
Abstract
Malignant brain tumors rank among the most challenging type of malignancies to manage. The current treatment protocol commonly entails surgery followed by radiotherapy and/or chemotherapy, however, the median patient survival rate is poor. Recent developments in immunotherapy for a variety of tumor types spark optimism that immunological strategies may help patients with brain cancer. Chimeric antigen receptor (CAR) T cells exploit the tumor-targeting specificity of antibodies or receptor ligands to direct the cytolytic capacity of T cells. Several molecules have been discovered as potential targets for immunotherapy-based targeting, including but not limited to EGFRvIII, IL13Rα2, and HER2. The outstanding clinical responses to CAR T cell-based treatments in patients with hematological malignancies have generated interest in using this approach to treat solid tumors. Research results to date support the astounding clinical response rates of CD19-targeted CAR T cells, early clinical experiences in brain tumors demonstrating safety and evidence for disease-modifying activity, and the promise for further advances to ultimately assist patients clinically. However, several variable factors seem to slow down the progress rate regarding treating brain cancers utilizing CAR T cells. The current study offers a thorough analysis of CAR T cells’ promise in treating brain cancer, including design and delivery considerations, current strides in clinical and preclinical research, issues encountered, and potential solutions.
Similar content being viewed by others

Introduction
Brain tumor, in general, is a mass/growth of abnormal brain cells. Malignant brain tumors can either originate primarily in the brain or as secondary or metastatic brain tumors that have metastasized from other types of cancer. Brain cancer patients usually survive only up to 5 years, that too with a significant compromise regarding their quality of life. Present therapeutic options are rarely curative and present with many undesired toxic effects. The success in T cell immunotherapy across a wide variety of tumor types has ignited the hope that the immune system can be strengthened to enhance outcomes for patients with brain tumors [1, 2]. Re-engineered T cells with a new selectivity towards specific tumor antigen(s) are emerging prospects for brain cancer management. One of the newest and most promising cancer treatments, chimeric antigen receptor (CAR) T cell therapy, boosts the body’s immune system to combat cancer. The advancement of CAR T treatment has been made possible by the convergence of gene sequencing, growing genetic knowledge, new methods of genome manipulation, and the development of novel gene transfer technologies. Prominent success for CAR T cell therapy was achieved earlier with lymphoma, leukemia, and other hematological cancers [3]. Relying upon the principle of specific antigen recognition, CAR T cells bear promise regarding the treatment of different solid tumors, including malignant tumors in the brain.
Access of the immune system to the brain is carefully regulated, and the immunosuppressive nature of CNS has developed to defend against immunologic attack. Approaches to boost endogenous T cell immune responses to treat brain tumors have been clinically proven to be effective despite significant difficulties. They are created by re-engineering T cells from the patient in the laboratory to produce proteins on their surface known as CARs. CARs recognize and bind to specific proteins on the surface of cancer cells. CAR-transplanted T cells gain the capacity to detect and lyse specific cancer cells. When used to treat tumors with minimal tumor mutational burden, such as the majority of brain tumors, CARs can target tumors without altering how the major histocompatibility complex (MHC) expresses its antigens [4, 5]. The fact that so many researchers are testing these strategies against brain cancer in both preclinical and clinical settings is a hint of their intriguing potential. The promise of CAR T cells in the treatment of brain cancer is emphasized in this review, along with considerations for their design and delivery, the recent therapeutic findings, issues encountered, and potential remedies.
Brain tumors: clinical constraints towards treatment
Brain tumors exhibit a poor prognosis and an extremely low patient survival rate. Combining the clinical features, genetic orientations, and degree of lethality, the World Health Organization (WHO) categorized CNS tumors into four grades [6]. Grade 1 refers to slow-growing benign tumors that heal after resection; grade 2 tumors are slow-growing tumors that often recur and sometimes tend to become malignant. Quickly proliferative grades 3 and 4 are histologically and cytologically malignant tumors, respectively. Every year, approximately 3 per 100,000 people worldwide acquire some form of brain tumors [7]. The classical therapeutic scheme of brain cancer includes surgical resection followed by chemotherapy and/or radiation therapy. However, a growing body of evidence revealed that the chances of relapse are very high for malignant brain tumors [8, 9].
