
Abstract
The RAS/mitogen-activated protein kinase (MAPK) signaling cascade is commonly dysregulated in human malignancies by processes driven by RAS or RAF oncogenes. Among the members of the RAF kinase family, CRAF plays an important role in the RAS-MAPK signaling pathway, as well as in the progression of cancer. Recent research has provided evidence implicating the role of CRAF in the physiological regulation and the resistance to BRAF inhibitors through MAPK-dependent and MAPK-independent mechanisms. Nevertheless, the effectiveness of solely targeting CRAF kinase activity remains controversial. Moreover, the kinase-independent function of CRAF may be essential for lung cancers with KRAS mutations. It is imperative to develop strategies to enhance efficacy and minimize toxicity in tumors driven by RAS or RAF oncogenes. The review investigates CRAF alterations observed in cancers and unravels the distinct roles of CRAF in cancers propelled by diverse oncogenes. This review also seeks to summarize CRAF-interacting proteins and delineate CRAF's regulation across various cancer hallmarks. Additionally, we discuss recent advances in pan-RAF inhibitors and their combination with other therapeutic approaches to improve treatment outcomes and minimize adverse effects in patients with RAF/RAS-mutant tumors. By providing a comprehensive understanding of the multifaceted role of CRAF in cancers and highlighting the latest developments in RAF inhibitor therapies, we endeavor to identify synergistic targets and elucidate resistance pathways, setting the stage for more robust and safer combination strategies for cancer treatment.
Similar content being viewed by others


Introduction
The RAS-RAF-MEK signaling cascade plays a pivotal role in modulating cellular processes such as proliferation, differentiation, and survival. However, this pathway is often constitutively activated in human malignancies characterized by RAS or RAF oncogenic drivers. RAS proteins activate many signaling pathways through direct interaction with effectors and guanosine triphosphate-bound RAS (GTP-RAS). CRAF (RAF1), a member of the RAF kinase family, is an effector of RAS signaling that was first discovered in 1988. CRAF contributes to RAS signaling and exhibits an array of kinase-dependent and kinase-independent activities. A comprehensive understanding regarding the implication of aberrant CRAF activity in tumors remains unclear. However, a series of distinct characteristics among the RAF proteins potentially accounts for their varied roles in oncogenesis. In addition, it has been reported that the associations between ROK-α, ASK1, and MST2 with CRAF illuminate their joint contribution to the anti-apoptotic function of CRAF.
Focusing on the kinase-dependent and –independent role of CRAF could facilitate the discovery of new potential therapeutic strategies for cancer treatment. As such, developing chemotherapeutic CRAF inhibitors is an attractive area of research. Several CRAF/pan-RAF inhibitors with diverse structural and biochemical properties have recently entered preclinical and clinical development. As highlighted in previous research, endeavors to inhibit CRAF kinase activity in human malignancies have produced inconclusive outcomes. Furthermore, it is noteworthy that no selective CRAF inhibitors have received regulatory approval.
This review describes documented RAF1 alterations observed in several cancer types (Fig. 1, Tables 1 and 2). It further explores the contribution of CRAF's role in various kinase-dependent and kinase-independent signaling pathways (Fig. 2), and CRAF-interacting proteins in varied cancer hallmarks (Fig. 3). Our coverage of recent developments regarding pan-RAF inhibitors (Fig. 4), including the combination of RAF inhibitors with other types of inhibitors or treatment strategies, to enhance anti-cancer efficacy in diverse clinical settings is of particular significance (Fig. 5, Table 3). Overall, the present review aims to explore the role of CRAF in cancer and highlights recent advances in RAF inhibitor combination therapies to improve treatment efficacy and mitigate toxicities in patients with RAF/RAS-mutant tumors.

Structure and molecular alteration of CRAF in TCGA patient cohorts. a Three conserved regions (CR1–CR3) are indispensable in activating CRAF by RAS-GTP. CR1, located at the N-terminus of CRAF, is comprised of the Ras-binding domain (RBD) and cysteine-rich domain (CRD). The CRD maintains the auto-inhibited state of CRAF through interacting with 14-3-3 and the C-terminal kinase domain. The CR2 region consists of a serine-threonine-rich segment and recognizes a series of regulators, including 14-3-3, Hsp90, CDC37, and prohibitin. The auto-inhibited CRAF monomer requires a 14-3-3 dimer to bind to phosphorylated Ser 259 in the CR2 region. The CR3 region is comprised of the protein kinase domain and a short C-terminal tail harboring the second binding site for 14-3-3 proteins. Point mutations are depicted as small colored dots in the graph. Blue dots represent point mutations in CRAF that result in inhibitory effects, while red dots represent point mutations that lead to activating effects. b The alteration of CRAF based on TCGA Pan-cancer Atlas studies as visualized on the UniProt data platform. In the figure, the "+" symbols below each tumor type indicate that the bar graph analysis incorporates "structural variants", "mutations", and "Copy Number Alterations (CNA)" for that specific tumor type. BLCA, Bladder Urothelial Carcinoma; SKCM, Skin Cutaneous Melanoma; DLBC, Lymphoid Neoplasm Diffuse Large B-cell Lymphoma; UCEC, Uterine Corpus Endometrial Carcinoma; STAD, Stomach adenocarcinoma; ESAD, Esophageal adenocarcinoma; SARC, Sarcoma; KIRC, Kidney renal clear cell carcinoma; COAD, Colon adenocarcinoma; THCA, Thyroid carcinoma; BRCA, Breast invasive carcinoma; LUAD, Lung adenocarcinoma; LIHC, Liver hepatocellular carcinoma; KIRP, Kidney renal papillary cell carcinoma; OV, Ovarian serous cystadenocarcinoma; CESC, Cervical squamous cell carcinoma and endocervical adenocarcinoma; LGG, Lower Grade Glioma; LUSC, Lung squamous cell carcinoma; THYM, Thymoma; PRAD, Prostate adenocarcinoma; GBM, Glioblastoma multiforme; TGCT, Testicular Germ Cell Tumor; HNSC, Head and Neck squamous cell carcinoma; PCPG, Pheochromocytoma and Paraganglioma

Kinase-Dependent and kinase-Independent Signaling Pathways Mediated by CRAF. a Role of CRAF in the kinase-dependent signaling pathway. As a cytosolic serine/threonine kinase, CRAF plays an important role in proliferation, migration invasion, EMT invasion, stem cell self-renewal, mitogen and stress-induced signaling responses, and cell apoptosis in the Ras-RAF-MEK-ERK cascade. β-arrestin mediates the active internalization of G protein-coupled receptors (GPCRs) and activates ERK1/2 through CRAF. GPCR also promotes Ca2+ mobilization and activation of protein kinase C (PKC) dependent of β-arrestin. Ca2+ signaling also promotes cAMP/protein kinase A (PKA) activity. PKA and PKC can activate B/CRAF, promoting the RAF/MEK/ERK MAPK signaling pathway. PKA can also facilitate ERK inhibition by forming an inactive complex with Rap1/CRAF. This complex disrupts the activation of MEK1 and MEK2 by sequestering CRAF activity. Similar to PKA, 14-3-3 proteins also contribute to the inactivation of CRAF. Upon activation of receptor tyrosine kinases (RTKs) by extracellular signals, CRAF dissociates from 14-3-3 and is recruited to the plasma membrane. PI3K-AKT is positioned downstream of RAS and interacts with CRAF through Polycystic Kidney Disease 1 (PKD1). The MAZ transcription factor is a downstream target of the oncoprotein Cyr61/CCN1 and promotes pancreatic cancer cell invasion via CRAF-ERK signaling. CRAF-MEK-ERK signaling pathway regulates numerous targets in the cytoplasm and nucleus, including c-FOS, c-JUN, E2F transcription factor, retinoblastoma protein (Rb), Bcl-2 interacting mediator of cell death (Bim) and Bcl-2 homologous killer (Bak), β-Catenin, Fos-related antigen 1(Fra1), ZEB1/ZEB2, and Pyruvate kinase M2 (PKM2). Glutathione S-transferase pi 1 (GSTP1) inhibits the CRAF pathway through an autocrine feedback loop. In addition, ERK can negatively regulate B/CRAF through the HSP90/ERK1/2/PP5 complex. Furthermore, Transforming Growth Factor-beta (TGF-β) regulates the AP-1-Snail involved in Epithelial-Mesenchymal Transition (EMT) through CRAF-MAPK signaling. b Role of CRAF in the kinase-independent signaling pathway. CRAF plays an important role in mitotic progression by promoting AURKA and Plk1 activation. Mitochondrial membrane-bound CRAF regulates cell apoptosis by recruiting Apoptosis Signal-Regulating Kinase 1 (ASK-1) and Bcl-2 phosphorylate homolog BAD. Moreover, mammalian sterile 20-like kinase (MST2)/Hippo signaling is also involved in anti-apoptotic. CRAF modifies T helper cell differentiation and enhances immune responses by antagonizing Spleen Tyrosine Kinase (Syk)-induced RelB activation. CRAF also induces acetylation of the Nuclear Factor-kappa B (NF-κB) p65 to modulate adaptive immunity by dendritic cells (DCs). Genotoxic stress also induces p21-activated protein kinase-1 (PAK-1) activity, activates CRAF at serine 338, and promotes DNA damage repair independent of MAPK pathway. GFR, Growth Factor Receptor; bFGF, basic Fibroblast Growth Factor; DC-SIGN, Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin; TLR, Toll-like Receptor; TRIF, TIR-domain-containing adapter-inducing interferon-β; NIK, NF-κB-Inducing Kinase

CRAF-mediated signal transduction promotes various cancer hallmarks. CRAF promotes seven features of malignant tumors, including self-sufficiency in growth signals, metabolic reprogramming (mainly glycolysis), tumor invasion and metastasis (EMT), evading cell death and senescence, sustaining angiogenesis, oxidative stress response, and avoiding immune destruction. The relevant upstream and downstream proteins are illustrated in the diagram. CAV1, Caveolin-1; HSP90, Heat Shock Protein 90; ROK-α, Rho-Associated Coiled-Coil Kinase Alpha; AURKA, Aurora Kinase A; PRMT6, Protein Arginine Methyltransferase 6; PKM2, Pyruvate Kinase M2; PHLPP1/2, PH Domain and Leucine-Rich Repeat Protein Phosphatases 1/2; PKA, Protein Kinase A; MAZ, MYC-Associated Zinc Finger Protein; PLK-1, Polo-Like Kinase 1; AKT, Protein Kinase B; TGF-β: Transforming Growth Factor-beta

Binding mode of type I & II RAF inhibitors. a DFG-in conformation for PDB 3OG7 (crystalized with vemurafenib, specifically targets BRAFV600E via selectively binding to the "active" DFG-in and αC-helix-out conformation of the ATP binding site); b DFG-out conformation for PDB 1UWJ (crystalized with sorafenib, "inactive" DFG-out and αC-helix-in conformation of the ATP binding site). This figure has been adapted from Wang, L. et al. [21]

Combination therapies of CRAF/pan-RAF inhibitors and other treatments. Target therapies for CRAF/pan-RAF kinases (also refer to Table 3), including CRAF/pan-RAF inhibitors, Scaffold/chaperone proteins inhibitors, RAF RBD-RAS binding inhibitors, dual EGFR-RAF inhibitors, dual RAF-MEK inhibitors are illustrated above. Additionally, combination therapies of other treatments with CRAF/pan-RAF inhibitors, including RASG12C covalent inhibitors, EGFR inhibitors, MEK inhibitors, CDK inhibitors, Rb-CRAF inhibitors, PI3K inhibitors, STAT inhibitors, mTOR inhibitors, PD-1/PD-L1 antibodies, and CTLA-4 antibody are shown. EGFR, Epidermal Growth Factor Receptor; PI3K, Phosphoinositide 3-Kinase; STAT, Signal Transducer and Activator of Transcription; mTOR: Mammalian Target of Rapamycin; PD-1/PD-L1: Programmed Cell Death Protein 1/Programmed Death-Ligand 1; CTLA-4: Cytotoxic T-Lymphocyte-Associated Protein 4
Structures of CRAF proteins
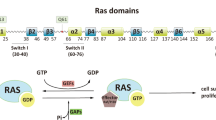
The RAF family, consisting of three RAF kinase paralogs: A-, B-, and CRAF, function as downstream effectors of RAS. Of the three RAF isoforms, CRAF is the earliest discovered RAF paralog. Three conserved regions (CR1–CR3) are indispensable for the recruitment and activation of CRAF by upstream effectors. Specifically, the CR1 region, comprised of the Ras-binding domain (RBD) and the cysteine-rich domain (CRD), is mainly responsible for binding to the RAS and membrane phospholipids. RBD and CRD of CRAF are associated with membrane-bound RAS via multivalent and dynamic interactions [51]. Cytosolic monomer RAF is auto-inhibited through the spatial conformation of the N-terminal regulatory region to the C-terminal kinase domain [52] and is activated by the recruitment of RAS-GTP to the plasma membrane. It is widely recognized that the RBD and CRD are two distinct globular domains that play crucial roles in the activation of CRAF. RBD binds to the interface of the RAS G domain, while CRD is responsible for the association with anionic lipid-rich membranes. Recent evidence has revealed synergistic influences of RBD and CRD on the dynamics of cellular membranes. The recruitment of RBD in proximity to the plasma membrane augments the local concentration of anionic lipids, thereby potentially intensifying the surface interaction between the RBD-CRD construct and the membrane [53]. Besides anchoring CRAF to the plasma membrane, CRD binds to RAS and stabilizes the active RAS-RAF complex in an RBD-independent manner [54]. A previously published report illustrated that CRD maintains the auto-inhibited state of CRAF through interacting with 14-3-3 and the C-terminal kinase domain [55]. CRD also plays a crucial role in RAF activation independently of its role in binding to RAS. Timothy et al. [56] revealed that the RAF1 T178A mutation located in the CRD domain diminished the interaction with RAS and inhibited CRAF kinase activity (~50%). Similarly, Daub et al. found that the CRD p.S177 and p.T182 mutation also resulted in impaired kinase activation [57].
The CR2 region, composed of a serine-threonine-rich segment, is recognized by various regulators, including 14-3-3, HSP90, CDC37, and prohibitin [58,59,60]. Auto-inhibited monomeric CRAF requires a 14-3-3 dimer binding to phosphorylated Ser259 in the CR2 region. Dephosphorylation of the CRAF Ser259 residue by HSP90, prohibitin, or protein phosphatase 2A (PP2A) abolishes the inhibitory effect of 14-3-3, resulting in its dissociation from the scaffold protein and its subsequent transfer to the plasma membrane for activation. CR3 contains the protein kinase domain and the remaining C-terminal tail, which harbors the second binding site that anchors the 14-3-3 scaffold protein. The catalytic kinase domain contains an αC-helix in the N-lobe, catalytic loop, and activation segment (AS) in the C-terminal, which spatially regulates CRAF kinase activity [61]. A recent study indicated that aside from classical catalytic activity, the CRAF kinase domain can also interact with the plasma membrane, thus coordinating CRAF recruitment and modulating its activation [62]. In summary, a detailed understanding of these conserved regions of CRAF is crucial for the development of more effective and safer CRAF inhibitors for cancer treatment. Key areas of focus include the synergistic effects of the RAS-binding domain and the cysteine-rich domain, the role of the CR2 region in binding to 14-3-3 proteins for activation, and the potential of the kinase domain to interact with the membrane.
CRAF functions and related pathways in oncogenic-driven cancers
Immature CRAF polypeptides are translated from the ribosome, followed by proper folding and stabilization by complex chaperones HSP90 and CDC37 [59]. Cytosol-localized monomeric RAF is auto-inhibited through the physical association of the N-terminal regulatory region to the C-terminal kinase domain. Moreover, 14-3-3 dimers contribute to steady-state regulation by binding to CRAF Ser259 and Ser621 residues located at the CR1 and CR3 regions, respectively. Upon activation of membrane-bound receptor tyrosine kinases (RTKs) by extracellular stimuli, CRAF becomes dissociated from 14-3-3 and is recruited to the plasma membrane to facilitate the propagation of downstream signaling [63]. The SHOC2–MRAS–PP1C complex facilitates the dissociation of 14-3-3 from CRAF through the dephosphorylation of Ser259 within the N-terminal domain. SHOC2-mediated dephosphorylation of CRAF is essential for RAF dimerization and efficient activation of the ERK pathway [64]. Additionally, the scaffold protein prohibitin facilitates the displacement of 14-3-3 from Ser259, further facilitating CRAF activation [65,66,67]. Several phosphorylation sites within or flanking the CRAF kinase domain are involved in its activation (Fig. 1). Thr491 and Ser494 sites within the activation segment are phosphorylated following CRAF membrane localization [68]. Ser338 and Tyr341 are considered the most essential phosphorylation sites for fully activating CRAF [69]. However, Oehrl W. et al. demonstrated that phosphorylation at Ser338 is not essential for CRAF activation, suggesting that CRAF activation can occur in a kinase-independent manner [70]. Taken together, CRAF acts as a key effector in the canonical RAS-MAPK cascade and plays a central role in kinase-independent signaling pathways in different cancers (as depicted in Fig. 2a and b).
The role of CRAF in cancers with mutant RAS
RAS mutations are the most common alterations in MAPK signaling and occur in nearly 30% of all human cancers. According to statistics, KRAS mutations exist in more than 90% of pancreatic ductal adenocarcinomas (PDACs), 40% of colorectal cancers, and 35% of non-small cell lung cancers (NSCLCs) [71]. Moreover, NRAS mutations occur in approximately 20% of malignant melanomas. Although RAS has historically been described as an "undruggable" target, allele-specific KRASG12C inhibitors have shown clinical benefits in lung cancer patients [72]. Additionally, non-covalent pan-KRAS inhibitors display promising therapeutic potential for patients with KRAS-driven malignancies [73].
Interestingly, a growing body of evidence coincides with the notion that the RAF family, particularly CRAF, assumes a pivotal role in oncogenic KRAS-driven cancers. An examination of CERES scores among KRAS, NRAS, and BRAFV600E mutant cancer cell lines indicated that KRAS and NRAS mutant cells had a heightened reliance on CRAF for proliferation, while BRAFV600E mutant cells primarily depended on BRAF for their growth [74]. Furthermore, genetic analysis of the RAS effectors within the MAPK pathway has revealed that ablation of CRAF exerts a promising therapeutic response with acceptable toxicities [75, 76]. Nonetheless, the impact of CRAF on tumorigenesis differs markedly across various KRAS-driven tumor models, and the exact role of CRAF in KRAS-mutant tumors remains to be elucidated.
KRAS-mutant lung cancer
Several studies have demonstrated that CRAF plays a crucial role in the development of lung carcinoma driven by the KRAS oncogene. Karreth et al. confirmed that CRAF, not BRAF, was essential for tumor initiation by resident KRASG12D oncogenes in non-small cell lung carcinoma. Interestingly, while BRAF has been proposed as the primary ERK activator due to its higher kinase activity [77], knock-out of BRAF and/or CRAF did not impact the phosphorylation of MEK [74, 78, 79]. Furthermore, systemic depletion of both CRAF and BRAF kinases in adult mice was found to be well-tolerated [80]. This suggests that in KRASG12V driven NSCLCs, all RAF proteins (A/B/CRAF) can sustain mitogenic signaling through the MAPK pathway.
Furthermore, recent research has shown that the KRAS mutant lung cancer growth is driven by heterodimerization of CRAF and ARAF, not merely by CRAF kinase activity. Remarkably, depletion of CRAF and ARAF inhibits sustained MAPK activation and alleviates cell-cycle arrest caused by CRAF ablation [74]. Moreover, CRAF ablation was shown to limit reactions detrimental to maintaining homeostasis [79, 80]. Concomitant suppression of CDK4 kinase activity and CRAF ablation effectively induced complete regression in 25% of KRAS/TP53-driven lung cancers [81]. Either CRAF depletion or sorafenib treatment decreased cyclin E expression and induced G1 arrest in KRAS mutant NSCLC cells [43]. Impairment of CRAF-MEK complex formation enhanced inhibition of CRAF-dependent ERK signaling in KRAS mutant tumors [82]. One promising hypothesis suggests that therapeutic effects derived from CRAF ablation may rely on other mechanisms aside from kinase inhibition. Although the contribution of RAF isoforms to the various stages of RAS-driven tumorigenesis and development remains unclear, the depletion of CRAF from KRASG12V expressing lung cells completely inhibited tumor development without inducing significant toxicities, suggesting a potential role for CRAF in modulating an alternative pathway essential for malignant transformation [43, 80]. Sanclemente et al. demonstrated that the anti-proliferative effect observed upon CRAF attenuation in lung adenocarcinoma cells occurs through occluding its interaction with the ASK1 or MST2 kinases [1]. Moreover, the enhanced apoptotic activity stemming from the loss of CRAF expression has a minimal impact on normal tissue homeostasis [83]. These studies suggest that targeting CRAF might be a beneficial therapeutic approach for KRAS mutant lung cancers. Moreover, dimerization of CRAF, rather than its kinase activity, is essential for KRAS mutant-driven lung cancer [83]. Depleting CRAF inhibited tumor growth in KRAS/p53-driven lung tumors. However, expressing kinase-dead CRAF variants (CRAFD468A and CRAFK375M) in KRAS/p53-driven lung GEMM models failed to achieve the same effect [1].
Above all, in RAS-driven lung cancer, inhibition of CRAF kinase activity with selective inhibitors remains suboptimal due to its less prominent role in the RAS-MAPK signaling pathway and paradoxical activation of other RAF subtypes. Regardless, the intervention of RAF dimers and promoting CRAF degradation may be an effective therapeutic strategy for KRAS mutant lung cancers.
