkpoint inhibitor toxicities: systems-based approaches to improve patient care and research. Lancet Oncol. 2020;21(8):e398–404.
Oliver AJ, Lau PKH, Unsworth AS, Loi S, Darcy PK, Kershaw MH, et al. Tissue-dependent tumor microenvironments and their impact on immunotherapy responses. Front Immunol. 2018;9:70.
Chaudhuri S, Thomas S, Munster P. Immunotherapy in breast cancer: a clinician’s perspective. J Natl Cancer Cent. 2021;1:47–57.
Gui C-P, Wei J-H, Zhang C, Tang Y-M, Shu G-N, Wu R-P, et al. Single-cell and spatial transcriptomics reveal 5-methylcytosine RNA methylation regulators immunologically reprograms tumor microenvironment characterizations, immunotherapy response and precision treatment of clear cell renal cell carcinoma. Translational Oncol. 2023;35:101726.
Acknowledgements
We acknowledge Ms. Angitha N. Nath for editing the manuscript.
Funding
This work is partly supported by Science and Engineering Research Board (SERB) Program, Govt. of India, (Project/Grant No. JCB/2023/000011) to GCK; Department of Biotechnology (DBT) Program (Project/Grant No. BT/PR-32388/TRM/120/242/2019), Govt of India to GCK; DBT-BUILDER Program (Project/Grant No. BT/INF/22/SP42155/2021), Govt of India to GCK and the DST INSPIRE Fellowship Program (Project/Grant No. DST/INSPIRE Fellowship/2021/IF210059), Govt of India to VKP.
Author information
Authors and Affiliations
Contributions
M.K., R.B., V.K.P., D.M. and G.C.K. conceptualized the study. M.K., R.B., V.K.P., D.M., S.D., T.M., P.K., S.W.G. and G.C.K. collected relevant literature, drafted the contents, wrote the original draft, and prepared the figures. M.K., R.B., D.M., VKP and G.C.K. significantly edited the manuscript text and figures. All authors have read and approved the content of the manuscript before the final submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Kundu, M., Butti, R., Panda, V.K. et al. Modulation of the tumor microenvironment and mechanism of immunotherapy-based drug resistance in breast cancer. Mol Cancer 23, 92 (2024). https://doi.org/10.1186/s12943-024-01990-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-024-01990-4