The presence of the blood-brain barrier (BBB) limits the therapeutic efficacy of certain chemotherapeutic drugs by restricting their penetration into the brain. Endothelial tight junctions in combination with efflux transporters, such as P-glycoprotein (P-gp), peptide transport system 6 (PTS6), and breast cancer-resistant protein (BCRP) restrict the entry of most of the drug molecules into the CNS [8, 10]. Enzymes, such as aminopeptidases and endopeptidases potentially render the cargo ineffective. Moreover, binding with non-transporter proteins can minimize available drug(s). Angiogenic vasculatures originating from tumors make up the blood-brain tumor barrier (BBTB) that influences the chemotherapeutic potential of a drug by regulating tight endothelial connections; Moreover, extracellular matrices in the tumor microenvironment are frequently regulated by tumor cells to encourage chemoresistance, and tumor cell proliferation and differentiation. Summarily, the poor prognosis of brain cancer is a combined result of limited access to drug delivery cargos, multi-drug resistance, and the critical ability of residual malignant cells to recur.
Current therapeutic tools used to treat brain cancer suffer from certain limitations. In case of surgery, accurate delineation of tumor boundary is difficult for proper resection, thus complete removal of malignant cells is rarely achieved. Resistance, and inability to precisely reach the CNS resulting in non-specific toxicities to other organs and tissues apprehend promises of chemotherapy. Radiation tends to exert effects largely on peripheral cells only. In addition, similar to surgery, radiotherapy also suffers from inaccurate delineation. Immunotherapy has come up with promising outputs to boost the immune system and outperform immune suppression imposed by tumor cells [11, 12].
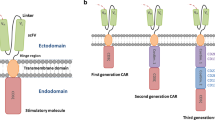
CAR T cells exhibit immunotherapeutic promise by utilizing the remarkable specificity of the antibody(ies) and/or target ligand(s) to guide the cytolytic ability of T cells. CARs mimic the canonical T cell pathway by virtue of the inclusion of either a single-chain variable fragment (ScFv) derived from a monoclonal antibody or a mutated ligand for target binding, one or more co-stimulatory domains, and the ξ chain of the CD3 complex within a single multi-domain receptor (Fig. 1) [13]. The introduction of CAR into T cells allows them to eliminate cells expressing specific target antigen(s) through the same effector functions utilized by wild-type T cells to kill infected or transformed cells (Fig. 1). CAR T cells can broadly be grouped into five generations based on how their intracellular signaling domains are organized, despite the fact that their essential modular structure has not changed since their inception (Fig. 1). The CD3ζ alone makes up the first generation of CARs, while additional co-stimulatory signaling domains are present in the second generation [14]. Two co-stimulatory domains are combined in the third generation. TRUCKs i.e. T cells redirected for antigen-unrestricted cytokine-initiated killing are fourth-generation CAR T cells that express chemokines such as (IL)-12 when activated. Compared to earlier generations, the fifth generation of CAR T cells contains an additional intracellular domain [15]. These CARs consist of truncated intracellular domains of cytokine receptors with a transcription factor-binding motif. The TRAC gene is inactivated in the fifth generation of CARs by virtue of gene editing methods, resulting in the removal of the T cell receptor α (TCR-α) and β (TCR-β) chains. Overexpression of CARs on endogenous T cells boosts their ability to target and kill specific cells.

Structural features of T cell receptor versus CAR T cell design. a Structural features of T cell receptor. b Production of CAR T cells from patient-derived T cells. c Structural features of CAR T cell design of different generations. The principal CAR structure includes an extracellular/binding domain, a transmembrane domain, and an intracellular/signaling domain. The extracellular portion of CAR is typically generated from a monoclonal antibody against the target, which is also known as a single-chain variable fragment (ScFv). The ScFv is affixed to the transmembrane domain that crosses the cell membrane via the hinge/spacer region. Following the recognition and binding of ScFv part of the CAR with tumor antigen, the intracellular/signaling domain comprised of co-stimulators and the CD3ζ chain initiates intracellular signaling. The first-generation CAR contains only immunoreceptor tyrosine-based activation motif (ITAM) motifs in the intracellular domain. The second-generation CAR includes one co-stimulatory molecule, and the third-generation CAR contains two co-stimulatory molecules. The fourth and fifth generations of CARs are based on second-generation CAR. The fourth-generation CAR contains 1–3 immunoreceptor tyrosine-based activation motifs combined with an inducible expressed cytokine; while the fifth-generation CAR is T cell receptor-deficient CAR T cells developed employing genome editing technologies
They even present with unique advantages of long residence time and proliferative abilities, making them comparable to ‘living drugs’ [16]. Certain chemokines and adhesion molecules crucial for T cell trafficking into the brain have been identified. The immunosuppressive microenvironment is shaped by mutations in the isocitrate dehydrogenase (IDH) genes IDH1 and IDH2 in glioma cells, which inhibit STAT1 expression and decrease the production of CD8 T cells, type 1 related effector molecules, and chemokines e.g. CXCL10 [17]. In line with the findings, T cell infiltration was significantly lower in IDH-mutant gliomas compared to IDH-wildtype gliomas [18]. Clearly, identifying and incorporating mechanisms that induce T cell surveillance and trafficking, into CAR T design might improve the systemic antitumor response against solid brain tumors. Stress responses arising from a lack of essential amino acids in the brain tumor microenvironment impact T cell functions negatively. Tumor-associated myeloid cells, assisted by regulatory T cells often encourage the production of immunosuppressive agents like ILs, especially IL-4 and IL-10, arginase 1, indoleamine 2,3-dioxygenase (IDO), etc. in the tumor microenvironment. Thus, CAR T therapy holds beneficial promises from the selective targeting of immunosuppressive myeloid cells in the tumor microenvironment. The therapeutic potential of CAR T cells would be significantly increased by introducing immunosuppressive resistance into the cells themselves, in combination with additional drugs that promote T cell activity.