KRAS-mutant pancreatic carcinomas
While some reports suggest that disrupting the PHB-CRAF interaction could impair oncogenic RAS-driven pancreatic cancer through the ERK-MAPK signaling pathway [67], it is widely believed that merely deleting CRAF produces minimal effects in KRAS-mutated pancreatic ductal adenocarcinomas (PDAC). Eser et al. found that CRAF expression was not essential for the initiation of KRAS-driven PDAC [84]. The function of CRAF in PDAC markedly differs from its role in KRAS mutant lung cancer, and the underlying mechanism for the disparity remains elusive. Cell proliferation defects in KRAS mutant pancreatic cancer cells in response to CRAF inhibition occur without p-ERK attenuation [74] and may be attributed to the differences in the kinase-dependent and kinase-independent roles of CRAF in KRAS mutant lung cancer compared to KRAS mutant PDAC. However, ablation of EGFR/CRAF resulted in complete regression of PDAC with mutant KRAS/TP53 [85]. Moreover, the adverse effects from concurrently depleting EGFR and CRAF mirrored those observed in EGFR-deficient mice, suggesting such an approach may be well-tolerated in vivo [86]. In a parallel study, Assi et al. also found that CRAF/EGFR signaling is crucial for pancreatic tumorigenesis in adult pancreas harboring KRAS mutations [87]. In addition, although pan-RAF inhibitors elicit unacceptable toxicities in the clinic when combined with MEK inhibitors by affecting the MAPK pathway [40], researchers have suggested that a low-dose intervention of pan-RAF and ERK inhibitors could provide an effective therapeutic alternative for KRAS mutant PDAC by circumventing harmful feedback mechanisms associated with ERK reactivation [88].
KRAS-mutant colorectal carcinomas
Solely inhibiting CRAF is insufficient to suppress the MAPK signaling and the proliferation of colon cancer cells harboring the KRASG13D mutation [89]. This suggests that the removal of CRAF has minimal impact on MAPK signaling, which is likely maintained by BRAF. However, Borovski et al. posited that CRAF is crucial for sustaining the transformed phenotype of KRAS mutant CRC cells, exerting its effects in a kinase-dependent manner but independently of MEK [90]. While KRAS mutations are notably frequent in lung adenocarcinomas (14%) and colorectal tumors (5%) [71], their dependence on the KRAS mutation seems to differ between these cancers. Specifically, a series of phase I/II clinical trials using AMG 510 or MRTX849, both KRASG12C inhibitors, produced significant responses in approximately half of the lung cancer patients, yet yielded no comparable results for those with colon tumors [91, 92]. Depletion of CRAF induces apoptosis in colon cancer cells by activating RAS mutations via a MEK-independent RAF signaling pathway. When combined with simultaneous MEK kinase inhibition, the pro-apoptotic effect is amplified [93]. The role of CRAF in mediating tumor growth in KRAS mutant lung cancer, pancreatic cancer, and colon cancers is gradually gaining consensus in the scientific community [74]. It is clear that the kinase domain of CRAF, independent of its catalytic activity, plays a significant role in this process. Interestingly, the mechanism that necessitates CRAF heterodimerization with ARAF is crucial for maintaining KRAS-driven tumors [74]. Consequently, the kinase-independent role of CRAF is pivotal when considering combination therapies targeting KRAS-driven colon cancer. The precise mechanisms behind the kinase-dependent and -independent activities of CRAF in RAS-driven cancers remain to be fully elucidated.
RAS-mutant skin cancer
CRAF was reported to play a vital cell-autonomous role in the development and maintenance of RAS-driven skin tumors. CRAF was reported as the primary RAS effector signaling through ERK specifically in melanoma cells harboring NRAS mutations [94, 95]. NRAS mutations in melanoma promote RAS-MEK signaling cascade by switching their signaling from BRAF to CRAF, facilitated by the disruption of the cAMP-PKA inhibitory pathway on CRAF activity [94]. However, two similar reports have indicated that BRAF but not CRAF plays a critical role in initiating NRAS-driven melanoma, even though both display compensatory functions in tumor progression [96, 97]. Dorard et al. indicated that BRAF is crucial during the early stages of NRAS-driven melanoma [97]. Besides, BRAF and CRAF collaborate to activate ERK and maintain proliferation in NRAS-mutated human melanoma cell lines. Furthermore, under certain conditions, ARAF also emerges as a significant player. Notably, in the absence of both BRAF and CRAF, ARAF can promote cell proliferation. Similarly, depletion of both BRAF and CRAF has shown promising effects on NRASQ61L/K mutant melanoma cells [89]. Independent of its kinase activity, CRAF modulates tumorigenesis by inhibiting Rok-α activity within the CRAF-Rok-α complex, facilitating STAT3 phosphorylation, Myc expression, and tumor cell dedifferentiation [98]. In addition, CRAF is not necessary for ERK activation in promoting skin homeostasis [99]. This implies that if CRAF drives Ras-induced skin cancer through interactions with Rok-α or other substrates, therapeutic approaches would need to focus beyond merely inhibiting CRAF catalytic activity.
While the specific role of CRAF dimerization-dependent activation in KRAS mutant tumors is evident, it remains intriguing that not every RAS-mutated tumor relies on CRAF activation. Additionally, the kinase-dependent and -independent actions of CRAF vary across different tumor types. Given the significance of CRAF in RAS mutant tumors, there is potential for the rapid translational application of pan-RAF inhibitors, either alone or in combination with other targeted therapies.
The role of CRAF in cancers with mutant BRAF
BRAF V600 mutant melanoma
The BRAFV600E mutation, the most common BRAF genetic alteration, occurs in 66% of cutaneous melanomas and 25% of colorectal cancers [100, 101]. Generally, BRAFV600E mutations cause sustained activation of the MAPK signaling pathway independent of the spatial activation and dimerization of RAF kinases. It has been observed that, compared to nevi tissues, melanomas exhibit elevated CRAF levels. Notably, depletion of CRAF levels compromises the viability of melanoma cells with either BRAFV600K mutation or wide-type BRAF [102]. Although CRAF has been reported to be required for non-V600E BRAF melanoma cell viability through an allosteric conformation mechanism or direct phosphorylation of its activation segment, its function in BRAFV600E melanoma is controversial [103,104,105]. Karreth et al. found that CRAF mRNA and protein levels in BRAFV600E melanoma cells are lower than in cells harboring wild-type BRAF, suggesting that transcriptional regulation plays a vital role in the reduction of CRAF expression. One promising mechanism is that melanoma cells expressing BRAFV600E bypass the antagonistic function of CRAF by reducing its expression. This, in turn, creates favorable conditions that promote MAPK pathway hyperactivation and cellular transformation [106].
While CRAF expression may differ in various types of BRAF mutant melanomas (BRAFV600E or non-BRAFV600E), it is worth noting that increased CRAF levels have been reported to promote resistance in a subset of BRAF mutant melanomas [107]. CRAF overexpression and dysregulation are critical mechanisms for RAF inhibitor resistance in melanoma via reactivation of MAPK signaling [108]. Elevated CRAF expression can result in reduced primary drug sensitivity or acquired resistance to AZ628 (a selective RAF inhibitor) in BRAF-driven mutant cells. This phenomenon is associated with a target shift from BRAF to CRAF, a process in which the kinase activity of CRAF appears to be dispensable [107]. There is little doubt that BRAFV600E mutations decrease their affinity with CRAF and the CRAF/BRAF ratio. Nevertheless, Karreth et al. discovered that CRAF elicited the inhibition of BRAFV600E kinase activity and MAPK activation by forming BRAFV600E–CRAF complexes [106]. Under these circumstances, oncogenic RAS could influence the MAPK signaling cascade by augmenting the stability of the CRAF-BRAFV600E complexes. The suppressive effect of CRAF on BRAFV600E may indicate why oncogenic RAS mutations and BRAFV600E have not been observed to occur concurrently. BRAF-mutant colorectal cancer is more prone to acquired resistance than BRAF-mutant melanoma, although CRAF was activated by oncogenic EGFR signaling in the former [109, 110]. Genetic ablation of RAF1 increases the activity of BRAF and MAPK signaling in fibroblasts [111]. Another study indicates that selective CRAF inhibition promotes paradoxical activation, which indicates that CRAF may negatively modulate MAPK signaling in some instances [112]. It is noteworthy that clinical sample analyses have revealed the emergence of secondary benign and malignant skin tumors in BRAFV600E melanoma patients undergoing BRAF inhibitor therapy. This phenomenon, linked to CRAF activation and BRAF-CRAF heterodimer formation, seems to be driven by RAS mutations. Specifically, oncogenic RAS mutations were detected in 58% of evaluated tumor samples (38/66) and 49% of control tumors from patients that had not received BRAFi therapy (30/62) [113]. These findings suggest a critical role for CRAF activation in acquired resistance to BRAF inhibitors in BRAF-driven tumors.
BRAF kinase-inactive mutation cancers
In addition to the dimerization-independent activation of BRAFV600, other BRAF mutants with impaired activity (also called class 3 BRAF mutants) were observed to stimulate MEK by alternatively activating CRAF via an allosteric or transphosphorylation mechanism [105]. Cytoplasmic mutant BRAFG596R/G466V was found to activate CRAF via transphosphorylate of its activation segment and 14-3-3-mediated hetero-oligomerization in an RAS-independent manner [114]. This type of mutation enhances the binding of BRAF mutants to activated RAS, leading to the increased formation of heterodimers between mutant BRAF and wild-type CRAF [115]. Likewise, CRAF appropriates the signal from low-activity BRAFG469E/D594G mutants and regulates apoptosis through mitochondrial localization via binding to Bcl-2 [116]. Moreover, ablation of CRAF suppresses MAPK signaling in cells harboring the impaired BRAF mutants but not BRAFV600E, which also indicates that sole inhibition of CRAF is not sufficient to abolish redundant activation of MEK by BRAFV600E. This finding suggests a critical role for CRAF kinase in enhancing resistance to BRAF inhibitors in BRAF-driven tumors. Additionally, certain inhibitory mutations in BRAF may result in CRAF assuming the mantle as the dominant driver of the MAPK signaling pathway. Heidorn et al. uncovered an intriguing phenomenon where kinase-dead BRAF (BRAFD594A) appears to necessitate the co-existence of oncogenic RAS to drive RAS-dependent CRAF activation and tumorigenesis [103]. This insight further highlights the connection between resistance to BRAF selective drugs and patients with RAS mutant tumors. However, BRAFD594A was unable to activate CRAF or stimulate MEK phosphorylation, rendering it catalytically and biologically inactive [105, 117]. In summary, kinase-dead BRAF mutants, apart from BRAFV600E, may still activate MEK by inducing CRAF through diverse mechanisms. However, solely inhibiting CRAF may not fully halt MEK activation, underscoring the intricate connection between BRAF mutations and CRAF in modulating the MAPK signaling pathway.
Key kinase-independent pathways of CRAF in cancers
While the kinase-dependent role of CRAF in the ERK-MAPK signaling pathway parallels that of BRAF, its kinase-independent function in oncogenic-driven cancers garners special attention. This unique role underpins the observed ability of prolonged CRAF ablation to prevent lung tumor initiation without inducing notable toxicity in adult mice. Such tumor regression likely arises from enhanced apoptosis rates combined with diminished cell proliferation. Yet, a clear understanding of the kinase-independent function of CRAF within KRAS and BRAF mutant tumors remains elusive. The therapeutic responses after CRAF suppression in lung cancer might be tied to the activation of pro-apoptotic pathways. In the following sections, we will dissect the central kinase-independent signaling pathways associated with CRAF (as depicted in Fig. 2).
p21-activated protein kinase-1 (PAK-1) was previously found to facilitate CRAF activation by direct phosphorylation of residues p.S338 and p.S339. The phosphorylated CRAF was subsequently translocated to the mitochondria and participated in protecting endothelial cells from intrinsic apoptosis in a kinase-independent manner [118, 119]. Further studies reveal that CRAF confers an anti-apoptotic effect when recruited to the mitochondrial membrane by Bcl-2. In contrast, CRAF recruited to the plasma membrane within the MAPK pathway does not manifest this anti-apoptotic effect [102, 120]. Based on this phenotype, Alavi et al. discovered that CRAF suppresses apoptosis by inhibiting stress-activated protein kinase ASK1, similar to the results observed by Chen et al. [121, 122]. Mammalian sterile 20-like kinase (MST2), a component of the Hippo signaling pathway was found to also contribute to the anti-apoptotic functions of CRAF independent of its kinase activity [123, 124]. Moreover, CRAF depletion promoted apoptosis by stimulating caspase-1 but not the MEK/ERK and NF-κB pathways [125]. Furthermore, the knock-down of CRAF inhibited the progression of RAS-driven and BRAFV600K mutant melanoma by mediating the inhibition of Bcl-2 rather than by inhibiting the mitogen-activated protein kinase pathway [102]. Similarly, CRAF knockout was shown to suppress the proliferation of fibroblasts and hematopoietic cells by increasing the apoptotic index rather than through cell cycle disruption [111]. The aforementioned studies highlight the anti-apoptotic function of CRAF rather than its role in accelerating cell proliferation. Nevertheless, knock-down of CRAF prevented the phosphorylation of Bcl-2 and apoptosis induced by taxol [126]. Moreover, further studies indicated that phosphorylation of CRAF and Bcl-2, but not ERK1/2, was crucial in taxol-induced apoptosis in breast cancer cells [127]. Although the induction of apoptosis by taxol is dependent upon CRAF and Bcl-2 phosphorylation and Bcl-2 cleavage, the kinase activity of CRAF may be dispensable in this process.
CRAF promotes T helper cell differentiation and enhances immune responses by antagonizing Syk-induced RelB activation [128]. Furthermore, CRAF partially reprograms gene expression and regulates the cell cycle by activating the transcription of NF-κB through phosphorylation of IκB [129]. Moreover, CRAF was also shown to induce acetylation of the NF-κB p65 to modulate adaptive immunity by dendritic cells (DCs) [130]. However, the inhibition of RAF, but not MEK1/2, results in partial activation of CD4+ T cells during DC differentiation, suggesting that CRAF regulates DC function in a different manner than MEK1/2 kinase [131]. CRAF also plays a vital role in cancer cell proliferation by facilitating AURKA and Plk1 activation, mitotic spindle location, and tumor progression in a kinase-independent function [132]. Similarly, Advani et al. revealed that CRAF promotes DNA damage response and tumor radioresistance by elevating CHK2 activation through a kinase-independent mechanism [133]. Additionally, CRAF has been reported to antagonize the Rok-α kinase domain within its cysteine-rich regulatory domain, resulting in increased migration of keratinocytes/fibroblasts and tumorigenesis [134,135,136]. Furthermore, the disturbance of the CRAF-Rb interaction is sufficient to inhibit MMP-associated migration of cancer in vitro and in vivo [137].
RAF1 alterations associated with cancer
Aberrant expression of CRAF
Elevated CRAF protein expression is correlated with poor prognosis in hepatocellular carcinoma (HCC) patients treated with sorafenib [133]. Mutations in RAF1 are extremely rare; however, overexpression of CRAF is correlated with disease progression in a subset of human cancers, including melanoma, non-small cell lung cancer (NSCLC), and hepatocellular carcinoma [102, 138, 139]. Overexpression of CRAF has been regarded as an early tumor marker for human lung adenocarcinoma [140]. Consistent with this observation, lung-restricted overexpression of full-length CRAF or its truncated kinase domain contributes to the MEK-dependent formation of lung adenomas [141, 142]. Moreover, increased CRAF levels have been reported to facilitate resistance in BRAF mutant melanomas [107]. However, the association between CRAF expression and tumor prognosis is controversial, and resistance mechanisms in vivo have not been demonstrated. According to the Gene Expression Profiling Interactive Analysis (GEPIA), an online tool for visualizing TCGA data, CRAF transcript expression does not completely align with the findings reported in the literature. For instance, CRAF expression in lung squamous cell carcinoma (LUSC) is significantly lower in tumors compared to normal tissues (http://gepia.cancer-pku.cn). Moreover, although CRAF overexpression is associated with tumor grade (p = 0.03), it appears that CRAF protein expression is not a reliable predictor of tumor progression [143].
Considering the intricate signaling biology of CRAF in the MAPK-dependent and MAPK-independent pathways, the diverse spectrum of alterations in CRAF and BRAF detected in cancer can manifest distinct functional attributes [82, 132]. Unlike BRAF, which is altered in up to 8% of all cancers, RAF1 has a notably lower alteration frequency of 0.7% in cancers. This disparity could be attributed to its reduced basal kinase activity compared to BRAF and the necessity for more intricate regulatory processes for its activation [144, 145]. According to TCGA pan-cancer atlas results (https://www.cbioportal.org/), we identified that RAF1 genetic mutations were present in 2.3% of all cancers. Specifically, RAF1 mutations were frequently observed in skin cutaneous melanoma (5.41%, 24/444) and uterine corpus endometrial carcinoma (4.54%, 24/529), whereas RAF1 amplification was highly concentrated in bladder tumors (10.71%, 44/411). Similar to Raie et al. [146], we observed that RAF1 mutations or copy number alterations were rare (<3%) or absent in other tumor types. Overall, developing methods for treating RAF1 mutant variants represent promising therapeutic targets in multiple cancer types.
Point mutation of RAF1
Patients harboring the BRAFV600E mutation have experienced clinical benefits from RAF inhibitors such as vemurafenib. However, with the observed limited median progression-free survival (less than 6 months) in melanoma treatments and the onset of rapid resistance, the focus has shifted to exploring combination therapy with MEK inhibitors [147]. Furthermore, numerous RAF1 mutations that facilitate biochemical and pharmacological resistance have been identified (summarized in Table 1). By understanding RAF1 mutations associated with drug resistance, we may enhance the likelihood of developing more effective therapeutic drugs [3, 7]. Demand for innovative treatments promotes the discovery of targetable chromosomal aberrances and mutations. For instance, a recent study demonstrated that RAF1P261A, located in the CR2 conserved region, promotes CRAF kinase activity in a dimer-dependent manner and benefits from the combination of LY3009120 and trametinib [4]. Another study demonstrated that RAF1 p.S257 and p.S259 enhance oncogenic activity and sensitivity to sorafenib [6]. Moreover, researchers have identified single amino acid substitutions (p.S257P, p.P261T, p.G361A, p.E478K) within RAF1 in melanoma cell lines resistant to RAF inhibitors [7]. In addition, Harms et al. identified a RAF1G361A amino acid substitution in patients with Noonan syndrome that may be associated with a significantly higher incidence of hypertrophic cardiomyopathy (HCM) [5]. Likewise, RAF1E478K mutation was found to constitutively heterodimerize and increase exogenous CRAF kinase activity. In contrast, another RAF1R401H mutation was observed to impair basal CRAF activity and enhance the inhibition of CRAF kinase by RAF inhibitors [2]. The levels of phosphor-MEK1/2 correlate positively with the efficiency of B/CRAF heterodimer formation, which is impaired by RAF1 interface mutations (p.E478K, p.R401H). Additionally, Atefi. M. identified a cancer-associated RAF1R391W mutation in melanoma, which conferred vemurafenib-resistant MAPK pathway activation in a dimerization-dependent manner [3]. A screening trial consisting of 82 acute myeloid leukemia (AML) patients revealed that the CRAF p.S427G mutation, rather than the p.I448V mutation, triggers constitutive activation of ERK by activating the CRAF-ERK signaling cascade, even though both mutations are associated with ERK activation [8]. The observations gleaned from the aforementioned studies lead to a fundamental question: can membrane recruitment or CRAF kinase activity be impeded or abolished by a specific site mutation? Sanclemente M. et al. demonstrated that, despite both RAF1D468A and RAF1K375M mutations completely abolishing CRAF kinase activity, the phosphorylation states of CRAF p.S338 and p.S621 in these kinase-inactive isoforms were inversely affected. This suggests that the reduced phosphorylation of CRAF p.S338 and p.S621 might be attributed to conformational changes rather than impaired kinase activity [1]. Moreover, despite the suppression of CRAF kinase activity, the phosphorylation of MEK1 remained unaffected, further suggesting that CRAF kinase activity is not essential for the activation of the MAPK signaling pathway. Hatzivassiliou et al. also confirmed that kinase-dead RAF1D486A was recruited to the plasma membrane in a kinase activity-independent manner [2]. Taken together, gain-of-function RAF1 point mutations may contribute to paradoxical activation caused by the type I1/2 inhibitors through activated dimerization. Nevertheless, oncogenic mutant RAF1 remains a rare target for the deployment of selective CRAF inhibitors in RAF or RAS-driven cancers.
Gene fusion of RAF1
Oncogenic RAF1 gene fusions have been observed in various cancers and RASopathies (summarized in Table 2). RAF1 gene fusions commonly occur in pancreatic acinar cell carcinomas (up to 18.5% in all cases) [11]. Although melanomas with RAF1 fusions are seldom observed (less than 1%), clinical sample analyses have consistently shown that melanomas harboring RAF1 fusions exhibit wide-type status for BRAF, RAS, and NF1 [12]. This finding implies that RAF1 fusions could potentially serve as therapeutic targets in melanoma patients lacking BRAFV600 or RAS mutations. Additionally, the prevalence of certain gene mutations, including TERTp (62%), CDKN2A (60%), TP53 (13%), ARID2 (10%), and PTEN (10%), within melanomas with active RAF1 fusions can aid in refining tumor classification strategies [12]. Phillips et al. corroborated this finding in anaplastic pleomorphic xanthoastrocytoma patients [20]. Two similar reports also described oncogenic RAF1 rearrangement in pilocytic astrocytoma with elevated CRAF kinase activity and MEK phosphorylation [14, 15]. In a cohort study comprised of 7119 melanoma patients, 40 cases (0.6%) were identified with activated RAF1 structural variants accompanied by mutations in TERTp and CDKN2A [12]. Another recent study identified a novel LRRFIP2-RAF1 fusion in wild-type BRAF acral melanoma with a concomitant KIT variant [16]. Comprehensive genomic profiling (CGP) of 3,633 pediatric cancer patients revealed RAF1 fusions in seven distinct pediatric tumor types. Within these fusions, RAF1 was found to associate with several gene partners, including MBNL1, TMF1, GOLGA4, SRGAP3, QKI, SOX6, and ATG7 [9]. Moreover, the RAF1 fusion is also found in a specific molecular subtype in spindle cell tumors that co-express S100 and CD34 [17, 18]. Similarly, a striking case report identified a MTAP–RAF1 gene fusion in an S100-positive soft tissue sarcoma [19].
Additionally, RAF1 fusions can also facilitate MAPK pathway activation in multiple tumor types [15]. However, Jain et al. reported that RAF1 fusions in pediatric low-grade gliomas (PLGGs) may not respond to type I and II RAF inhibitors previously proven effective in tumors harboring BRAF fusions [148]. The group also developed a heterologous RAF1 fusion model and identified that the PLGGs are sensitive to pan-RAF and combinatorial inhibitors of the MAPK/PI3K signaling pathway. Clinical trials have indicated that RAF1 gene fusions frequently occur in cases of acquired resistance to KRAS inhibitors (i.e., adagrasib and sotorasib); however, the underlying mechanisms contributing to this trend are currently unclear [149, 150]. Of note, results obtained from several preclinical studies investigating metastatic melanoma have indicated that activating RAF1 fusions are sensitive to MEK inhibitors [10, 13, 151]. This evidence suggests that dimer-dependent activation of CRAF induced by RAF1 fusions can be blocked by MEK inhibitors (i.e. selumetinib and trametinib). However, Jain et al. discovered that tumors with RAF1 fusions only partially respond to MEK inhibitors [148]. Therefore, additional studies are needed to evaluate the efficacy of combination therapies that target RAF dimerization and MEK in malignancies harboring RAF1 fusions.
Multiple studies have emphasized that the CRAF kinase domain–but not its kinase activity–plays a more pivotal role in KRAS-driven tumorigenesis [1, 74]. Therefore, other pathogenic oncogene fusions involving the CRAF kinase domain might serve as potential therapeutic targets. Research highlighting constitutive transformational activation of CRAF kinase induced by RAF1 fusion with other truncated kinases may aid in identifying efficacious multi-target therapies.
Other alterations of RAF1
RAF1 amplifications, which enhance RAF/MEK/ERK signaling pathway activation, have recently been reported in various tumors. For instance, using data obtained from the GENIE v3 cohort, we observed that RAF1 amplifications occur in bladder tumors at a frequency of 3.8% (139 out of 3844 patients), a rate higher than that for RAF1 amplifications in any other tumor type within this cohort. As a result of RAF1 amplification, bladder tumors with RAS oncogenic mutations are sensitive to RAF and MEK inhibitors.
Similarly, a randomized phase III clinical trial including 119 melanoma patients revealed that RAF1 amplification elevated the efficacy of carboplatin and paclitaxel with sorafenib (CPS) in terms of progression-free survival (PFS) compared with carboplatin and paclitaxel treatment alone (CP) (HR, 0.372; P = 0.025) [152]. Additionally, a high level of RAF1 amplification was observed in recurred/metastasized phyllode tumors of the breast compared with patients without recurrence/metastasis [153]. Coincidentally, a breast cancer study also indicated that dysregulation of the MAPK pathway due to RAF1 amplification is associated with poor outcomes and resistance to PD-1/PD-L1 therapy. However, RAF1 amplification is highly correlated with the genomically unstable (GU) Lund classification subtype, which responds best to the PD-L1 antibody atezolizumab, with approximately 50% of patients demonstrating a partial or complete response [154]. In terms of other rare genetic alterations, a recent report described a novel human truncated form of RAF1 (RAF1-tr) that exhibited increased nuclear localization and enhanced the double-stranded DNA damage response through the modulation of PRKDC function in a RAS-MAPK independent manner [132]. In summary, CRAF amplification is associated with the activation of the RAF/MEK/ERK pathway.
Molecular regulators of CRAF
Upstream molecular regulation of CRAF
Epigenetic regulation of CRAF
miRNAs modulate target gene expression by interacting with the 3′-UTR region, resulting in mRNA degradation or inhibition of translation. Using luciferase reporter assays, researchers have identified a large category of miRNAs that interact with the 3′-UTR of RAF1, including miR-15a/b, miR-16, and miR-195. Consequently, processes such as cell proliferation, migration, senescence, and drug resistance are subject to modulation via miRNAs. Notably, miR-195 was found to significantly inhibit thyroid cancer cell proliferation by suppressing CRAF protein expression [155]. Evidence also suggests that miR-16 interacts with the 3′-UTR of IGF1R, KRAS, and RAF1, thereby reducing osteosarcoma cell proliferation through the CRAF–MAPK pathway [156]. Moreover, miR-424 was shown to trigger apoptosis and cell-cycle arrest in glioblastoma cells by directly targeting the RAF1 and AKT1 oncogenes [157]. Ghousein et al. also found that miR-4510 functions as a tumor suppressor in hepatocellular carcinoma (HCC) by directly targeting and inhibiting RAF1 mRNA [158].
Moreover, ceRNAs can regulate CRAF expression through sequestering RAF1 targeting miRNAs. LINC00460 was shown to enhance papillary thyroid cancer progression by targeting and neutralizing the suppression of miR-485-5p, a RAF1-targeting miRNA [159]. The lncRNA ITGB2-AS1 promoted pancreatic ductal adenocarcinoma progression by upregulating RAF1 through sequestering miR-4319 [160]. Moreover, the LINC01559/miR-1343-3p/CRAF axis was found to promote pancreatic cancer progression [161]. ciRS-7, a potential miR-7 sponge, enhanced EGFR and CRAF activation, leading to a more aggressive colorectal cancer phenotype [162]. In addition, CircAGFG1/miR-370-3p and CircCDR1/miR-1287 were reported to regulate the transcription of RAF1 in cervical cancer and hepatocellular carcinoma respectively [163, 164].
Regulation by transcription factors or transcription activators
A series of transcription factors regulate the transcription-mediated activation of the CRAF signaling pathway. AP-2α modulates the transcription of RAF1 by amplifying its promoter transcriptional activity in HBV-expressing cells [165]. Similarly, bromodomain PHD finger transcription factor (BPTF) activates the MAPK pathway and is coexpressed with CRAF in T-cell lymphoma tissues [166]. Likewise, EZH2 contributes to impaired DNA damage repair and RAF1 amplification. RAF1 amplification leads to CRAF-β-catenin pathway activation and promotes stem cell self-renewal through the negative regulation of RAD51 [167]. Additionally, a report suggested that miR-493-3p inhibits RAF1 transcription by potentially decreasing the transcription of ETS1 [168]. Another study demonstrated that ETS2-mediated transcription of RAF1 promotes MAPK pathway activation [169].
Regulators of CRAF protein modification
Posttranslational regulation of CRAF is vital for CRAF stability and catalytic activity.
The RanBPM/CTLH complex promotes the ubiquitination and degradation of CRAF through its direct interaction with the C-terminus of CRAF [170]. Furthermore, CRAF protein stability is maintained by physical interaction with USP13, USP15 and inhibitors of apoptosis proteins (IAPs) at the post-translational level [171,172,173]. O-GlcNAcylation of CRAF promotes epithelial-mesenchymal transition (EMT) via inhibiting ubiquitination of CRAF, which is involved in the progression of renal interstitial fibrosis [174]. PRMT5 mediates the methylation of CRAF and promotes CRAF degradation and RAS-driven MAPK signaling [175]. Protein arginine N-methyltransferase 6 (PRMT6) inhibits aerobic glycolysis and cell stemness through the methylation of CRAF R100. This posttranslational modification of CRAF subsequently impedes PKM2 nuclear translocation and stem cell marker (CD133, SOX2, and NANOG) expression, respectively [176, 177].
Activating stimuli on CRAF
Scaffolding and chaperone proteins
In addition to being stimulated by the previously mentioned small GTP-RAS proteins, CRAF activity is also regulated by several MAPK scaffolding proteins, including KSR1/2, arrestins-2, SHOC2, 14-3-3 and PHB [67, 178,179,180,181]. The specific role of scaffolding proteins in a signal transduction cascade can vary depending on the specific target proteins involved. Unlike CRAF, KSR1/2 are characterized as pseudokinases owing to mutations in the active site. A previous report showed that KSR1 functions as an allosteric activator to promote CRAF catalytic function [182]. It is widely believed that KSR functions similarly to that of CRAF in the ERK pathway, as it competes with CRAF for binding to inhibited BRAF, resulting in allosteric activation [183]. In this context, KSR1 competes with CRAF for dimerization with BRAF in the presence of BRAF inhibitors. Given that CRAF-BRAF dimerization augments ERK signaling, KSR1 might effectively reduce the paradoxical activation of ERK signaling by promoting the complex formation between KSR and BRAF [184]. As a scaffold protein in the MAPK signaling cascade, arrestin-2 primarily interacts with CRAF but not MEK1 and ERK2 [178]. Additionally, β-arrestins promote phosphorylation of Src and thus enhance E2F expression driven by the CRAF-Rb complexes [185]. SHOC2 and the catalytic subunit of protein phosphatase 1 (PP1c) serve as highly specific effectors of M-Ras, critically influencing the activation of the MAPK pathway. Importantly, SHOC2 acts as a scaffold protein, mediating interactions between PP1C and M-Ras to specifically dephosphorylate the inhibitory p.S259 site on CRAF. This modulation enhances CRAF activity within distinct signaling complexes [186]. Doudican et al. found that the PHB1-CRAF complex mediates type I1/2 RAF inhibitor resistance; additionally, the group discovered that the conformational inhibitor rocaglamide A interrupts the interaction between PHB and CRAF, thus inhibiting the reactivation of MAPK signaling [108]. Most scaffolding proteins activate MAPK signaling by scaffolding kinase cascades; however, 14-3-3 constrains CRAF in an inactive conformation within the cytosol. Moreover, MAST1, another scaffold protein, contributes to cisplatin resistance by promoting CRAF-mediated activation of MEK, thereby exerting an anti-apoptotic effect [187]. Chaperone proteins, such as HSP90 and CDC37, play a role in maturing and moderating CRAF, subsequently facilitating mitogen-activated protein kinase pathway activation [59, 188]. Additionally, CNK1 regulates the activation of CRAF in a concentration-dependent manner by forming a trimeric complex with pre-activated CRAF and activated Src [189].
Other proteins
RIPK4 was shown to activate the CRAF-MEK-ERK pathway by promoting the degradation of proteasome-mediated phosphatidylethanolamine binding protein 1 (PEBP1) in pancreatic cancer [190]. PDK1 regulates P2Y receptor agonists-induced platelet activation via directly activating CRAF, which indicates that PDK1 regulates crosstalk between the canonical PI3K and MAPK pathways [191]. MAZ positively regulates CRAF signaling in pancreatic cancer by promoting PAK activation and AKT suppression through phosphorylation at p.S338 and dephosphorylation at p.S259, respectively. This regulation promotes epithelial-mesenchymal transition (EMT) [192]. In prostate cancer, PLK1 induces autophosphorylation at CRAF p.S621, which is crucial for protecting against degradation and regulating EMT and cellular motility [193, 194]. A study has shown that PHB1 and PHB2 interact with CRAF to facilitate chronic Hepatitis C Virus (HCV) infection. Notably, the group indicated that knock down of CRAF blocks HCV infection, whereas solely inhibiting RAF kinase fails to achieve the same outcome [195].
CRAF activity is indispensable in Lasonolide A (LSA) induced protein hyperphosphorylation and premature chromosome condensation independent of the MAPK pathway [196]. The activation of PAK1 and CHK2 is triggered through the p.S338 site of CRAF via a mechanism independent of its kinase activity [197]. It was reported that G protein-coupled receptors (GPCRs) activate CRAF through guanine nucleotide-binding G-proteins and the β-arrestins signaling pathway [198]. Interestingly, β-arrestins specifically bind to the Ras-binding domain of CRAF to balance CRAF activation due to stimuli from G-protein coupled receptors (GPCRs) and the EGFR-RAS signaling cascade. Several studies have reported that TM7SF2, PDCD6, p21-activated kinase (PAK3), and serine/threonine kinase 3 (STK3) contribute to tumorigenesis via direct binding and activation of CRAF [199, 200]. It was also reported that CRAF-ERK is the dominant pathway involved in HER-2-mediated tumor progression [201]. As a complex regulator of the MAPK signaling cascade, PP2A positively regulates this pathway by catalyzing the dephosphorylation CRAF Ser259 [202].
Negative regulation of CRAF
The unexpected regulation of MAPK signaling by cAMP/PKA is partly due to Rap1-mediated suppression of CRAF [203]. Although Rap1 activates ERK signaling through BRAF, the overall effect of cAMP/PKA on this pathway is determined by the ratio of CRAF, BRAF, and PKA isoforms [204,205,206]. A study has reported that CRAF reverts to a signaling-competent state through interactions with protein phosphatase PP2A and prolyl isomerase Pin1. PP2A dephosphorylates CRAF, while Pin1 catalyzes the isomerization of its phosphorylated residues. This process facilitates the efficient recycling of CRAF within the MAPK/ERK signaling pathway [207]. The cross-talk between the PI3K-AKT and RAF-MAPK pathways in cell proliferation, metabolism, and motility is apparent due to the interaction between AKT and CRAF. It has been observed that AKT (also known as protein kinase B) suppresses CRAF at the S259 site, resulting in cross-inhibition between the AKT and ERK pathways [208, 209]. Serine/threonine protein phosphatase 5 (PP5) is traditionally thought to negatively regulate MAPK signaling by dephosphorylating CRAF at p.S338. However, a study by Matthew et al. recently shed light on a potentially contradictory role of PP5. Their work indicates that PP5's influence on CRAF's feedback phosphorylation is also contingent upon forming PP5-ERK1/2 complexes, a process driven by active Rac1 [210]. By binding to the N-terminal region of CRAF, the CRAF kinase inhibitor protein (RKIP) negatively regulates CRAF, a process vital for cell growth and differentiation [211, 212]. A previous study showed that the RKIP inhibitor suramin enhances the MAPK pathway by preventing RKIP from binding to CRAF [213]. EphA2 inhibitor dasatinib interferes with the BRAF/CRAF heterodimer activity via elevating caveolin-1 (CAV-1) in uterine carcinoma [214]. Another report reveals that PHLPP1/2 dephosphorylates CRAF, diminishing colorectal cancer cell invasion and migration [215]. SPRY2 attenuates B-cell receptor (BCR) and MAPK-ERK signaling by binding to CRAF and BRAF in normal B cells and chronic lymphocytic leukemia (CLL) cells [216].
Downstream effectors of CRAF
Catalytic effects of RAF kinase on MEK
Under physiological conditions, RAS-driven activation of RAF proteins occurs on the plasma membrane where activated RAS promotes RAF dimerization, a pivotal event to trigger the kinase activity of RAF proteins. The observation that kinase-dead BRAF was able to activate ERK signaling through dimerizing with and activating CRAF provided further support for the role of RAF dimerization and raised awareness that catalysis-dependent and -independent functions of RAF are functionally important [103, 217, 218]. In addition to the BRAF-CRAF heterodimer, respective homodimers of the two isoforms have also been detected but were noted to exhibit lower kinase activity. Notably, RAF family members can form physiologically relevant heterodimers and homodimers, resulting in their transactivation [180, 219]. Once RAF adopts an active conformation, its dimer interface is further stabilized by the hydrophobic Rspine residue in the αC-helix (p.L505 for BRAF, p.L397 for CRAF, and p.L358 for ARAF) located adjacent to the conserved RKTR motif [220]. Upon the relocation of R509 to the center of the dimer interface, αC-helix interacts with the NTA motif of the trans-RAF molecule and adopts the “IN” conformation [221, 222].
RAF phosphorylates MEK p.S218 and p.S222 within the activation loop; however, this activation also necessitates the prior association of MEK with RAF [223]. In a quiescent state, BRAF and MEK coalesce to form a heterodimer within the cytosol. Under these conditions, CRAF and ARAF abstain from interactions with MEK, prompting questions about their recruitment strategies for MEK [224]. Protein crystallography studies have indicated that BRAF directly interacts with MEK1, establishing contact predominantly through the αG helices and the activation loop. Concurrently, RAF proteins showcase a propensity to self-dimerize in a side-to-side fashion [223]. Studies have highlighted that RAF dimers or the homodimer of MEK itself predominantly phosphorylate the MEK homodimer but not its monomeric counterpart [225, 226]. During the activation phase, RAF and MEK collaboratively form a tetrameric complex, illustrated as MEK-RAF-RAF-MEK in crystallographic studies [100]. Subsequently, the phosphorylated MEK activates ERK.
Beyond the four standard components of the Ras-Raf-MEK-ERK signaling pathway, KSR1/2 serves as a pivotal scaffolding protein, facilitating the assembly of Raf-MEK-ERK complexes. Brennan and colleagues observed that BRAF enhances KSR2 activity by forming BRAF-KSR2 heterodimers, subsequently promoting the phosphorylation of MEK1 [224]. Notably, Lavoie et al. discovered that MEK facilitates the side-to-side dimerization of the BRAF–KSR1 kinase domain independently of MEK's catalytic activity [227].
CRAF's non-catalytic target regulation
Activated CRAF was previously reported as a potential therapeutic target against immune escape via stimulating TLR4-mediated inflammatory responses [228]. Moreover, it has been observed that the interaction between CRAF, Aurora-A, and Plk-1 at the centrosomes and spindle poles plays a pivotal role in promoting mitosis. Similarly, allosteric inhibitors of CRAF, but not ATP-competitive inhibitors, induce G2/M phase arrest by impairing the activation of Plk1 [132]. Through suppressing the pro-apoptotic kinases BAD [229], ASK1 [122], and MST2 [123], CRAF exerts a significant influence on apoptosis in a MAPK-independent manner. Another report reveals that crosstalk between the MAPK and Hippo signaling pathways depends on the CRAF/MST-2 complex [230]. Furthermore, CRAF facilitates the recruitment of Rokα, a function intriguingly not reliant on its kinase activity [231]. Direct inhibition of Rokα-mediated keratinocyte dedifferentiation by blocking CRAF prevented GDC-0879 induced tumorigenesis [136, 232]. Furthermore, CRAF was found to promote cell proliferation and migration in human lung fibroblasts through the TGF-β1/CRAF/Smad pathway [233]. A previously published report indicated that CRAF promotes the transformation of fibroblast cells through MEKK1-mediated NF-kB activation [234]. Upstream and downstream molecular regulation of CRAF across different cancer hallmarks has been summarized in Fig. 3.
Combination therapy and related anti-tumor applications
Strategies for CRAF inhibition
Since the discovery of oncogenic RAF1, there has been a concerted effort to develop therapeutic inhibitors to attenuate its aberrant activity in tumor cells. Recent studies have confirmed CRAF as a promising therapeutic target in KRAS-driven NSCLC [80]. Depletion of CRAF has been shown to decrease tumor size without notably affecting MAPK signaling in KRAS-driven lung cancer [83]. These findings have spurred a strong interest in selectively targeting CRAF as a potential treatment for KRAS mutant lung cancer. Current research indicates that CRAF has diverse functions in cancer, encompassing both kinase-dependent and kinase-independent mechanisms. The absence of significant toxicity upon CRAF depletion suggests that its primary mode of action might extend beyond the MAPK signaling pathway. Additionally, given the marked structural and functional similarities between BRAF and CRAF proteins, devising specific CRAF kinase inhibitors is inherently challenging. However, there still exist potential strategies worthy of exploration.
Inhibitors for selective CRAF kinase
Sorafenib, designed initially as a CRAF kinase inhibitor, has shown limited efficacy in clinical trials for melanoma, with favorable clinical responses less than 5% [235]. While sorafenib inhibits CRAF, wild-type BRAF, and BRAFV600E kinases, it also targets other kinases such as Flt3, Kit, and VEGFR. A series of selective CRAF inhibitors have been successfully developed in vitro by modifying the structure of existing BRAF and pan-RAF inhibitors, enabling them to specifically target CRAF. The selective CRAF inhibitor ZM336372 significantly reduces bioactive hormone levels and human achaete-scute homologue-1 (ASH-1) expression in carcinoid tumor cells, leading to pronounced suppression of cellular proliferation and the cell cycle [25]. Recently, Zhao et al. identified a novel spirocyclic CRAF inhibitor, SHR902275, which has exhibited excellent drug metabolism and pharmacokinetic properties in vivo [26]. GW5074, a CRAF inhibitor, was found to enhance the anticancer effects of sorafenib by inducing mitochondrial dysfunction [22, 23]. The pyrimidin-4-yl-1H-imidazol-2-yl derivative 7a showed potent and selective inhibition of CRAF with an IC50 value of 0.62 μM and demonstrated superior antiproliferative activity compared to Sorafenib [29]. Several compounds have been reported as highly potent and selective CRAF inhibitors, including (4-aminobenzyl/benzoyl)-1H-imidazol-1-yl pyrimidin-2-yl derivatives 10c, with an IC50 of 8.79 nM [28]. Other promising compounds include 1,4-dihydropyrazolo [4,3-d]imidazole phenyl derivatives 2t, with IC50 values ranging from 0.56 to 0.86 μM in WM3629 cell lines [27], and pyrimidin-4-yl-1H-imidazol-2-yl derivatives 7a, showing IC50 values of 0.62 and 4.49 μM in A375P and WM3629 cell lines respectively [29]. 3-carboxamido-2Hindazole-6-arylamide 10d is also a potent CRAF inhibitor, which exhibits an IC50 of 38.6 nM [30]. Various natural small molecules have also been identified as selective inhibitors of CRAF. One such example is gallic acid, which inhibits MMP-1 expression through targeting CRAF [236]. Luteolin, a natural CRAF inhibitor, reduces inflammatory responses in human neutrophils by inhibiting the MAPK signaling pathway [237]. Another natural compound, erianin, the main component of Dendrobium chrysotoxum, has been found to inhibit the progression of melanoma and colorectal cancer by targeting CRAF and downstream MEK1/2 [50]. In a previous study, researchers have demonstrated that the targeted delivery of mutant RAF1 to the neovasculature using nanocrystals exhibited anti-angiogenic effects. These findings suggest novel prospects for targeting tumor neovasculature with small-molecule drugs that act specifically on CRAF. Such targeted interventions may induce apoptosis in endothelial cells and lead to regression of tumor vasculature [118, 238]. Despite being developed as specific CRAF inhibitors, many compounds still exert inhibitory effects on BRAF kinase due to the highly homologous protein structures of B/CRAF.
Inhibitors for the scaffold proteins or partners of CRAF
Scaffolding proteins are central to orchestrating MAPK pathway activity. MAPK scaffold proteins notably (i) connect directly with various MAPK signaling components, (ii) coordinate or segregate protein interactions, and (iii) modulate signal intensity to specific stimuli, ensuring precise and timely MAPK signal relay. Importantly, therapeutic strategies are available to target scaffolding and chaperone proteins that interact with CRAF. Scaffold protein HSP90 was reported vital for CRAF activation via dephosphorylation of the p.S259 residue. KBU2046 selectively inhibits the activation of CRAF and modulates cell motility by binding to the interface of HSP90/CDC37, thereby disturbing the interaction between the CRAF and HSP90/CDC37 heterocomplex [33]. It has been reported that radicicol and novobiocin induce the degradation of the HSP90 client protein CRAF but do not degrade BRAFV600E or inhibit MEK1/2 activation in HT29 human colon cancer cells [34]. Peptide R18 is found to effectively block the interaction between CRAF and the physiological ligand of 14-3-3, thereby inhibiting the protective effect of 14-3-3 against phosphatase-induced inactivation of CRAF [239]. RKIP has been reported to inhibit the phosphorylation of CRAF at S338 and Y341. Additionally, small molecule ligands such as DHPE and Locostatin interfere with the interaction between CRAF kinase and RKIP [35]. Moreover, suramin directly binds to RKIP and prevents its inhibitory effect on the MAPK signal pathway [213]. These scaffolding proteins, when bound to CRAF, influence the activation of the MAPK cascade and also facilitate CRAF degradation. However, due to the nonspecific nature of client proteins, inhibitors might counteract oncogene switching, a key mechanism by which tumors evade kinase inhibitors [240].
Inhibitors for the upstream and downstream protein of CRAF
Inhibitors for KRAS
As essential upstream regulators of CRAF, members of the RAS family of GTPases, which include KRAS, NRAS, and HRAS, undergo a transition between the GTP-loaded “on” state and the GDP-loaded “off” state. This transition is orchestrated through the activity of RAS guanine nucleotide exchange factors (RAS-GEFs) and RAS GTPase-activating proteins (RAS-GAPs), respectively [241, 242]. Considerable efforts have been devoted to suppressing RAS oncogenic signals by addressing upstream proteins, downstream proteins, and directly targeting RAS itself. During the activation-inactivation process, the importance of conformational alterations in two specific regions of the RAS protein, notably in switch II, has become apparent and has played a pivotal role in the eventual progression of RAS inhibitors [243]. Efforts are currently in progress to create mutant-specific RAS inhibitors that target KRASG12C in the switch-II region. The approval of Sotorasib for treating KRASG12C NSCLC represents a noteworthy achievement as the initial targeted therapy for tumors harboring KRAS mutations, offering hopeful prospects for the advancement of similar allele-specific treatments for mutant RAS [244]. Several potent covalent inhibitors of KRASG12C acting through a similar mechanism have entered clinical development. Adagrasib has demonstrated a significant reduction in cellular viability exclusively in KRASG12C cell lines and induced tumor regression in xenograft models [92]. Notably, Adagrasib has received FDA approval for the treatment of previously treated advanced-stage KRASG12C mutant NSCLC, based on the results of a phase I/II clinical trial (NCT03785249). Furthermore, both monotherapy and combination therapies involving covalent irreversible KRASG12C inhibitors, in conjunction with other targeted agents, are currently undergoing clinical trials for patients with advanced-stage KRASG12C mutant solid tumors. Examples include GDC-6036 in combination with the SHP2 inhibitor GDC-1971 (NCT04449874), JDQ443 in combination with another SHP2 inhibitor TNO155 (NCT04699188), and LY3537982 in combination with the CDK4/6 inhibitor Abemaciclib or the PD-1 inhibitor Pembrolizumab (NCT04956640).
Mutations in RAS proteins also influence their binding affinity with downstream effectors. For instance, KRASG12D exhibits a notably fivefold weaker binding to the CRAF-RBD compared to wild-type KRAS [245]. Thus alternative therapeutic strategies exist to inhibit CRAF activity, such as targeting its interaction with upstream KRAS oncoproteins. By disrupting the interaction between RAS and its downstream effector CRAF, B4-27 has demonstrated potent inhibition of RAS signaling in RAS-mutant cancer cells [246]. The compounds Kobe0065 and Kobe2602 have been identified as potential inhibitors that interrupt the binding between HRAS and CRAF. Furthermore, the compounds effectively suppressed the activity of the kinases located downstream of MEK at a concentration of 20 μM in HRASG12V mutant NIH3T3 cells [31]. MCP110 effectively blocks RAS-induced activation of CRAF in vitro, resulting in reduced anchorage-independent cell growth, the induction of G1 cell cycle arrest, and decreased cyclin D expression in A549 cells [32]. Moreover, rigosertib has been characterized as a RAS mimetic compound with the ability to disrupt the interaction between the RAF and PI3K protein families with KRAS [247].
Inhibitors for downstream apoptotic effectors
Indeed, although targeting the interaction between CRAF and its upstream activator KRAS can be a promising strategy, selectivity remains a pressing concern. A more selective approach can be achieved by targeting the specific interaction between CRAF and its apoptotic effectors, such as ROK-α, ASK1, and MST2, which operate independently of its kinase activity [99, 122, 124]. Nevertheless, further efforts are required to fully validate the therapeutic potential of targeting CRAF effectors. Structural studies and mapping of protein-protein interaction interfaces can provide valuable insights into the molecular mechanisms underlying these interactions.
pan-RAF inhibitor therapy
In the past few decades, the discovery of BRAFV600E mutations, which are oncogenic and highly active in most melanomas, has spurred significant interest in targeting this particular kinase [248, 249]. Type I1/2 inhibitors (Fig. 4), including Vemurafenib and Dabrafenib, selectively associate with the "active" DFG-in and αC-helix-out conformation of the ATP binding site, thereby specifically targeting BRAFV600E [250]. However, these RAF inhibitors often paradoxically activate the MAPK signaling pathway in RAS-driven tumors by promoting dimerization of inhibited BRAF with CRAF. To overcome the activation of RAF homo- and heterodimers, further development of type II pan-RAF inhibitors able to bind with the "inactive" DFG-out and αC-helix-in conformation at the ATP binding site has been pursued [251]. These inhibitors exhibit comparable potencies in stabilizing the αC-helix-in conformation of RAF proteins, effectively targeting both active RAF dimers and monomers. However, due to their similar potencies in targeting BRAF and CRAF, the process of transactivation between dimer partners is minimized [252].
Taking these findings into consideration, a class of RAF inhibitors, which utilize the structure of Type I1/2 inhibitors and are referred to as paradox breakers, such as PLX8394, have been developed. These inhibitors counteract the paradoxical activation of ERK by targeting BRAF-containing dimers, while preserving RAF function in normal cells where CRAF homodimers facilitate signaling, and selectively disrupt RAS-independent BRAF-driven signaling. Concerning the mechanism, these inhibitors demonstrate a stronger affinity for both BRAF homodimers and BRAF-CRAF heterodimers, yet they are less effective against CRAF, potentially suggesting reduced impact on KRAS-driven cancers [253]. In addition, several "type II" RAF inhibitors, including TAK580, TAK632, LHX254, BGB283, and RAF709, have been developed as potent inhibitors of RAF dimer activity. By displaying similar activity against monomeric and dimeric forms of RAF and minimizing off-target activation of wild-type RAF, these inhibitors have proven effective in blocking MAPK signaling in tumors harboring BRAF or RAS mutations [36,37,38, 254,255,256]. LY3009120, another pan-RAF inhibitor, preferentially inhibits the kinase activity of RAF dimers [257]. Additional selective RAF dimer inhibitors such as belvarafenib (GDC-5573) have shown preliminary efficacy in BRAFV600E and RAS-mutated advanced solid tumors in the early clinical phases. According to these findings, the development of DFG-out-type pan-RAF inhibitors holds greater potential for treating patients with cancers carrying oncogenic BRAFV600E or NRAS mutations. Moreover, several DFG-out type pan-RAF inhibitors (such as RAF265, TAK632, and LY3009120) have entered clinical research programs [40, 44]. Interestingly, ARAF mutations were shown to promote resistance to belvarafenib through dimerization-dependent and kinase activity-dependent mechanisms [42]. It is important to note that although pan-RAF inhibitors have shown promising efficacy in vitro, non-specificity for BRAF mutations could also suppress wild-type RAF dimer activity in normal cells [40, 258]. Additionally, the inherent limitation of target-based therapy lies in the narrow specificity of the agents used, which can be circumvented by activating alternative survival pathways in cancer cells. This concept is based on the understanding that survival pathways have pleiotropic effects. Over time, cancer cells have evolved within the host, enabling them to activate multiple signaling pathways to evade apoptosis and promote proliferation. Consequently, utilizing multi-target agents presents a promising approach to overcome these limitations.
Resistance mechanisms to RAF inhibitor
Resistance mechanisms in cancer with mutant BRAF
Recent studies have identified multiple primary mechanisms of resistance to RAF inhibitors in BRAFV600E melanoma. Lito et al. reported that cancers with BRAFV600E mutations develop resistance to I1/2 RAF inhibitors primarily through elevating active RAS-GTP levels and altering BRAFV600E splicing [259]. Alterations in BRAFV600E splicing variants without N terminus (V600E/ΔNT) and BRAF(ΔVNTAP) were discovered to promote the formation of protein homodimers and diminish the efficacy of the type I1/2 and II RAF inhibitors [225, 260, 261]. Additionally, several studies suggest that diverse oncogenic modifications stabilize the R-spine of BRAF, leading to constitutively active kinases resistant to RAF inhibitors [221, 225, 262]. Yap et al. found that dimer affinity is not directly tied to drug resistance in BRAF mutant cancers [263]. The group also observed that the enhanced stability of the R-spine in BRAF mutants with LLRins506/VLRins506 insertions drives resistance to both types I1/2 and II RAF inhibitors. Interestingly, these specific mutations significantly decrease the dimerization of oncogenic BRAF mutants. Intriguingly, drug-resistant cells become dependent on RAF inhibitors, and discontinuing the treatment slows the growth of resistant tumors [264].
Another avenue involves the activation of parallel (or "bypass") signaling pathways. For instance, the diminished efficacy of the I1/2 inhibitor in BRAF-mutated CRC is mostly linked to enhanced MAP kinase pathway-independent mechanisms involving EGFR signaling or the PTEN-PI3K-AKT signaling axis [109, 265, 266]. Interestingly, under basal conditions, RAF-MAPK signaling inhibits RTK–EGFR signaling via a negative feedback loop [110]. Moreover, several oncogenic mutations contribute to acquired resistance against BRAF inhibitors. Specifically, even after administering BRAF inhibitors, activating mutations in RAS and MAP2K1/2 can still reactivate the MAPK kinase pathway [267,268,269]. Furthermore, acquired resistance can also arise from the reactivation of MAPK signaling due to CRAF overexpression/mutation and COT overexpression [7, 270]. Studies have pinpointed elevated CRAF protein levels in melanoma cell culture models as a potential mechanism for BRAF inhibitor resistance [107]. COT diminishes the sensitivity of BRAFV600E melanoma cells to vemurafenib via MEK, bypassing the RAF signaling [271]. Dimerization of BRAF and CRAF results in increased accumulation of nuclear β-catenin in cancer-associated fibroblasts (CAFs), which further contributes to resistance against BRAF inhibitors [272]. In light of these insights, delving deeper into the clinical significance of increased CRAF protein levels becomes crucial when addressing BRAF inhibitor resistance.
Resistance mechanisms in cancer with wild-type BRAF
In wild-type BRAF isoforms, RAF inhibitor-induced paradoxical activation arises due to enhanced dimerization of BRAF and CRAF [2, 105]. One mechanism is that the inhibitor binding to CRAF facilitates the formation of CRAF homodimers, activating CRAF and triggering downstream MEK-ERK activation. Another potential, yet nonconflicting, mechanism involves the inhibitor binding to BRAF, resulting in a BRAF-CRAF heterodimer and subsequent CRAF activation. Correspondingly, pan-RAF inhibitors effectively target both protomers of the RAF dimers. Meanwhile, upon binding, paradox breakers induce a transition to the αC-helix out conformation that blocks dimerization-driven transactivation. Deepening our understanding of these inhibitor-RAF interactions and the reactions of wild-type RAF can pave the way for reducing off-target effects in patients.
Combination therapy of pan-RAF inhibitors
Until now, type I1/2 RAF inhibitors have exhibited limited efficacy when utilized in the context of colorectal and thyroid tumors harboring BRAF mutations [100, 273]. With advances in cancer treatment, single therapeutic strategies no longer suffice in effectively tackling the complex and diverse mechanisms that drive tumor growth and progression. Therefore, combination therapy is considered a more effective cancer treatment strategy as it targets multiple molecular targets simultaneously. For instance, pan-RAF inhibitor LXH254 blocks dimeric BRAF and CRAF, which can provide a potential clinical strategy when combined with MEK or ERK inhibitors to treat KRAS mutant NSCLC or NRAS mutant melanoma [38]. In addition, EGFR-mediated activation of RAS and RAF serves as the impetus for the reactivation of MAPK signaling in a subset of BRAF-mutant CRCs [109, 110]. The findings emphasize the value of combining RAF inhibitors with immune checkpoint inhibitors, tyrosine kinase inhibitors, or MEK inhibitors to enhance clinical outcomes and delay drug resistance [274].
Combination of MEK and pan-RAF inhibitors
It is important to note that the efficacy of therapeutic strategies relying on CRAF inhibition has been limited to inducing complete tumor regression in a small proportion of cases. Consequently, the successful development of clinically effective therapies targeting KRAS-mutant tumors may require the discovery of potent inhibitors targeting CRAF and the identification of additional targets to expand the spectrum of responsive tumors.
A phase I trial (NCT02407509) revealed that the novel MEK-RAF inhibitor, CH5126766, demonstrated significant efficacy in treating solid tumors and multiple myeloma with MAPK pathway mutations, with 27% of the 26 assessed patients achieving objective responses [46]. Erianin, a MEK-CRAF inhibitor, was reported to suppress the constitutive activation of the MAPK signaling pathway and exhibit anti-tumor effects in melanoma and colorectal cancer PDX models [50]. RAF709 has demonstrated superior antitumor activity in cell line and tumor xenograft models with BRAF or RAS mutations. Moreover, when combined with MEK inhibitor trametinib, RAF709 produced a heightened antitumor response in RAS-mutant models compared to RAF709 treatment alone [254]. It was previously shown that the selective pan-RAF inhibitor TAK-632 exhibits synergistic effects with the MEK inhibitor TAK-733 in BRAF inhibitor-resistant melanoma [37]. In a Phase Ib upgrade/expansion study, LXH254 was evaluated in conjunction with trametinib among patients grappling with advanced/metastatic non-small cell lung cancer harboring KRAS or BRAF mutations and NRAS-mutated melanoma (NCT02974725). Importantly, LXH254 produced promising preliminary antitumor efficacy in NRAS-mutated melanoma patients [39]. Additionally, the synergistic modulation of the MAPK pathway was identified in an HCT116 xenograft mouse model upon co-administration of GNE-9815 (or GNE-0749) and cobimetinib [275, 276].
Specific ARAF mutations may contribute to acquired resistance to RAF dimer inhibitors, such as belvarafenib. A promising clinical strategy is to combine RAF and MEK inhibitors, as demonstrated in ongoing clinical trials (NCT02405065, NCT03118817), which was recently proposed to delay the onset of ARAF-driven resistance [42]. Based on structural analysis, belvarafenib and GW5074 may serve as promising templates for developing covalent inhibitors that selectively target pan-RAF or CRAF. Notably, cobimetinib, a type III MEK inhibitor, holds the potential for modification to selectively inhibit the MEK1/2 allosteric activity. These insights offer a basis for medicinal chemists to design novel covalent inhibitors for targeting the MAPK pathway [277].
Combination of EGFR tyrosine kinase inhibitors and pan-RAF inhibitors
Temporary suppression of phospho-ERK through the use of BRAF inhibitors is seen in CRCs harboring BRAF mutations. However, a reactivation of ERK takes place due to EGFR-mediated activation of RAS and CRAF. Interestingly, BRAF mutant CRCs exhibit elevated levels of phosphor-EGFR compared to BRAF mutant melanomas. This phenomenon suggests that CRCs are particularly inclined towards developing EGFR-mediated drug resistance. Concurrent inhibition of RAF and EGFR has demonstrated the ability to prevent the reactivation of MAPK signaling in BRAF mutant CRC cells [109]. Likewise, the simultaneous suppression of CRAF and EGFR expression, which is crucial for the onset of pancreatic metaplasia, effectively halted tumorigenesis [278, 279]. Of greater significance, simultaneous excision of CRAF and EGFR alleles in mice with existing tumors resulted in complete tumor regression in a subset of the mice. However, the exact mechanism by which EGFR ablation collaborates with the loss of CRAF expression to achieve this outcome is not fully understood. Importantly, when CRAF and EGFR were simultaneously targeted, no additional toxicities were observed beyond the skin alterations [85]. As previously mentioned, studies have indicated that acquired resistance to the RAF inhibitor PLX8394 occurs through EGFR-mediated RAS-mTOR signaling. However, early combination therapy of PLX8394 with EGFR or mTOR inhibitors can prevent resistance to PLX8394. These findings provide a sound biological rationale and a potential combinatorial treatment strategy to facilitate the application of PLX8394 in BRAF mutant lung cancer patients [280]. In addition, results from a phase I study have demonstrated that Lifirafenib, a newly developed inhibitor targeting RAF and EGFR kinases, exhibited a favorable risk-benefit profile and has shown antitumor activity in patients with solid tumors harboring BRAFV600 mutations [281].
Combination of immune checkpoint inhibitors and pan-RAF inhibitors
In recent years, increasing evidence has suggested that combining immune checkpoint inhibitors (ICIs) and BRAF/MEK inhibitors can enhance the efficacy of cancer treatment, especially for patients who are resistant to monotherapy of ICIs. Similarly, clinical trials (NCT02224781) have demonstrated that sequential use of immune therapy (Ipilimumab and Nivolumab) followed by BRAF-targeted treatment (Dabrafenib and Cobimetinib) achieves superior therapeutic effects compared to the reverse sequence [282]. However, the combination of pan-RAF inhibitors and immune checkpoint inhibitors is still in the early exploratory stage.
An ongoing phase I clinical trial is actively exploring the therapeutic dose of oral pan-RAF inhibitor LXH254 in combination with PDR001 PD-1 monoclonal antibody in patients with advanced solid tumors (including NSCLC, ovarian cancer, and melanoma) with MAPK pathway alterations [283]. Furthermore, a phase Ib clinical trial evaluating the combination of regorafenib and nivolumab demonstrated that the aforementioned dual therapy has a manageable safety profile and exhibits promising antitumor activity [47]. Overall, the combination of pan-RAF and immune checkpoint inhibitors has produced promising results in preclinical and clinical studies, offering new therapeutic options for patients with various solid tumors. Further studies are warranted to optimize treatment regimens and identify patient populations that can benefit the most.
Combination therapy of pan-RAF inhibitors with other inhibitors
Dysregulation of MAPK and PI3K/Akt signaling pathways plays a critical role in the pathogenesis and progression of various cancers, particularly those driven by oncogenic RAS mutations. The therapeutic efficacy of CDK4 kinase inhibition is restricted in the treatment of KRAS-driven lung adenocarcinomas; however, an intriguing approach involving the combination of CDK4 kinase inhibition and elimination of CRAF expression has demonstrated acceptable toxicities in preclinical in vivo studies [81]. Disrupting the interaction between Rb and CRAF by RRD-251 was found to substantially reduce the malignant characteristics of pancreatic cancer cells, regardless of their sensitivity to gemcitabine [284]. A combination index analysis revealed a notable synergistic effect of the RAF265/SB590885 + ZSTK474 treatment regimen in papillary thyroid cancer cell lines [45]. The combination of AZ628 and BP-1-102 significantly suppressed MEK/ERK signaling pathway activation in lung cancer cells harboring KRAS mutations. This suggests that a combination of pan-RAF and STAT3 inhibitors could be an effective treatment for lung cancer cells with KRAS mutations [285]. Another study illustrated that combining pan-RAF (RAF265) and mTOR inhibitors (RAD001) enhanced the anti-tumor effects through the RAS-RAF and PI3K pathways, possibly through targeting the 4EBP1 and S6 protein [286]. The combination of RAF265 and BEZ 235 (a PI3K inhibitor) significantly inhibited the growth of xenograft tumors with KRAS and RET mutations, suggesting that blocking the ERK and PI3K signaling pathways can effectively inhibit tumor progression in differentiated and medullary thyroid cancer [287]. Our previous research indicated that TOPK activates the AKT/mTOR signal pathway and ERK signaling pathways in different esophageal cancers, suggesting potential opportunities for combination therapies with TOPK inhibitors [288, 289]. By targeting TOPK kinase, ADA-07 inhibits AP-1 activity by suppressing the phosphorylation of ERK1/2, p38, and JNKs [290]. Indeed, the results of the previous investigation were so supportive that a combined study with RAF inhibitors is currently underway. A recent study showed that inhibiting MAP4K2 with BAY61-3606 can sensitize KRAS wild-type colorectal cancer cells to AZ628, a RAF kinase inhibitor, suggesting a potential therapeutic strategy for treating colorectal cancer [291]. Moreover, a noteworthy finding is that the combination of volasertib and LXH254 has shown superiority over LXH254 monotherapy in suppressing long-term cell viability [283]. Furthermore, combination therapy of Raf265 and 5-FU promotes anti-tumor and anti-metastatic activity in colorectal cancer by targeting CD26+ tumor stem cells [292]. Regorafenib, a multi-target inhibitor of VEGFR1/2/3, PDGFRβ, and CRAF, has emerged as a notable systemic treatment for HCC patients who had recently received sorafenib treatment, indicating that multi-target inhibition holds tremendous potential in reducing the emergence of resistance [48].
Conclusions and outlook
Significant progress has been made in the field of CRAF research over the past three decades since its initial discovery. We possess a firm intellectual foundation regarding its involvement in RAS-MAPK signaling and an extensive familiarity with the primary signaling inputs that regulate its function. Although current selective small-molecule CRAF inhibitors possess limitations, they offer hope for controlling unchecked CRAF catalytic/allosteric activity to improve the anti-cancer effect. The rapid accumulation of structural data on members of the RAF family has revealed the intricacies of catalytic switching and may facilitate the discovery of highly specific RAF inhibitors in the future.
Since its discovery, the intricate regulation of CRAF and its crucial role in human health have captivated researchers. Given the challenges and potential benefits, we expect continued intense exploration in the coming years. Concerning unresolved issues, ongoing efforts in the following areas are expected to yield fruitful results. One area of research focuses on understanding the structure of CRAF proteins, including the role of the N-terminal region and the kinase structural domain in the interaction with RAS-RAF RBD. Furthermore, the exploration of allosteric inhibitors with the capability to disrupt RAF/MEK interactions holds promise as an attractive avenue for future research. In contrast to the first- and second-generation RAF inhibitors, next-generation allosteric inhibitors should have many more advantages, such as circumventing the paradoxical effect and producing fewer off-target effects. Recent findings have underscored the significance of RAF dimerization in various cellular contexts, particularly with its role in resistance linked to inactivation and signal transduction mechanisms. The CRAF DIF mutation (p.R401H), which mirrors the BRAF p.R509H mutation, was found to significantly reduce MEK activation despite facilitating the formation of CRAF homodimers. This finding suggests that RAF-mediated MEK activation relies on a dual-pronged mechanism that involves both dimerization and DIF-dependent transactivation. From a structural standpoint, understanding the RAF dimer interface (DIF) mechanism is crucial. This insight is essential not only in understanding how dimerization affects inhibitor activation but also in guiding the design of future ATP-competitive and allosteric RAF kinase inhibitors. Furthermore, a comprehensive understanding of RAF homo- and hetero-dimerization is crucial. Future studies could guide the design of specialized RAF dimer interface inhibitors that selectively target heterodimers vital for oncogenic signaling while preserving those essential for physiological processes.
Regulation of CRAF by HSP90 and 14-3-3 proteins, and the modulation of downstream apoptotic effectors, such as ROK-α, ASK1, and MST2 by CRAF, are also important areas of investigation. Although CRAF is not crucial for ERK activation, its significance in tumorigenesis and cancer progression largely arises from interactions and cross-talk with other signaling pathways, culminating in non-oncogene addiction in RAS-driven lung cancers and PDAC [74, 85]. Although these alternative inhibitors that target kinase-independent functions may present a safer and more effective therapeutic option, the multi-targeted nature of scaffold proteins introduces a challenge: how to strike a balance between potential multi-targeting toxicities and leveraging their tumor-suppressive attributes. Another pertinent field of research that warrants attention is combining pan-RAF inhibitors with other targeted therapies, such as immune checkpoint inhibitors, EGFR tyrosine kinase inhibitors, or MEK inhibitors. By investigating the aforementioned combination strategies, researchers will likely develop more effective therapeutic approaches for cancer treatment.
The importance of targeting CRAF therapeutically has been substantiated through the validation of new mouse models for KRAS/Trp53-mutant pancreatic and lung cancers. Moreover, systemic elimination of CRAF in adult mice has shown no substantial toxicities, which contrasts with previous reports of toxicity observed upon ablation of its downstream MEK1/2 and ERK1/2 kinases [81, 80, 85]. However, it is crucial to note that this approach may not universally apply to all KRAS-driven cancers. This has been demonstrated with the use of KRASG12C inhibitors, which have shown limited efficacy in treating colon cancer. Given the well-known challenges of blocking protein-protein interactions with small molecules, pharmacological degradation of CRAF may produce more favorable outcomes. Recent research in targeted protein degradation may present opportunities for the selective degradation of CRAF. Promising prospects include using molecular chaperone-mediated protein degraders (CHAMP) and proteolysis-targeting chimera (PROTAC). Identifying compounds that can bind to domains exclusive to CRAF isoforms makes it possible to target and specifically degrade CRAF [293]. In sum, although the inhibition of CRAF has exhibited promising results in preclinical studies, the clinical development of CRAF inhibitors is still in its early stages. However, with ongoing progress in innovative technologies and increasing comprehension of the intricacies of KRAS signaling, there is growing optimism that targeting CRAF may constitute a pivotal component of cancer therapies.
Availability of data and materials
Not applicable.
Abbreviations
- BLCA:
-
Bladder Urothelial Carcinoma
- SKCM:
-
Skin Cutaneous Melanoma
- DLBC:
-
Lymphoid Neoplasm Diffuse Large B-cell Lymphoma
- UCEC:
-
Uterine Corpus Endometrial Carcinoma
- STAD:
-
Stomach adenocarcinoma
- ESAD:
-
Esophageal adenocarcinoma
- SARC:
-
Sarcoma
- KIRC:
-
Kidney renal clear cell carcinoma
- COAD:
-
Colon adenocarcinoma
- THCA:
-
Thyroid carcinoma
- BRCA:
-
Breast invasive carcinoma
- LUAD:
-
Lung adenocarcinoma
- LIHC:
-
Liver hepatocellular carcinoma
- KIRP:
-
Kidney renal papillary cell carcinoma
- OV:
-
Ovarian serous cystadenocarcinoma
- CESC:
-
Cervical squamous cell carcinoma and endocervical adenocarcinoma
- LGG:
-
Brain Lower Grade Glioma
- LUSC:
-
Lung squamous cell carcinoma
- THYM:
-
Thymoma
- PRAD:
-
Prostate adenocarcinoma
- GBM:
-
Glioblastoma multiforme
- TGCT:
-
Testicular Germ Cell Tumor
- HNSC:
-
Head and Neck squamous cell carcinoma
- PCPG:
-
Pheochromocytoma and Paraganglioma
- GFR:
-
Growth Factor Receptor
- bFGF:
-
basic Fibroblast Growth Factor
- DC-SIGN:
-
Dendritic Cell-Specific Intercellular adhesion molecule-3-Grabbing Non-integrin
- TLR:
-
Toll-like Receptor
- TRIF:
-
TIR-domain-containing Adapter-inducing Interferon-β
- NIK:
-
NF-κB-Inducing Kinase
- CAV1:
-
Caveolin-1
- HSP90:
-
Heat Shock Protein 90
- ROK-α:
-
Rho-Associated Coiled-Coil Kinase Alpha
- AURKA:
-
Aurora Kinase A
- PRMT6:
-
Protein Arginine Methyltransferase 6
- PKM2:
-
Pyruvate Kinase M2
- PHLPP1/2:
-
PH Domain and Leucine-Rich Repeat Protein Phosphatases 1/2
- PKA:
-
Protein Kinase A
- MAZ:
-
MYC-Associated Zinc Finger Protein
- PLK-1:
-
Polo-Like Kinase 1
- AKT:
-
Protein Kinase B
- TGF-β:
-
Transforming Growth Factor-beta
- EGFR:
-
Epidermal Growth Factor Receptor
- PI3K:
-
Phosphoinositide 3-Kinase
- STAT:
-
Signal Transducer and Activator of Transcription
- mTOR:
-
Mammalian Target of Rapamycin
- PD-1/PD-L1:
-
Programmed Cell Death Protein 1/Programmed Death-Ligand 1
- CTLA-4:
-
Cytotoxic T-Lymphocyte-Associated Protein 4
References
Sanclemente M, Nieto P, Garcia-Alonso S, Fernandez-Garcia F, Esteban-Burgos L, Guerra C, Drosten M, Caleiras E, Martinez-Torrecuadrada J, Santamaria D, et al. RAF1 kinase activity is dispensable for KRAS/p53 mutant lung tumor progression. Cancer Cell. 2021;39:294–6.
Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, Ludlam MJC, Stokoe D, Gloor SL, Vigers G, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–5.
Atefi M, Titz B, Tsoi J, Avramis E, Le A, Ng C, Lomova A, Lassen A, Friedman M, Chmielowski B, et al. CRAF R391W is a melanoma driver oncogene. Sci Rep. 2016;6:27454.
Noeparast A, Giron P, Noor A, Bahadur Shahi R, De Brakeleer S, Eggermont C, Vandenplas H, Boeckx B, Lambrechts D, De Greve J, Teugels E. CRAF mutations in lung cancer can be oncogenic and predict sensitivity to combined type II RAF and MEK inhibition. Oncogene. 2019;38:5933–41.
Harms FL, Alawi M, Amor DJ, Tan TY, Cuturilo G, Lissewski C, Brinkmann J, Schanze D, Kutsche K, Zenker M. The novel RAF1 mutation p.(Gly361Ala) located outside the kinase domain of the CR3 region in two patients with Noonan syndrome, including one with a rare brain tumor. Am J Med Genet A. 2018;176:470–6.
Imielinski M, Greulich H, Kaplan B, Araujo L, Amann J, Horn L, Schiller J, Villalona-Calero MA, Meyerson M, Carbone DP. Oncogenic and sorafenib-sensitive ARAF mutations in lung adenocarcinoma. J Clin Invest. 2014;124:1582–6.
Antony R, Emery C, Sawyer A, Garraway L. C-RAF mutations confer resistance to RAF inhibitors. Cancer research. 2013;73:4840–51.
Zebisch A, Staber P, Delavar A, Bodner C, Hiden K, Fischereder K, Janakiraman M, Linkesch W, Auner H, Emberger W, et al. Two transforming C-RAF germ-line mutations identified in patients with therapy-related acute myeloid leukemia. Cancer research. 2006;66:3401–8.
Rankin A, Johnson A, Roos A, Kannan G, Knipstein J, Britt N, Rosenzweig M, Haberberger J, Pavlick D, Severson E, et al. Targetable BRAF and RAF1 Alterations in Advanced Pediatric Cancers. Oncologist. 2021;26:e153–63.
Pacaud A, Amintas S, Boussemart L, Cappellen D, Gerard E. A case of multi-metastatic melanoma with RAF1 fusion: a surprising response to anti-MEK therapy. Eur J Cancer. 2021;147:161–3.
Prall OWJ, Nastevski V, Xu H, McEvoy CRE, Vissers JHA, Byrne DJ, Takano E, Yerneni S, Ellis S, Green T, et al. RAF1 rearrangements are common in pancreatic acinar cell carcinomas. Mod Pathol. 2020;33:1811–21.
Williams EA, Shah N, Montesion M, Sharaf R, Pavlick DC, Sokol ES, Alexander BM, Venstrom JM, Elvin JA, Ross JS, et al. Melanomas with activating RAF1 fusions: clinical, histopathologic, and molecular profiles. Mod Pathol. 2020;33:1466–74.
McEvoy CR, Xu H, Smith K, Etemadmoghadam D, San Leong H, Choong DY, Byrne DJ, Iravani A, Beck S, Mileshkin L, et al. Profound MEK inhibitor response in a cutaneous melanoma harboring a GOLGA4-RAF1 fusion. J Clin Invest. 2019;129:1940–5.
Yde CW, Sehested A, Mateu-Regue A, Ostrup O, Scheie D, Nysom K, Nielsen FC, Rossing M. A new NFIA:RAF1 fusion activating the MAPK pathway in pilocytic astrocytoma. Cancer Genet. 2016;209:440–4.
Jones DT, Kocialkowski S, Liu L, Pearson DM, Ichimura K, Collins VP. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene. 2009;28:2119–23.
LeBlanc RE, Lefferts JA, Baker ML, Linos KD. Novel LRRFIP2-RAF1 fusion identified in an acral melanoma: A review of the literature on melanocytic proliferations with RAF1 fusions and the potential therapeutic implications. J Cutan Pathol. 2020;47:1181–6.
Suurmeijer AJH, Dickson BC, Swanson D, Zhang L, Sung YS, Cotzia P, Fletcher CDM, Antonescu CR. A novel group of spindle cell tumors defined by S100 and CD34 co-expression shows recurrent fusions involving RAF1, BRAF, and NTRK1/2 genes. Genes Chromosomes Cancer. 2018;57:611–21.
Mok Y, Kimpo MS, Chen H, Kuick CH, Chang KT, Lee VKM. Spindle cell tumour with S100 and CD34 co-expression showing PDZRN3-RAF1 rearrangement - a recently described entity. Histopathology. 2019;74:1109–11.
Hicks JK, Henderson-Jackson E, Duggan J, Joyce DM, Brohl AS. Identification of a novel MTAP-RAF1 fusion in a soft tissue sarcoma. Diagn Pathol. 2018;13:77.
Phillips JJ, Gong H, Chen K, Joseph NM, van Ziffle J, Jin LW, Bastian BC, Bollen AW, Perry A, Nicolaides T, et al. Activating NRF1-BRAF and ATG7-RAF1 fusions in anaplastic pleomorphic xanthoastrocytoma without BRAF p.V600E mutation. Acta Neuropathol. 2016;132:757–60.
Wang L, Zhang Q, Zhu G, Zhang Z, Zhi Y, Zhang L, Mao T, Zhou X, Chen Y, Lu T, Tang W. Design, synthesis and evaluation of derivatives based on pyrimidine scaffold as potent Pan-Raf inhibitors to overcome resistance. Eur J Med Chem. 2017;130:86–106.
Hu JM, Chang YL, Hsieh CC, Huang SM. The Synergistic Cytotoxic Effects of GW5074 and Sorafenib by Impacting Mitochondrial Functions in Human Colorectal Cancer Cell Lines. Front Oncol. 2022;12: 925653.
Tsai YT, Chuang MJ, Tang SH, Wu ST, Chen YC, Sun GH, Hsiao PW, Huang SM, Lee HJ, Yu CP, et al. Novel Cancer Therapeutics with Allosteric Modulation of the Mitochondrial C-Raf-DAPK Complex by Raf Inhibitor Combination Therapy. Cancer Res. 2015;75:3568–82.
Kao CC, Ho CL, Yang MH, Tsai YT, Liu SY, Chang PY, Wu YY, Chen JH, Huang TC, Yehn RH, et al. Phase I Targeted Combination Trial of Sorafenib and GW5074 in Patients with Advanced Refractory Solid Tumors. J Clin Med. 2022;11(8):2183.
Van Gompel JJ, Kunnimalaiyaan M, Holen K, Chen H. ZM336372, a Raf-1 activator, suppresses growth and neuroendocrine hormone levels in carcinoid tumor cells. Mol Cancer Ther. 2005;4:910–7.
Zhao P, Zhuang L, Wang X, Huang S, Wu H, Zhou Y, Yan Y, Zhang F, Shen R, Li J, et al. Discovery of spiro amide SHR902275: A potent, selective, and efficacious RAF inhibitor targeting RAS mutant cancers. Eur J Med Chem. 2022;228: 114040.
Yu H, Jung Y, Kim H, Lee J, Oh CH, Yoo KH, Sim T, Hah JM. 1,4-dihydropyrazolo[4,3-d]imidazole phenyl derivatives: a novel type II Raf kinase inhibitors. Bioorg Med Chem Lett. 2010;20:3805–8.
Kim M, Lee J, Jung K, Kim H, Aman W, Ryu JS, Hah JM. Design, synthesis and biological evaluation of benzyl 2-(1H-imidazole-1-yl) pyrimidine analogues as selective and potent Raf inhibitors. Bioorg Med Chem Lett. 2014;24:3600–4.
Lee J, Kim H, Yu H, Chung JY, Oh CH, Yoo KH, Sim T, Hah JM. Discovery and initial SAR of pyrimidin-4-yl-1H-imidazole derivatives with antiproliferative activity against melanoma cell lines. Bioorg Med Chem Lett. 2010;20:1573–7.
Aman W, Lee J, Kim M, Yang S, Jung H, Hah JM. Discovery of highly selective CRAF inhibitors, 3-carboxamido-2H-indazole-6-arylamide: In silico FBLD design, synthesis and evaluation. Bioorg Med Chem Lett. 2016;26:1188–92.
Shima F, Yoshikawa Y, Ye M, Araki M, Matsumoto S, Liao J, Hu L, Sugimoto T, Ijiri Y, Takeda A, et al. In silico discovery of small-molecule Ras inhibitors that display antitumor activity by blocking the Ras-effector interaction. Proc Natl Acad Sci U S A. 2013;110:8182–7.
Kato-Stankiewicz J, Hakimi I, Zhi G, Zhang J, Serebriiskii I, Guo L, Edamatsu H, Koide H, Menon S, Eckl R, et al. Inhibitors of Ras/Raf-1 interaction identified by two-hybrid screening revert Ras-dependent transformation phenotypes in human cancer cells. Proc Natl Acad Sci U S A. 2002;99:14398–403.
Zhang L, Pattanayak A, Li W, Ko HK, Fowler G, Gordon R, Bergan R. A Multifunctional Therapy Approach for Cancer: Targeting Raf1- Mediated Inhibition of Cell Motility, Growth, and Interaction with the Microenvironment. Mol Cancer Ther. 2020;19:39–51.
Fukuyo Y, Inoue M, Nakajima T, Higashikubo R, Horikoshi NT, Hunt C, Usheva A, Freeman ML, Horikoshi N. Oxidative stress plays a critical role in inactivating mutant BRAF by geldanamycin derivatives. Cancer Res. 2008;68:6324–30.
Janjusevic M, Greco S, Islam MS, Castellucci C, Ciavattini A, Toti P, Petraglia F, Ciarmela P. Locostatin, a disrupter of Raf kinase inhibitor protein, inhibits extracellular matrix production, proliferation, and migration in human uterine leiomyoma and myometrial cells. Fertil Steril. 2016;106(1530–1538):e1531.
Nishiguchi GA, Rico A, Tanner H, Aversa RJ, Taft BR, Subramanian S, Setti L, Burger MT, Wan L, Tamez V, et al. Design and Discovery of N-(2-Methyl-5’-morpholino-6’-((tetrahydro-2H-pyran-4-yl)oxy)-[3,3’-bipyridin]-5-yl)-3-(trifluoromethyl)benzamide (RAF709): A Potent, Selective, and Efficacious RAF Inhibitor Targeting RAS Mutant Cancers. J Med Chem. 2017;60:4869–81.
Nakamura A, Arita T, Tsuchiya S, Donelan J, Chouitar J, Carideo E, Galvin K, Okaniwa M, Ishikawa T, Yoshida S. Antitumor activity of the selective pan-RAF inhibitor TAK-632 in BRAF inhibitor-resistant melanoma. Cancer Res. 2013;73:7043–55.
Monaco KA, Delach S, Yuan J, Mishina Y, Fordjour P, Labrot E, McKay D, Guo R, Higgins S, Wang HQ, et al. LXH254, a Potent and Selective ARAF-Sparing Inhibitor of BRAF and CRAF for the Treatment of MAPK-Driven Tumors. Clin Cancer Res. 2021;27:2061–73.
de Braud F, Dooms C, Heist RS, Lebbe C, Wermke M, Gazzah A, Schadendorf D, Rutkowski P, Wolf J, Ascierto PA, et al. Initial Evidence for the Efficacy of Naporafenib in Combination With Trametinib in NRAS-Mutant Melanoma: Results From the Expansion Arm of a Phase Ib, Open-Label Study. J Clin Oncol. 2023;41:2651–60.
Sullivan RJ, Hollebecque A, Flaherty KT, Shapiro GI, Rodon Ahnert J, Millward MJ, Zhang W, Gao L, Sykes A, Willard MD, et al. A Phase I Study of LY3009120, a Pan-RAF Inhibitor, in Patients with Advanced or Metastatic Cancer. Mol Cancer Ther. 2020;19:460–7.
Peng SB, Henry JR, Kaufman MD, Lu WP, Smith BD, Vogeti S, Rutkoski TJ, Wise S, Chun L, Zhang Y, et al. Inhibition of RAF Isoforms and Active Dimers by LY3009120 Leads to Anti-tumor Activities in RAS or BRAF Mutant Cancers. Cancer Cell. 2015;28:384–98.
Yen I, Shanahan F, Lee J, Hong YS, Shin SJ, Moore AR, Sudhamsu J, Chang MT, Bae I, Dela Cruz D, et al. ARAF mutations confer resistance to the RAF inhibitor belvarafenib in melanoma. Nature. 2021;594:418–23.
Takezawa K, Okamoto I, Yonesaka K, Hatashita E, Yamada Y, Fukuoka M, Nakagawa K. Sorafenib inhibits non-small cell lung cancer cell growth by targeting B-RAF in KRAS wild-type cells and C-RAF in KRAS mutant cells. Cancer Res. 2009;69:6515–21.
Izar B, Sharfman W, Hodi FS, Lawrence D, Flaherty KT, Amaravadi R, Kim KB, Puzanov I, Sosman J, Dummer R, et al. A first-in-human phase I, multicenter, open-label, dose-escalation study of the oral RAF/VEGFR-2 inhibitor (RAF265) in locally advanced or metastatic melanoma independent from BRAF mutation status. Cancer Med. 2017;6:1904–14.
Barollo S, Bertazza L, Baldini E, Ulisse S, Cavedon E, Boscaro M, Pezzani R, Mian C. The combination of RAF265, SB590885, ZSTK474 on thyroid cancer cell lines deeply impact on proliferation and MAPK and PI3K/Akt signaling pathways. Invest New Drugs. 2014;32:626–35.
Guo C, Chenard-Poirier M, Roda D, de Miguel M, Harris SJ, Candilejo IM, Sriskandarajah P, Xu W, Scaranti M, Constantinidou A, et al. Intermittent schedules of the oral RAF-MEK inhibitor CH5126766/VS-6766 in patients with RAS/RAF-mutant solid tumours and multiple myeloma: a single-centre, open-label, phase 1 dose-escalation and basket dose-expansion study. Lancet Oncol. 2020;21:1478–88.
Fukuoka S, Hara H, Takahashi N, Kojima T, Kawazoe A, Asayama M, Yoshii T, Kotani D, Tamura H, Mikamoto Y, et al. Regorafenib Plus Nivolumab in Patients With Advanced Gastric or Colorectal Cancer: An Open-Label, Dose-Escalation, and Dose-Expansion Phase Ib Trial (REGONIVO, EPOC1603). J Clin Oncol. 2020;38:2053–61.
Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, Pracht M, Yokosuka O, Rosmorduc O, Breder V, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389:56–66.
Wu CS, Wu SY, Chen HC, Chu CA, Tang HH, Liu HS, Hong YR, Huang CF, Huang GC, Su CL. Curcumin functions as a MEK inhibitor to induce a synthetic lethal effect on KRAS mutant colorectal cancer cells receiving targeted drug regorafenib. J Nutr Biochem. 2019;74: 108227.
Wang P, Jia X, Lu B, Huang H, Liu J, Liu X, Wu Q, Hu Y, Li P, Wei H, et al. Erianin suppresses constitutive activation of MAPK signaling pathway by inhibition of CRAF and MEK1/2. Signal Transduct Target Ther. 2023;8:96.
Fang Z, Lee KY, Huo KG, Gasmi-Seabrook G, Zheng L, Moghal N, Tsao MS, Ikura M, Marshall CB. Multivalent assembly of KRAS with the RAS-binding and cysteine-rich domains of CRAF on the membrane. Proc Natl Acad Sci U S A. 2020;117:12101–8.
Cutler RE Jr, Stephens RM, Saracino MR, Morrison DK. Autoregulation of the Raf-1 serine/threonine kinase. Proc Natl Acad Sci U S A. 1998;95:9214–9.
Travers T, Lopez CA, Agamasu C, Hettige JJ, Messing S, Garcia AE, Stephen AG, Gnanakaran S. Anionic Lipids Impact RAS-Binding Site Accessibility and Membrane Binding Affinity of CRAF RBD-CRD. Biophys J. 2020;119:525–38.
Williams JG, Drugan JK, Yi GS, Clark GJ, Der CJ, Campbell SL. Elucidation of binding determinants and functional consequences of Ras/Raf-cysteine-rich domain interactions. J Biol Chem. 2000;275:22172–9.
Park E, Rawson S, Li K, Kim BW, Ficarro SB, Pino GG, Sharif H, Marto JA, Jeon H, Eck MJ. Architecture of autoinhibited and active BRAF-MEK1-14-3-3 complexes. Nature. 2019;575:545–50.
Tran TH, Chan AH, Young LC, Bindu L, Neale C, Messing S, Dharmaiah S, Taylor T, Denson JP, Esposito D, et al. KRAS interaction with RAF1 RAS-binding domain and cysteine-rich domain provides insights into RAS-mediated RAF activation. Nat Commun. 2021;12:1176.
Daub M, Jockel J, Quack T, Weber CK, Schmitz F, Rapp UR, Wittinghofer A, Block C. The RafC1 cysteine-rich domain contains multiple distinct regulatory epitopes which control Ras-dependent Raf activation. Mol Cell Biol. 1998;18:6698–710.
Morrison DK, Cutler RE. The complexity of Raf-1 regulation. Curr Opin Cell Biol. 1997;9:174–9.
Mitra S, Ghosh B, Gayen N, Roy J, Mandal AK. Bipartite Role of Heat Shock Protein 90 (Hsp90) Keeps CRAF Kinase Poised for Activation. J Biol Chem. 2016;291:24579–93.
Rajalingam K, Rudel T. “Prohibitin”g CRAF/MAPK activation with rocaglamides. Chem Biol. 2012;19:1077–8.
Roskoski R Jr. RAF protein-serine/threonine kinases: structure and regulation. Biochem Biophys Res Commun. 2010;399:313–7.
Prakash P, Hancock JF, Gorfe AA. Three distinct regions of cRaf kinase domain interact with membrane. Sci Rep. 2019;9:2057.
Terai K, Matsuda M. Ras binding opens c-Raf to expose the docking site for mitogen-activated protein kinase kinase. EMBO Rep. 2005;6:251–5.
Jones GG, Del Rio IB, Sari S, Sekerim A, Young LC, Hartig N, Areso Zubiaur I, El-Bahrawy MA, Hynds RE, Lei W, et al. SHOC2 phosphatase-dependent RAF dimerization mediates resistance to MEK inhibition in RAS-mutant cancers. Nat Commun. 2019;10:2532.
Yoshiki S, Matsunaga-Udagawa R, Aoki K, Kamioka Y, Kiyokawa E, Matsuda M. Ras and calcium signaling pathways converge at Raf1 via the Shoc2 scaffold protein. Mol Biol Cell. 2010;21:1088–96.
Simanshu DK, Morrison DK. A Structure Is Worth a Thousand Words: New Insights for RAS and RAF Regulation. Cancer Discov. 2022;12(4):899–912.
Luan Z, He Y, Alattar M, Chen Z, He F. Targeting the prohibitin scaffold-CRAF kinase interaction in RAS-ERK-driven pancreatic ductal adenocarcinoma. Mol Cancer. 2014;13:38.
Chong H, Lee J, Guan KL. Positive and negative regulation of Raf kinase activity and function by phosphorylation. EMBO J. 2001;20:3716–27.
Diaz B, Barnard D, Filson A, MacDonald S, King A, Marshall M. Phosphorylation of Raf-1 serine 338-serine 339 is an essential regulatory event for Ras-dependent activation and biological signaling. Mol Cell Biol. 1997;17:4509–16.
Oehrl W, Rubio I, Wetzker R. Serine 338 phosphorylation is dispensable for activation of c-Raf1. J Biol Chem. 2003;278:17819–26.
Drosten M, Barbacid M. Targeting the MAPK Pathway in KRAS-Driven Tumors. Cancer Cell. 2020;37:543–50.
Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, Italiano A, Schuler M, Borghaei H, Barlesi F, et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N Engl J Med. 2021;384:2371–81.
Kim D, Herdeis L, Rudolph D, Zhao Y, Bottcher J, Vides A, Ayala-Santos CI, Pourfarjam Y, Cuevas-Navarro A, Xue JY, et al. Pan-KRAS inhibitor disables oncogenic signalling and tumour growth. Nature. 2023;619:160–6.
Venkatanarayan A, Liang J, Yen I, Shanahan F, Haley B, Phu L, Verschueren E, Hinkle TB, Kan D, Segal E, et al. CRAF dimerization with ARAF regulates KRAS-driven tumor growth. Cell Rep. 2022;38: 110351.
Bromberg-White JL, Duesbery NS. Biological and biochemical characterization of anthrax lethal factor, a proteolytic inhibitor of MEK signaling pathways. Methods Enzymol. 2008;438:355–65.
Yufune S, Satoh Y, Akai R, Yoshinaga Y, Kobayashi Y, Endo S, Kazama T. Suppression of ERK phosphorylation through oxidative stress is involved in the mechanism underlying sevoflurane-induced toxicity in the developing brain. Sci Rep. 2016;6:21859.
Desideri E, Cavallo AL, Baccarini M. Alike but Different: RAF Paralogs and Their Signaling Outputs. Cell. 2015;161:967–70.
Sanclemente M, Francoz S, Esteban-Burgos L, Bousquet-Mur E, Djurec M, Lopez-Casas PP, Hidalgo M, Guerra C, Drosten M, Musteanu M, Barbacid M. c-RAF Ablation Induces Regression of Advanced Kras/Trp53 Mutant Lung Adenocarcinomas by a Mechanism Independent of MAPK Signaling. Cancer Cell. 2018;33(217–228):e214.
Karreth FA, Frese KK, DeNicola GM, Baccarini M, Tuveson DA. C-Raf is required for the initiation of lung cancer by K-Ras(G12D). Cancer Discov. 2011;1:128–36.
Blasco RB, Francoz S, Santamaria D, Canamero M, Dubus P, Charron J, Baccarini M, Barbacid M. c-Raf, but not B-Raf, is essential for development of K-Ras oncogene-driven non-small cell lung carcinoma. Cancer Cell. 2011;19:652–63.
Esteban-Burgos L, Wang H, Nieto P, Zheng J, Blanco-Aparicio C, Varela C, Gomez-Lopez G, Fernandez-Garcia F, Sanclemente M, Guerra C, et al. Tumor regression and resistance mechanisms upon CDK4 and RAF1 inactivation in KRAS/P53 mutant lung adenocarcinomas. Proc Natl Acad Sci U S A. 2020;117:24415–26.
Lito P, Saborowski A, Yue J, Solomon M, Joseph E, Gadal S, Saborowski M, Kastenhuber E, Fellmann C, Ohara K, et al. Disruption of CRAF-mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer cell. 2014;25:697–710.
Sanclemente M, Francoz S, Esteban-Burgos L, Bousquet-Mur E, Djurec M, Lopez-Casas P, Hidalgo M, Guerra C, Drosten M, Musteanu M, Barbacid M. c-RAF Ablation Induces Regression of Advanced Kras/Trp53 Mutant Lung Adenocarcinomas by a Mechanism Independent of MAPK Signaling. Cancer cell. 2018;33:217-228.e214.
Eser S, Reiff N, Messer M, Seidler B, Gottschalk K, Dobler M, Hieber M, Arbeiter A, Klein S, Kong B, et al. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell. 2013;23:406–20.
Blasco MT, Navas C, Martin-Serrano G, Grana-Castro O, Lechuga CG, Martin-Diaz L, Djurec M, Li J, Morales-Cacho L, Esteban-Burgos L, et al. Complete Regression of Advanced Pancreatic Ductal Adenocarcinomas upon Combined Inhibition of EGFR and C-RAF. Cancer Cell. 2019;35(573–587):e576.
Franzke CW, Cobzaru C, Triantafyllopoulou A, Loffek S, Horiuchi K, Threadgill DW, Kurz T, van Rooijen N, Bruckner-Tuderman L, Blobel CP. Epidermal ADAM17 maintains the skin barrier by regulating EGFR ligand-dependent terminal keratinocyte differentiation. J Exp Med. 2012;209:1105–19.
Assi M, Achouri Y, Loriot A, Dauguet N, Dahou H, Baldan J, Libert M, Fain JS, Guerra C, Bouwens L, et al. Dynamic Regulation of Expression of KRAS and Its Effectors Determines the Ability to Initiate Tumorigenesis in Pancreatic Acinar Cells. Cancer Res. 2021;81:2679–89.
Ozkan-Dagliyan I, Diehl JN, George SD, Schaefer A, Papke B, Klotz-Noack K, Waters AM, Goodwin CM, Gautam P, Pierobon M, et al. Low-Dose Vertical Inhibition of the RAF-MEK-ERK Cascade Causes Apoptotic Death of KRAS Mutant Cancers. Cell Rep. 2020;31:107764.
Jaiswal BS, Janakiraman V, Kljavin NM, Eastham-Anderson J, Cupp JE, Liang Y, Davis DP, Hoeflich KP, Seshagiri S. Combined targeting of BRAF and CRAF or BRAF and PI3K effector pathways is required for efficacy in NRAS mutant tumors. PLoS One. 2009;4:e5717.
Borovski T, Vellinga TT, Laoukili J, Santo EE, Fatrai S, van Schelven S, Verheem A, Marvin DL, Ubink I, Borel Rinkes IHM, Kranenburg O. Inhibition of RAF1 kinase activity restores apicobasal polarity and impairs tumour growth in human colorectal cancer. Gut. 2017;66:1106–15.
Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, Gaida K, Holt T, Knutson CG, Koppada N, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575:217–23.
Hallin J, Engstrom LD, Hargis L, Calinisan A, Aranda R, Briere DM, Sudhakar N, Bowcut V, Baer BR, Ballard JA, et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020;10:54–71.
Subramanian RR, Yamakawa A. Combination therapy targeting Raf-1 and MEK causes apoptosis of HCT116 colon cancer cells. Int J Oncol. 2012;41:1855–62.
Dumaz N, Hayward R, Martin J, Ogilvie L, Hedley D, Curtin JA, Bastian BC, Springer C, Marais R. In melanoma, RAS mutations are accompanied by switching signaling from BRAF to CRAF and disrupted cyclic AMP signaling. Cancer Res. 2006;66:9483–91.
Smalley KS, Nathanson KL, Flaherty KT. Genetic subgrouping of melanoma reveals new opportunities for targeted therapy. Cancer Res. 2009;69:3241–4.
Druillennec S, Pouponnot C, Eychene A. NRAS-driven melanoma: A RAF can hide another. Mol Cell Oncol. 2017;4:e1344758.
Dorard C, Estrada C, Barbotin C, Larcher M, Garancher A, Leloup J, Beermann F, Baccarini M, Pouponnot C, Larue L, et al. RAF proteins exert both specific and compensatory functions during tumour progression of NRAS-driven melanoma. Nat Commun. 2017;8:15262.
Ehrenreiter K, Kern F, Velamoor V, Meissl K, Galabova-Kovacs G, Sibilia M, Baccarini M. Raf-1 addiction in Ras-induced skin carcinogenesis. Cancer Cell. 2009;16:149–60.
Grabocka E, Bar-Sagi D. Raf-1 and squamous cell carcinoma: Rok-ing the boat. Cancer Cell. 2009;16:85–6.
Roskoski R. Targeting oncogenic Raf protein-serine/threonine kinases in human cancers. Pharmacological Research. 2018;135:239–58.
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54.
Jilaveanu LB, Zito CR, Aziz SA, Conrad PJ, Schmitz JC, Sznol M, Camp RL, Rimm DL, Kluger HM. C-Raf is associated with disease progression and cell proliferation in a subset of melanomas. Clin Cancer Res. 2009;15:5704–13.
Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, Hussain J, Reis-Filho JS, Springer CJ, Pritchard C, Marais R. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–21.
Hingorani SR, Jacobetz MA, Robertson GP, Herlyn M, Tuveson DA. Suppression of BRAF(V599E) in human melanoma abrogates transformation. Cancer Res. 2003;63:5198–202.
Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–67.
Karreth FA, DeNicola GM, Winter SP, Tuveson DA. C-Raf inhibits MAPK activation and transformation by B-Raf(V600E). Mol Cell. 2009;36:477–86.
Montagut C, Sharma SV, Shioda T, McDermott U, Ulman M, Ulkus LE, Dias-Santagata D, Stubbs H, Lee DY, Singh A, et al. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68:4853–61.
Doudican NA, Orlow SJ. Inhibition of the CRAF/prohibitin interaction reverses CRAF-dependent resistance to vemurafenib. Oncogene. 2017;36:423–8.
Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP, Brown RD, Della Pelle P, Dias-Santagata D, Hung KE, et al. EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov. 2012;2:227–35.
Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, Beijersbergen RL, Bardelli A, Bernards R. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. 2012;483:100–3.
Mikula M, Schreiber M, Husak Z, Kucerova L, Rüth J, Wieser R, Zatloukal K, Beug H, Wagner E, Baccarini M. Embryonic lethality and fetal liver apoptosis in mice lacking the c-raf-1 gene. The EMBO journal. 2001;20:1952–62.
Morgan CW, Dale IL, Thomas AP, Hunt J, Chin JW. Selective CRAF Inhibition Elicits Transactivation. J Am Chem Soc. 2021;143:4600–6.
Boussemart L, Girault I, Malka-Mahieu H, Mateus C, Routier E, Rubington M, Kamsu-Kom N, Thomas M, Tomasic G, Agoussi S, et al. Secondary Tumors Arising in Patients Undergoing BRAF Inhibitor Therapy Exhibit Increased BRAF-CRAF Heterodimerization. Cancer Res. 2016;76:1476–84.
Garnett MJ, Rana S, Paterson H, Barford D, Marais R. Wild-type and mutant B-RAF activate C-RAF through distinct mechanisms involving heterodimerization. Mol Cell. 2005;20:963–9.
Yao Z, Yaeger R, Rodrik-Outmezguine VS, Tao A, Torres NM, Chang MT, Drosten M, Zhao H, Cecchi F, Hembrough T, et al. Tumours with class 3 BRAF mutants are sensitive to the inhibition of activated RAS. Nature. 2017;548:234–8.
Smalley KS, Xiao M, Villanueva J, Nguyen TK, Flaherty KT, Letrero R, Van Belle P, Elder DE, Wang Y, Nathanson KL, Herlyn M. CRAF inhibition induces apoptosis in melanoma cells with non-V600E BRAF mutations. Oncogene. 2009;28:85–94.
Ikenoue T, Hikiba Y, Kanai F, Tanaka Y, Imamura J, Imamura T, Ohta M, Ijichi H, Tateishi K, Kawakami T, et al. Functional analysis of mutations within the kinase activation segment of B-Raf in human colorectal tumors. Cancer Res. 2003;63:8132–7.
Alavi A, Hood JD, Frausto R, Stupack DG, Cheresh DA. Role of Raf in vascular protection from distinct apoptotic stimuli. Science. 2003;301:94–6.
Liu H, Liu K, Dong Z. The Role of p21-Activated Kinases in Cancer and Beyond: Where Are We Heading? Front Cell Dev Biol. 2021;9:641381.
Wang HG, Rapp UR, Reed JC. Bcl-2 targets the protein kinase Raf-1 to mitochondria. Cell. 1996;87:629–38.
Chen J, Fujii K, Zhang L, Roberts T, Fu H. Raf-1 promotes cell survival by antagonizing apoptosis signal-regulating kinase 1 through a MEK-ERK independent mechanism. Proc Natl Acad Sci U S A. 2001;98:7783–8.
Alavi AS, Acevedo L, Min W, Cheresh DA. Chemoresistance of endothelial cells induced by basic fibroblast growth factor depends on Raf-1-mediated inhibition of the proapoptotic kinase, ASK1. Cancer Res. 2007;67:2766–72.
O’Neill E, Rushworth L, Baccarini M, Kolch W. Role of the kinase MST2 in suppression of apoptosis by the proto-oncogene product Raf-1. Science. 2004;306:2267–70.
Romano D, Nguyen LK, Matallanas D, Halasz M, Doherty C, Kholodenko BN, Kolch W. Protein interaction switches coordinate Raf-1 and MST2/Hippo signalling. Nat Cell Biol. 2014;16:673–84.
Jesenberger V, Procyk KJ, Ruth J, Schreiber M, Theussl HC, Wagner EF, Baccarini M. Protective role of Raf-1 in Salmonella-induced macrophage apoptosis. J Exp Med. 2001;193:353–64.
Blagosklonny M, Schulte T, Nguyen P, Trepel J, Neckers L. Taxol-induced apoptosis and phosphorylation of Bcl-2 protein involves c-Raf-1 and represents a novel c-Raf-1 signal transduction pathway. Cancer research. 1996;56:1851–4.
Huang Y, Sheikh MS, Fornace AJ Jr, Holbrook NJ. Serine protease inhibitor TPCK prevents Taxol-induced cell death and blocks c-Raf-1 and Bcl-2 phosphorylation in human breast carcinoma cells. Oncogene. 1999;18:3431–9.
Gringhuis SI, den Dunnen J, Litjens M, van der Vlist M, Wevers B, Bruijns SC, Geijtenbeek TB. Dectin-1 directs T helper cell differentiation by controlling noncanonical NF-kappaB activation through Raf-1 and Syk. Nat Immunol. 2009;10:203–13.
Li S, Sedivy JM. Raf-1 protein kinase activates the NF-kappa B transcription factor by dissociating the cytoplasmic NF-kappa B-I kappa B complex. Proc Natl Acad Sci U S A. 1993;90:9247–51.
Gringhuis SI, den Dunnen J, Litjens M, van Het Hof B, van Kooyk Y, Geijtenbeek TB. C-type lectin DC-SIGN modulates Toll-like receptor signaling via Raf-1 kinase-dependent acetylation of transcription factor NF-kappaB. Immunity. 2007;26:605–16.
Riegel K, Schloder J, Sobczak M, Jonuleit H, Thiede B, Schild H, Rajalingam K. RAF kinases are stabilized and required for dendritic cell differentiation and function. Cell Death Differ. 2020;27:1300–15.
Mielgo A, Seguin L, Huang M, Camargo MF, Anand S, Franovic A, Weis SM, Advani SJ, Murphy EA, Cheresh DA. A MEK-independent role for CRAF in mitosis and tumor progression. Nat Med. 2011;17:1641–5.
Mo Z, Ding H, Zhou X, Zeng Z, Long L. Gd-EOB-DTPA-enhanced magnetic resonance imaging may help identify patients with hepatocellular carcinoma eligible for treatment targeted at RAF1. Abdom Radiol (NY). 2021;47(1):209–20.
Ehrenreiter K, Piazzolla D, Velamoor V, Sobczak I, Small JV, Takeda J, Leung T, Baccarini M. Raf-1 regulates Rho signaling and cell migration. J Cell Biol. 2005;168:955–64.
Niault T, Sobczak I, Meissl K, Weitsman G, Piazzolla D, Maurer G, Kern F, Ehrenreiter K, Hamerl M, Moarefi I, et al. From autoinhibition to inhibition in trans: the Raf-1 regulatory domain inhibits Rok-alpha kinase activity. J Cell Biol. 2009;187:335–42.
Varga A, Ehrenreiter K, Aschenbrenner B, Kocieniewski P, Kochanczyk M, Lipniacki T, Baccarini M. RAF1/BRAF dimerization integrates the signal from RAS to ERK and ROKalpha. Sci Signal. 2017;10(469):eaai8482.
Johnson JL, Pillai S, Pernazza D, Sebti SM, Lawrence NJ, Chellappan SP. Regulation of matrix metalloproteinase genes by E2F transcription factors: Rb-Raf-1 interaction as a novel target for metastatic disease. Cancer Res. 2012;72:516–26.
Hwang YH, Choi JY, Kim S, Chung ES, Kim T, Koh SS, Lee B, Bae SH, Kim J, Park YM. Over-expression of c-raf-1 proto-oncogene in liver cirrhosis and hepatocellular carcinoma. Hepatol Res. 2004;29:113–21.
Tian H, Yin L, Ding K, Xia YY, Wang XH, Wu JZ, He X. Raf1 is a prognostic factor for progression in patients with non-small cell lung cancer after radiotherapy. Oncol Rep. 2018;39:1966–74.
Cekanova M, Majidy M, Masi T, Al-Wadei HA, Schuller HM. Overexpressed Raf-1 and phosphorylated cyclic adenosine 3’-5’-monophosphatate response element-binding protein are early markers for lung adenocarcinoma. Cancer. 2007;109:1164–73.
Kiefer PE, Bepler G, Kubasch M, Havemann K. Amplification and expression of protooncogenes in human small cell lung cancer cell lines. Cancer Res. 1987;47:6236–42.
Kiefer PE, Wegmann B, Bacher M, Erbil C, Heidtmann H, Havemann K. Different pattern of expression of cellular oncogenes in human non-small-cell lung cancer cell lines. J Cancer Res Clin Oncol. 1990;116:29–37.
Mhawech-Fauceglia P, Fischer G, Beck A, Cheney RT, Herrmann FR. Raf1, Aurora-A/STK15 and E-cadherin biomarkers expression in patients with pTa/pT1 urothelial bladder carcinoma; a retrospective TMA study of 246 patients with long-term follow-up. Eur J Surg Oncol. 2006;32:439–44.
Emuss V, Garnett M, Mason C, Marais R. Mutations of C-RAF are rare in human cancer because C-RAF has a low basal kinase activity compared with B-RAF. Cancer Res. 2005;65:9719–26.
Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–9.
Bekele RT, Samant AS, Nassar AH, So J, Garcia EP, Curran CR, Hwang JH, Mayhew DL, Nag A, Thorner AR, et al. RAF1 amplification drives a subset of bladder tumors and confers sensitivity to MAPK-directed therapeutics. J Clin Invest. 2021;131(22):e147849.
Zhao Y, Adjei AA. The clinical development of MEK inhibitors. Nat Rev Clin Oncol. 2014;11:385–400.
Jain P, Fierst TM, Han HJ, Smith TE, Vakil A, Storm PB, Resnick AC, Waanders AJ. CRAF gene fusions in pediatric low-grade gliomas define a distinct drug response based on dimerization profiles. Oncogene. 2017;36:6348–58.
Nussinov R, Tsai CJ, Jang H. Anticancer drug resistance: An update and perspective. Drug Resist Updat. 2021;59: 100796.
Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, Johnson ML, Heist RS, Patil T, Riely GJ, et al. Acquired Resistance to KRAS(G12C) Inhibition in Cancer. N Engl J Med. 2021;384:2382–93.
Nakama K, Ogata D, Nakano E, Tsutsui K, Jinnai S, Namikawa K, Takahashi A, Yamazaki N. Clinical response to a MEK inhibitor in a patient with metastatic melanoma harboring an RAF1 gene rearrangement detected by cancer gene panel testing. J Dermatol. 2021;48:e256–7.
Wilson MA, Zhao F, Khare S, Roszik J, Woodman SE, D’Andrea K, Wubbenhorst B, Rimm DL, Kirkwood JM, Kluger HM, et al. Copy Number Changes Are Associated with Response to Treatment with Carboplatin, Paclitaxel, and Sorafenib in Melanoma. Clin Cancer Res. 2016;22:374–82.
Tan WJ, Lai JC, Thike AA, Lim JC, Tan SY, Koh VC, Lim TH, Bay BH, Tan MH, Tan PH. Novel genetic aberrations in breast phyllodes tumours: comparison between prognostically distinct groups. Breast Cancer Res Treat. 2014;145:635–45.
Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–8.
Wang F, Jiang C, Sun Q, Yan F, Wang L, Fu Z, Liu T, Hu F. miR-195 is a key regulator of Raf1 in thyroid cancer. Onco Targets Ther. 2015;8:3021–8.
Chen L, Wang Q, Wang GD, Wang HS, Huang Y, Liu XM, Cai XH. miR-16 inhibits cell proliferation by targeting IGF1R and the Raf1-MEK1/2-ERK1/2 pathway in osteosarcoma. FEBS Lett. 2013;587:1366–72.
Gheidari F, Arefian E, Adegani FJ, Kalhori MR, Seyedjafari E, Kabiri M, Teimoori-Toolabi L, Soleimani M. miR-424 induces apoptosis in glioblastoma cells and targets AKT1 and RAF1 oncogenes from the ERBB signaling pathway. Eur J Pharmacol. 2021;906: 174273.
Ghousein A, Mosca N, Cartier F, Charpentier J, Dupuy JW, Raymond AA, Bioulac-Sage P, Grosset CF. miR-4510 blocks hepatocellular carcinoma development through RAF1 targeting and RAS/RAF/MEK/ERK signalling inactivation. Liver Int. 2020;40:240–51.
Li G, Kong Q. LncRNA LINC00460 promotes the papillary thyroid cancer progression by regulating the LINC00460/miR-485-5p/Raf1 axis. Biol Res. 2019;52:61.
Yang M, Qin Q, Zhu J, Guo Y, Yin T, Wu H, Wang C. Long noncoding RNA ITGB2-AS1 promotes growth and metastasis through miR-4319/RAF1 axis in pancreatic ductal adenocarcinoma. J Cell Physiol. 2020;235:1–14.
Chen X, Wang J, Xie F, Mou T, Zhong P, Hua H, Liu P, Yang Q. Long noncoding RNA LINC01559 promotes pancreatic cancer progression by acting as a competing endogenous RNA of miR-1343-3p to upregulate RAF1 expression. Aging (Albany NY). 2020;12:14452–66.
Weng W, Wei Q, Toden S, Yoshida K, Nagasaka T, Fujiwara T, Cai S, Qin H, Ma Y, Goel A. Circular RNA ciRS-7-A Promising Prognostic Biomarker and a Potential Therapeutic Target in Colorectal Cancer. Clin Cancer Res. 2017;23:3918–28.
Wu F, Zhou J. CircAGFG1 promotes cervical cancer progression via miR-370-3p/RAF1 signaling. BMC Cancer. 2019;19:1067.
Zhang B, Li F, Zhu Z, Ding A, Luo J. CircRNA CDR1as/miR-1287/Raf1 Axis Modulates Hepatocellular Carcinoma Progression Through MEK/ERK Pathway. Cancer Manag Res. 2020;12:8951–64.
Qu J, Li J, Chen K, Qin D, Li K, Sheng Y, Zou C, Wang S, Huang A, Tang H. Hepatitis B virus regulation of Raf1 promoter activity through activation of transcription factor AP-2alpha. Arch Virol. 2013;158:887–94.
Bai D, Zhou Y, Shen F, Gao D, Suo W, Zhang H, Li H. BPTF activates the MAPK pathway through coexpression with Raf1 to promote proliferation of T-cell lymphoma. Oncol Lett. 2022;24:223.
Chang CJ, Yang JY, Xia W, Chen CT, Xie X, Chao CH, Woodward WA, Hsu JM, Hortobagyi GN, Hung MC. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer Cell. 2011;19:86–100.
Kleemann M, Schneider H, Unger K, Bereuther J, Fischer S, Sander P, Marion Schneider E, Fischer-Posovszky P, Riedel CU, Handrick R, Otte K. Induction of apoptosis in ovarian cancer cells by miR-493-3p directly targeting AKT2, STK38L, HMGA2, ETS1 and E2F5. Cell Mol Life Sci. 2019;76:539–59.
Iorns E, Turner NC, Elliott R, Syed N, Garrone O, Gasco M, Tutt AN, Crook T, Lord CJ, Ashworth A. Identification of CDK10 as an important determinant of resistance to endocrine therapy for breast cancer. Cancer Cell. 2008;13:91–104.
McTavish CJ, Berube-Janzen W, Wang X, Maitland MER, Salemi LM, Hess DA, Schild-Poulter C. Regulation of c-Raf Stability through the CTLH Complex. Int J Mol Sci. 2019;20(4):934.
Wang X, Wang X, Zhang X, Zhang Y, Zhu Z, Li Y, Zhang M, Ji J, Yu Y, Ye SD. Inhibition of ubiquitin-specific protease 13-mediated degradation of Raf1 kinase by Spautin-1 has opposing effects in naive and primed pluripotent stem cells. J Biol Chem. 2021;297: 101332.
Oberoi-Khanuja TK, Karreman C, Larisch S, Rapp UR, Rajalingam K. Role of melanoma inhibitor of apoptosis (ML-IAP) protein, a member of the baculoviral IAP repeat (BIR) domain family, in the regulation of C-RAF kinase and cell migration. J Biol Chem. 2012;287:28445–55.
Hayes SD, Liu H, MacDonald E, Sanderson CM, Coulson JM, Clague MJ, Urbe S. Direct and indirect control of mitogen-activated protein kinase pathway-associated components, BRAP/IMP E3 ubiquitin ligase and CRAF/RAF1 kinase, by the deubiquitylating enzyme USP15. J Biol Chem. 2012;287:43007–18.
Feng D, Sheng-Dong L, Tong W, Zhen-Xian D. O-GlcNAcylation of RAF1 increases its stabilization and induces the renal fibrosis. Biochim Biophys Acta Mol Basis Dis. 2020;1866: 165556.
Andreu-Perez P, Esteve-Puig R, de Torre-Minguela C, Lopez-Fauqued M, Bech-Serra JJ, Tenbaum S, Garcia-Trevijano ER, Canals F, Merlino G, Avila MA, Recio JA. Protein arginine methyltransferase 5 regulates ERK1/2 signal transduction amplitude and cell fate through CRAF. Sci Signal. 2011;4:ra58.
Wong TL, Ng KY, Tan KV, Chan LH, Zhou L, Che N, Hoo RLC, Lee TK, Richard S, Lo CM, et al. CRAF Methylation by PRMT6 Regulates Aerobic Glycolysis-Driven Hepatocarcinogenesis via ERK-Dependent PKM2 Nuclear Relocalization and Activation. Hepatology. 2020;71:1279–96.
Chan LH, Zhou L, Ng KY, Wong TL, Lee TK, Sharma R, Loong JH, Ching YP, Yuan YF, Xie D, et al. PRMT6 Regulates RAS/RAF Binding and MEK/ERK-Mediated Cancer Stemness Activities in Hepatocellular Carcinoma through CRAF Methylation. Cell Rep. 2018;25:690–701 (e698).
Qu C, Park JY, Yun MW, He QT, Yang F, Kim K, Ham D, Li RR, Iverson TM, Gurevich VV, et al. Scaffolding mechanism of arrestin-2 in the cRaf/MEK1/ERK signaling cascade. Proc Natl Acad Sci U S A. 2021;118(37):e2026491118.
Tzivion G, Luo Z, Avruch J. A dimeric 14-3-3 protein is an essential cofactor for Raf kinase activity. Nature. 1998;394:88–92.
Hu J, Yu H, Kornev AP, Zhao J, Filbert EL, Taylor SS, Shaw AS. Mutation that blocks ATP binding creates a pseudokinase stabilizing the scaffolding function of kinase suppressor of Ras, CRAF and BRAF. Proc Natl Acad Sci U S A. 2011;108:6067–72.
Kaplan FM, Kugel CH 3rd, Dadpey N, Shao Y, Abel EV, Aplin AE. SHOC2 and CRAF mediate ERK1/2 reactivation in mutant NRAS-mediated resistance to RAF inhibitor. J Biol Chem. 2012;287:41797–807.
Michaud NR, Therrien M, Cacace A, Edsall LC, Spiegel S, Rubin GM, Morrison DK. KSR stimulates Raf-1 activity in a kinase-independent manner. Proc Natl Acad Sci U S A. 1997;94:12792–6.
Shi F, Lemmon MA. Biochemistry. KSR plays CRAF-ty. Science. 2011;332:1043–4.
McKay MM, Freeman AK, Morrison DK. Complexity in KSR function revealed by Raf inhibitor and KSR structure studies. Small GTPases. 2011;2:276–81.
Dasgupta P, Rastogi S, Pillai S, Ordonez-Ercan D, Morris M, Haura E, Chellappan S. Nicotine induces cell proliferation by beta-arrestin-mediated activation of Src and Rb-Raf-1 pathways. J Clin Invest. 2006;116:2208–17.
Rodriguez-Viciana P, Oses-Prieto J, Burlingame A, Fried M, McCormick F. A phosphatase holoenzyme comprised of Shoc2/Sur8 and the catalytic subunit of PP1 functions as an M-Ras effector to modulate Raf activity. Mol Cell. 2006;22:217–30.
Jin L, Chun J, Pan C, Li D, Lin R, Alesi GN, Wang X, Kang HB, Song L, Wang D, et al. MAST1 Drives Cisplatin Resistance in Human Cancers by Rewiring cRaf-Independent MEK Activation. Cancer Cell. 2018;34(315–330): e317.
Ding H, Peterson KL, Correia C, Koh B, Schneider PA, Nowakowski GS, Kaufmann SH. Histone deacetylase inhibitors interrupt HSP90*RASGRP1 and HSP90*CRAF interactions to upregulate BIM and circumvent drug resistance in lymphoma cells. Leukemia. 2017;31:1593–602.
Ziogas A, Moelling K, Radziwill G. CNK1 is a scaffold protein that regulates Src-mediated Raf-1 activation. J Biol Chem. 2005;280:24205–11.
Qi ZH, Xu HX, Zhang SR, Xu JZ, Li S, Gao HL, Jin W, Wang WQ, Wu CT, Ni QX, et al. RIPK4/PEBP1 axis promotes pancreatic cancer cell migration and invasion by activating RAF1/MEK/ERK signaling. Int J Oncol. 2018;52:1105–16.
Manne BK, Munzer P, Badolia R, Walker-Allgaier B, Campbell RA, Middleton E, Weyrich AS, Kunapuli SP, Borst O, Rondina MT. PDK1 governs thromboxane generation and thrombosis in platelets by regulating activation of Raf1 in the MAPK pathway. J Thromb Haemost. 2018;16:1211–25.
Maity G, Haque I, Ghosh A, Dhar G, Gupta V, Sarkar S, Azeem I, McGregor D, Choudhary A, Campbell DR, et al. The MAZ transcription factor is a downstream target of the oncoprotein Cyr61/CCN1 and promotes pancreatic cancer cell invasion via CRAF-ERK signaling. J Biol Chem. 2018;293:4334–49.
Wu J, Ivanov AI, Fisher PB, Fu Z. Polo-like kinase 1 induces epithelial-to-mesenchymal transition and promotes epithelial cell motility by activating CRAF/ERK signaling. Elife. 2016;5:e10734.
Noble C, Mercer K, Hussain J, Carragher L, Giblett S, Hayward R, Patterson C, Marais R, Pritchard CA. CRAF autophosphorylation of serine 621 is required to prevent its proteasome-mediated degradation. Mol Cell. 2008;31:862–72.
Liu S, Wang W, Brown LE, Qiu C, Lajkiewicz N, Zhao T, Zhou J, Porco JA Jr, Wang TT. A Novel Class of Small Molecule Compounds that Inhibit Hepatitis C Virus Infection by Targeting the Prohibitin-CRaf Pathway. EBioMedicine. 2015;2:1600–6.
Josse R, Zhang YW, Giroux V, Ghosh AK, Luo J, Pommier Y. Activation of RAF1 (c-RAF) by the Marine Alkaloid Lasonolide A Induces Rapid Premature Chromosome Condensation. Mar Drugs. 2015;13:3625–39.
Advani SJ, Camargo MF, Seguin L, Mielgo A, Anand S, Hicks AM, Aguilera J, Franovic A, Weis SM, Cheresh DA. Kinase-independent role for CRAF-driving tumour radioresistance via CHK2. Nat Commun. 2015;6:8154.
Zang Y, Kahsai AW, Pakharukova N, Huang LY, Lefkowitz RJ. The GPCR-beta-arrestin complex allosterically activates C-Raf by binding its amino terminus. J Biol Chem. 2021;297: 101369.
Xu Y, Chen X, Pan S, Wang ZW, Zhu X. TM7SF2 regulates cell proliferation and apoptosis by activation of C-Raf/ERK pathway in cervical cancer. Cell Death Discov. 2021;7:299.
Wang X, Wu F, Wang H, Duan X, Huang R, Tuersuntuoheti A, Su L, Yan S, Zhao Y, Lu Y, et al. PDCD6 cooperates with C-Raf to facilitate colorectal cancer progression via Raf/MEK/ERK activation. J Exp Clin Cancer Res. 2020;39:147.
Hausherr CK, Schiffer IB, Gebhard S, Banic A, Tanner B, Kolbl H, Thoenes E, Beckers T, Spangenberg C, Prawitt D, et al. Dephosphorylation of p-ERK1/2 in relation to tumor remission after HER-2 and Raf1 blocking therapy in a conditional mouse tumor model. Mol Carcinog. 2006;45:302–8.
Adams DG, Coffee RL Jr, Zhang H, Pelech S, Strack S, Wadzinski BE. Positive regulation of Raf1-MEK1/2-ERK1/2 signaling by protein serine/threonine phosphatase 2A holoenzymes. J Biol Chem. 2005;280:42644–54.
Nussinov R, Jang H, Zhang M, Tsai CJ, Sablina AA. The Mystery of Rap1 Suppression of Oncogenic Ras. Trends Cancer. 2020;6:369–79.
Stork PJ, Dillon TJ. Multiple roles of Rap1 in hematopoietic cells: complementary versus antagonistic functions. Blood. 2005;106:2952–61.
Spirli C, Morell CM, Locatelli L, Okolicsanyi S, Ferrero C, Kim AK, Fabris L, Fiorotto R, Strazzabosco M. Cyclic AMP/PKA-dependent paradoxical activation of Raf/MEK/ERK signaling in polycystin-2 defective mice treated with sorafenib. Hepatology. 2012;56:2363–74.
Yuan Q, Dong CD, Ge Y, Chen X, Li Z, Li X, Lu Q, Peng F, Wu X, Zhao J, Liu K. Proteome and phosphoproteome reveal mechanisms of action of atorvastatin against esophageal squamous cell carcinoma. Aging (Albany NY). 2019;11:9530–43.
Dougherty MK, Muller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, Conrads TP, Veenstra TD, Lu KP, Morrison DK. Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell. 2005;17:215–24.
Xu B, Washington AM, Hinton BT. PTEN signaling through RAF1 proto-oncogene serine/threonine kinase (RAF1)/ERK in the epididymis is essential for male fertility. Proc Natl Acad Sci U S A. 2014;111:18643–8.
Zhao S, Jiang Y, Zhao J, Li H, Yin X, Wang Y, Xie Y, Chen X, Lu J, Dong Z, Liu K. Quercetin-3-methyl ether inhibits esophageal carcinogenesis by targeting the AKT/mTOR/p70S6K and MAPK pathways. Mol Carcinog. 2018;57:1540–52.
Mazalouskas MD, Godoy-Ruiz R, Weber DJ, Zimmer DB, Honkanen RE, Wadzinski BE. Small G proteins Rac1 and Ras regulate serine/threonine protein phosphatase 5 (PP5).extracellular signal-regulated kinase (ERK) complexes involved in the feedback regulation of Raf1. J Biol Chem. 2014;289:4219–32.
Wu Z, Fu C, Shi L, Ruan L, Lin D, Guo C. Structural basis for RKIP binding with its substrate Raf1 kinase. Biotechnol Lett. 2014;36:1869–74.
Parate S, Rampogu S, Lee G, Hong JC, Lee KW. Exploring the Binding Interaction of Raf Kinase Inhibitory Protein With the N-Terminal of C-Raf Through Molecular Docking and Molecular Dynamics Simulation. Front Mol Biosci. 2021;8: 655035.
Guo C, Wu Z, Lin W, Xu H, Chang T, Dai Y, Lin D. Suramin Targets the Conserved Ligand-Binding Pocket of Human Raf1 Kinase Inhibitory Protein. Molecules. 2021;26(4):1151.
Huang J, Hu W, Bottsford-Miller J, Liu T, Han HD, Zand B, Pradeep S, Roh JW, Thanapprapasr D, Dalton HJ, et al. Cross-talk between EphA2 and BRaf/CRaf is a key determinant of response to Dasatinib. Clin Cancer Res. 2014;20:1846–55.
Li X, Stevens PD, Liu J, Yang H, Wang W, Wang C, Zeng Z, Schmidt MD, Yang M, Lee EY, Gao T. PHLPP is a negative regulator of RAF1, which reduces colorectal cancer cell motility and prevents tumor progression in mice. Gastroenterology. 2014;146(1301–1312):e1301-1310.
Shukla A, Rai K, Shukla V, Chaturvedi NK, Bociek RG, Pirruccello SJ, Band H, Lu R, Joshi SS. Sprouty 2: a novel attenuator of B-cell receptor and MAPK-Erk signaling in CLL. Blood. 2016;127:2310–21.
Rajakulendran T, Sahmi M, Lefrancois M, Sicheri F, Therrien M. A dimerization-dependent mechanism drives RAF catalytic activation. Nature. 2009;461:542–5.
Kwong LN, Chin L. The brothers RAF. Cell. 2010;140:180–2.
Taylor SS, Kornev AP. Protein kinases: evolution of dynamic regulatory proteins. Trends Biochem Sci. 2011;36:65–77.
Baljuls A, Mahr R, Schwarzenau I, Muller T, Polzien L, Hekman M, Rapp UR. Single substitution within the RKTR motif impairs kinase activity but promotes dimerization of RAF kinase. J Biol Chem. 2011;286:16491–503.
Hu J, Stites EC, Yu H, Germino EA, Meharena HS, Stork PJS, Kornev AP, Taylor SS, Shaw AS. Allosteric activation of functionally asymmetric RAF kinase dimers. Cell. 2013;154:1036–46.
Jambrina PG, Rauch N, Pilkington R, Rybakova K, Nguyen LK, Kholodenko BN, Buchete NV, Kolch W, Rosta E. Phosphorylation of RAF Kinase Dimers Drives Conformational Changes that Facilitate Transactivation. Angew Chem Int Ed Engl. 2016;55:983–6.
Haling JR, Sudhamsu J, Yen I, Sideris S, Sandoval W, Phung W, Bravo BJ, Giannetti AM, Peck A, Masselot A, et al. Structure of the BRAF-MEK complex reveals a kinase activity independent role for BRAF in MAPK signaling. Cancer Cell. 2014;26:402–13.
Brennan DF, Dar AC, Hertz NT, Chao WC, Burlingame AL, Shokat KM, Barford D. A Raf-induced allosteric transition of KSR stimulates phosphorylation of MEK. Nature. 2011;472:366–9.
Yuan J, Ng WH, Lam PYP, Wang Y, Xia H, Yap J, Guan SP, Lee ASG, Wang M, Baccarini M, Hu J. The dimer-dependent catalytic activity of RAF family kinases is revealed through characterizing their oncogenic mutants. Oncogene. 2018;37:5719–34.
Yuan J, Ng WH, Tian Z, Yap J, Baccarini M, Chen Z, Hu J. Activating mutations in MEK1 enhance homodimerization and promote tumorigenesis. Sci Signal. 2018;11(554):eaar6795.
Lavoie H, Sahmi M, Maisonneuve P, Marullo SA, Thevakumaran N, Jin T, Kurinov I, Sicheri F, Therrien M. MEK drives BRAF activation through allosteric control of KSR proteins. Nature. 2018;554:549–53.
Hou J, Li L, Dong D, Wang L, Wang X, Yang K, Xu X, Chen C, Wu X, Chen X. Glycomolecules in Echinococcus granulosus cyst fluid inhibit TLR4-mediated inflammatory responses via c-Raf. Cell Mol Immunol. 2020;17:423–5.
Kebache S, Ash J, Annis MG, Hagan J, Huber M, Hassard J, Stewart CL, Whiteway M, Nantel A. Grb10 and active Raf-1 kinase promote Bad-dependent cell survival. J Biol Chem. 2007;282:21873–83.
Feng R, Gong J, Wu L, Wang L, Zhang B, Liang G, Zheng H, Xiao H. MAPK and Hippo signaling pathways crosstalk via the RAF-1/MST-2 interaction in malignant melanoma. Oncol Rep. 2017;38:1199–205.
Piazzolla D, Meissl K, Kucerova L, Rubiolo C, Baccarini M. Raf-1 sets the threshold of Fas sensitivity by modulating Rok-alpha signaling. J Cell Biol. 2005;171:1013–22.
Doma E, Rupp C, Varga A, Kern F, Riegler B, Baccarini M. Skin tumorigenesis stimulated by Raf inhibitors relies upon Raf functions that are dependent and independent of ERK. Cancer Res. 2013;73:6926–37.
Li S, Liu J, Tan J, Li L, Kaltreider MJ, Zhao J, Kass DJ, Shang D, Zhao Y. Inhibition of Raf1 ameliorates bleomycin-induced pulmonary fibrosis through attenuation of TGF-beta1 signaling. Am J Physiol Lung Cell Mol Physiol. 2018;315:L241–7.
Baumann B, Weber CK, Troppmair J, Whiteside S, Israel A, Rapp UR, Wirth T. Raf induces NF-kappaB by membrane shuttle kinase MEKK1, a signaling pathway critical for transformation. Proc Natl Acad Sci U S A. 2000;97:4615–20.
Mangana J, Levesque MP, Karpova MB, Dummer R. Sorafenib in melanoma. Expert Opin Investig Drugs. 2012;21:557–68.
Jo S, Jung Y, Cho Y, Seo J, Lim W, Nam T, Lim T, Byun S. Rosa gallicaOral Administration of Prevents UVB-Induced Skin Aging through Targeting the c-Raf Signaling Axis. Antioxidants (Basel, Switzerland). 2021;10(11):1663.
Yang SC, Chen PJ, Chang SH, Weng YT, Chang FR, Chang KY, Chen CY, Kao TI, Hwang TL. Luteolin attenuates neutrophilic oxidative stress and inflammatory arthritis by inhibiting Raf1 activity. Biochem Pharmacol. 2018;154:384–96.
Hood JD, Bednarski M, Frausto R, Guccione S, Reisfeld RA, Xiang R, Cheresh DA. Tumor regression by targeted gene delivery to the neovasculature. Science. 2002;296:2404–7.
Wang B, Yang H, Liu YC, Jelinek T, Zhang L, Ruoslahti E, Fu H. Isolation of high-affinity peptide antagonists of 14-3-3 proteins by phage display. Biochemistry. 1999;38:12499–504.
Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer. 2010;10:537–49.
Simanshu DK, Nissley DV, McCormick F. RAS Proteins and Their Regulators in Human Disease. Cell. 2017;170:17–33.
Hobbs GA, Der CJ, Rossman KL. RAS isoforms and mutations in cancer at a glance. J Cell Sci. 2016;129:1287–92.
Moore AR, Rosenberg SC, McCormick F, Malek S. RAS-targeted therapies: is the undruggable drugged? Nat Rev Drug Discov. 2020;19:533–52.
Hong DS, Fakih MG, Strickler JH, Desai J, Durm GA, Shapiro GI, Falchook GS, Price TJ, Sacher A, Denlinger CS, et al. KRAS(G12C) Inhibition with Sotorasib in Advanced Solid Tumors. N Engl J Med. 2020;383:1207–17.
Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol Cancer Res. 2015;13:1325–35.
Buyanova M, Cai S, Cooper J, Rhodes C, Salim H, Sahni A, Upadhyaya P, Yang R, Sarkar A, Li N, et al. Discovery of a Bicyclic Peptidyl Pan-Ras Inhibitor. J Med Chem. 2021;64:13038–53.
Athuluri-Divakar SK, Vasquez-Del Carpio R, Dutta K, Baker SJ, Cosenza SC, Basu I, Gupta YK, Reddy MV, Ueno L, Hart JR, et al. A Small Molecule RAS-Mimetic Disrupts RAS Association with Effector Proteins to Block Signaling. Cell. 2016;165:643–55.
Dalle S, Poulalhon N, Thomas L. Vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;365:1448–9 (author reply 1450).
Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH Jr, Kaempgen E, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012;380:358–65.
Hertzman Johansson C, Egyhazi Brage S. BRAF inhibitors in cancer therapy. Pharmacol Ther. 2014;142:176–82.
Arora R, Di Michele M, Stes E, Vandermarliere E, Martens L, Gevaert K, Van Heerde E, Linders JT, Brehmer D, Jacoby E, Bonnet P. Structural investigation of B-Raf paradox breaker and inducer inhibitors. J Med Chem. 2015;58:1818–31.
Cook FA, Cook SJ. Inhibition of RAF dimers: it takes two to tango. Biochem Soc Trans. 2021;49:237–51.
Yao Z, Gao Y, Su W, Yaeger R, Tao J, Na N, Zhang Y, Zhang C, Rymar A, Tao A, et al. RAF inhibitor PLX8394 selectively disrupts BRAF dimers and RAS-independent BRAF-mutant-driven signaling. Nature medicine. 2019;25:284–91.
Shao W, Mishina YM, Feng Y, Caponigro G, Cooke VG, Rivera S, Wang Y, Shen F, Korn JM, Mathews Griner LA, et al. Antitumor Properties of RAF709, a Highly Selective and Potent Inhibitor of RAF Kinase Dimers, in Tumors Driven by Mutant RAS or BRAF. Cancer Res. 2018;78:1537–48.
Tkacik E, Li K, Gonzalez-Del Pino G, Ha BH, Vinals J, Park E, Beyett TS, Eck MJ. Structure and RAF family kinase isoform selectivity of type II RAF inhibitors tovorafenib and naporafenib. J Biol Chem. 2023;299: 104634.
BGB-283 Deemed Effective in Phase I Study. Cancer Discov. 2016;6(7):OF1.
Durrant DE, Morrison DK. Targeting the Raf kinases in human cancer: the Raf dimer dilemma. Br J Cancer. 2018;118:3–8.
Girotti MR, Lopes F, Preece N, Niculescu-Duvaz D, Zambon A, Davies L, Whittaker S, Saturno G, Viros A, Pedersen M, et al. Paradox-breaking RAF inhibitors that also target SRC are effective in drug-resistant BRAF mutant melanoma. Cancer Cell. 2015;27:85–96.
Lito P, Pratilas CA, Joseph EW, Tadi M, Halilovic E, Zubrowski M, Huang A, Wong WL, Callahan MK, Merghoub T, et al. Relief of profound feedback inhibition of mitogenic signaling by RAF inhibitors attenuates their activity in BRAFV600E melanomas. Cancer Cell. 2012;22:668–82.
Poulikakos PI, Persaud Y, Janakiraman M, Kong X, Ng C, Moriceau G, Shi H, Atefi M, Titz B, Gabay MT, et al. RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature. 2011;480:387–90.
Chen SH, Zhang Y, Van Horn RD, Yin T, Buchanan S, Yadav V, Mochalkin I, Wong SS, Yue YG, Huber L, et al. Oncogenic BRAF Deletions That Function as Homodimers and Are Sensitive to Inhibition by RAF Dimer Inhibitor LY3009120. Cancer Discov. 2016;6:300–15.
Hu J, Ahuja LG, Meharena HS, Kannan N, Kornev AP, Taylor SS, Shaw AS. Kinase regulation by hydrophobic spine assembly in cancer. Mol Cell Biol. 2015;35:264–76.
Yap J, Deepak R, Tian Z, Ng WH, Goh KC, Foo A, Tee ZH, Mohanam MP, Sim YRM, Degirmenci U, et al. The stability of R-spine defines RAF inhibitor resistance: A comprehensive analysis of oncogenic BRAF mutants with in-frame insertion of alphaC-beta4 loop. Sci Adv. 2021;7(24):eabg0390.
Wang L, Leite de Oliveira R, Huijberts S, Bosdriesz E, Pencheva N, Brunen D, Bosma A, Song JY, Zevenhoven J, Los-de Vries GT, et al. An Acquired Vulnerability of Drug-Resistant Melanoma with Therapeutic Potential. Cell. 2018;173:1413–25 (e1414).
Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu X, Gimotty PA, Kee D, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18:683–95.
Su F, Bradley WD, Wang Q, Yang H, Xu L, Higgins B, Kolinsky K, Packman K, Kim MJ, Trunzer K, et al. Resistance to selective BRAF inhibition can be mediated by modest upstream pathway activation. Cancer Res. 2012;72:969–78.
Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–7.
Emery CM, Vijayendran KG, Zipser MC, Sawyer AM, Niu L, Kim JJ, Hatton C, Chopra R, Oberholzer PA, Karpova MB, et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc Natl Acad Sci U S A. 2009;106:20411–6.
Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014;4:94–109.
Spagnolo F, Ghiorzo P, Orgiano L, Pastorino L, Picasso V, Tornari E, Ottaviano V, Queirolo P. BRAF-mutant melanoma: treatment approaches, resistance mechanisms, and diagnostic strategies. Onco Targets Ther. 2015;8:157–68.
Johannessen CM, Boehm JS, Kim SY, Thomas SR, Wardwell L, Johnson LA, Emery CM, Stransky N, Cogdill AP, Barretina J, et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–72.
Liu T, Zhou L, Xiao Y, Andl T, Zhang Y. BRAF Inhibitors Reprogram Cancer-Associated Fibroblasts to Drive Matrix Remodeling and Therapeutic Escape in Melanoma. Cancer Res. 2022;82:419–32.
Karoulia Z, Gavathiotis E, Poulikakos P. New perspectives for targeting RAF kinase in human cancer. Nature reviews Cancer. 2017;17:676–91.
Schadendorf D, Fisher DE, Garbe C, Gershenwald JE, Grob JJ, Halpern A, Herlyn M, Marchetti MA, McArthur G, Ribas A, et al. Melanoma. Nat Rev Dis Primers. 2015;1:15003.
Huestis MP, Durk MR, Eigenbrot C, Gibbons P, Hunsaker TL, La H, Leung DH, Liu W, Malek S, Merchant M, et al. Targeting KRAS Mutant Cancers via Combination Treatment: Discovery of a Pyridopyridazinone pan-RAF Kinase Inhibitor. ACS Med Chem Lett. 2021;12:791–7.
Huestis MP, Dela Cruz D, DiPasquale AG, Durk MR, Eigenbrot C, Gibbons P, Gobbi A, Hunsaker TL, La H, Leung DH, et al. Targeting KRAS Mutant Cancers via Combination Treatment: Discovery of a 5-Fluoro-4-(3H)-quinazolinone Aryl Urea pan-RAF Kinase Inhibitor. J Med Chem. 2021;64:3940–55.
Romany A, Liu R, Zhan S, Clayton J, Shen J. Analysis of the ERK Pathway Cysteinome for Targeted Covalent Inhibition of RAF and MEK Kinases. J Chem Inf Model. 2023;63:2483–94.
Ardito CM, Gruner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, Delgiorno KE, Carpenter ES, Halbrook CJ, Hall JC, et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell. 2012;22:304–17.
Navas C, Hernandez-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22:318–30.
Okimoto RA, Lin L, Olivas V, Chan E, Markegard E, Rymar A, Neel D, Chen X, Hemmati G, Bollag G, Bivona TG. Preclinical efficacy of a RAF inhibitor that evades paradoxical MAPK pathway activation in protein kinase BRAF-mutant lung cancer. Proc Natl Acad Sci U S A. 2016;113:13456–61.
Desai J, Gan H, Barrow C, Jameson M, Atkinson V, Haydon A, Millward M, Begbie S, Brown M, Markman B, et al. Phase I, Open-Label, Dose-Escalation/Dose-Expansion Study of Lifirafenib (BGB-283), an RAF Family Kinase Inhibitor, in Patients With Solid Tumors. J Clin Oncol. 2020;38:2140–50.
Atkins MB, Lee SJ, Chmielowski B, Tarhini AA, Cohen GI, Truong TG, Moon HH, Davar D, O’Rourke M, Stephenson JJ, et al. Combination Dabrafenib and Trametinib Versus Combination Nivolumab and Ipilimumab for Patients With Advanced BRAF-Mutant Melanoma: The DREAMseq Trial-ECOG-ACRIN EA6134. J Clin Oncol. 2023;41:186–97.
Park S, Kim TM, Cho SY, Kim S, Oh Y, Kim M, Keam B, Kim DW, Heo DS. Combined blockade of polo-like kinase and pan-RAF is effective against NRAS-mutant non-small cell lung cancer cells. Cancer Lett. 2020;495:135–44.
Trevino JG, Verma M, Singh S, Pillai S, Zhang D, Pernazza D, Sebti SM, Lawrence NJ, Centeno BA, Chellappan SP. Selective disruption of rb-raf-1 kinase interaction inhibits pancreatic adenocarcinoma growth irrespective of gemcitabine sensitivity. Mol Cancer Ther. 2013;12:2722–34.
Wang Z, Yin M, Chu P, Lou M. STAT3 inhibitor sensitized KRAS-mutant lung cancers to RAF inhibitor by activating MEK/ERK signaling pathway. Aging (Albany NY). 2019;11:7187–96.
Mordant P, Loriot Y, Leteur C, Calderaro J, Bourhis J, Wislez M, Soria JC, Deutsch E. Dependence on phosphoinositide 3-kinase and RAS-RAF pathways drive the activity of RAF265, a novel RAF/VEGFR2 inhibitor, and RAD001 (Everolimus) in combination. Mol Cancer Ther. 2010;9:358–68.
Jin N, Jiang T, Rosen DM, Nelkin BD, Ball DW. Synergistic action of a RAF inhibitor and a dual PI3K/mTOR inhibitor in thyroid cancer. Clin Cancer Res. 2011;17:6482–9.
Wu W, Xu J, Gao D, Xie Z, Chen W, Li W, Yuan Q, Duan L, Zhang Y, Yang X, et al. TOPK promotes the growth of esophageal cancer in vitro and in vivo by enhancing YB1/eEF1A1 signal pathway. Cell Death Dis. 2023;14:364.
Jiang Y, Zhang J, Zhao J, Li Z, Chen H, Qiao Y, Chen X, Liu K, Dong Z. TOPK promotes metastasis of esophageal squamous cell carcinoma by activating the Src/GSK3beta/STAT3 signaling pathway via gamma-catenin. BMC Cancer. 2019;19:1264.
Gao G, Zhang T, Wang Q, Reddy K, Chen H, Yao K, Wang K, Roh E, Zykova T, Ma W, et al. ADA-07 Suppresses Solar Ultraviolet-Induced Skin Carcinogenesis by Directly Inhibiting TOPK. Mol Cancer Ther. 2017;16:1843–54.
Lau KS, Zhang T, Kendall KR, Lauffenburger D, Gray NS, Haigis KM. BAY61-3606 affects the viability of colon cancer cells in a genotype-directed manner. PLoS One. 2012;7: e41343.
Chow AK, Cheng NS, Lam CS, Ng L, Wong SK, Wan TM, Man JH, Cheung AH, Yau TC, Poon JT, et al. Preclinical analysis of the anti-tumor and anti-metastatic effects of Raf265 on colon cancer cells and CD26(+) cancer stem cells in colorectal carcinoma. Mol Cancer. 2015;14:80.
Alabi S, Jaime-Figueroa S, Yao Z, Gao Y, Hines J, Samarasinghe KTG, Vogt L, Rosen N, Crews CM. Mutant-selective degradation by BRAF-targeting PROTACs. Nat Commun. 2021;12:920.
Acknowledgments
We would like to acknowledge the assistance of BioRender in creating cartoon illustrations.
Funding
This work was supported by the National Natural Science Foundations of China (No. 81872335) and the Central Plains Science and Technology Innovation Leading Talents (No. 224200510015).
Author information
Authors and Affiliations
Contributions
All authors made substantial contributions to the review. P.L.W organized and wrote this review, K.L and X.C.J contributed substantially to check the language and corresponding authors Z.G.D and K.D.L provided editorial assistance.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, P., Laster, K., Jia, X. et al. Targeting CRAF kinase in anti-cancer therapy: progress and opportunities. Mol Cancer 22, 208 (2023). https://doi.org/10.1186/s12943-023-01903-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-023-01903-x