Understanding the complexity of tumor heterogeneity is crucial in order to increase the repertoire of antigens for CAR T cells and find optimal targets to put an end to brain tumor antigen escape. Studies have revealed that the leading edge of tumors exhibits a proneural-like characteristic, whereas the tumor core poses with a more mesenchymal-like appearance [19]. It is also interesting to note that single-cell RNA sequencing has revealed a non-autonomous regulatory mechanism for brain tumor heterogeneity [20]. Recurrent glioblastomas are known to demonstrate reduced monocytes compared to primary glioblastoma. Advancing CAR therapy against brain tumors requires strategies to minimize antigen escape caused by tumor heterogeneity.
Clinical successes of CAR T in hematological cancers igniting the hope
As a result of the impressive clinical success achieved by CD19-specific CAR T cells in the field of hematological malignancies, the United States Food and Drug Administration (FDA) has approved some CAR T cell-based therapeutics against leukemia, multiple myeloma, and lymphoma (Table 1) [21, 22]. While T cells require both the shape of the T cell receptor and the presentation of the MHCs in order to recognize tumor-associated antigens produced by cancer cells, CAR T cells exclusively rely on the CAR structure. Both the antigen-binding specificity of CARs and the cytotoxicity of T cells are present in CAR T cells. CD19 is used most frequently to treat hematological cancers. When injected into the body, CD19 CAR T cells target all CD19-positive cells. Through extensive proliferation and phosphorylation, CAR T cells start the activation process. Cytotoxicity and the release of cytokines are the major anticancer response mechanisms. CD8+ CAR T cells are crucial to the process of eliminating tumor cells. CD4+ CAR T cells provide a supporting role that can improve the immune response against tumors. Granzyme and perforin, which are capable of harming tumor cells, are secreted by CAR T cells to exert cytotoxicity [23]. The other method of cytotoxicity involves inducing apoptosis in cancer cells by endorsing apoptotic signal transduction. CAR T cell-released cytokines improve tumor clearance by stimulating a variety of immune cells and producing synergistic effects.
CAR T cell therapy is undeniably a promising tool for future cancer immunotherapy. Longer persistence of CAR T cells has been demonstrated to improve anti-cancer response to hematological cancers, which seems to be replicated for CAR-mediated treatment of other types of cancers too [24]. In many cases, brain malignancies share common targetable tumor antigens and peptides with hematological cancers e.g. B7-H3, CD70, etc. [25]. Moreover, CD19 and/or CD28-based CAR moieties with co-stimulatory domains for 4-1BB seem equally promising for both hematological and brain cancers [26]. The fact that CAR T cells, modified immune cells from the patient body, are not constrained by CNS barriers fuels more optimism for a cure for brain cancer.
CAR T cells for brain tumors
Following promising clinical outcomes with CD19-CAR T therapy against hematological cancers, the implication of CAR T cell-based therapeutic approaches for the treatment of solid tumors, including brain tumor has sparked huge interest among the scientific community. After treatment, analysis of the cerebrospinal fluid (CSF) revealed that the systemically administered CAR T cells frequently travelled to the CNS [27, 28]. Due to factors such as the distinct tumor microenvironment, the challenge of accessing the tumor(s), and heterogeneity in the expression of the target antigen, CAR T cells initially failed to replicate their effectiveness against hematological cancers in the brain. The subsequent generations of CAR T cells have been prepared in an effort to get beyond the obstacles faced by initial CAR T cell technologies in the treatment of brain cancers. Rapid developments in gene editing technology have opened up a number of possibilities for CAR T cell modification e.g. cytokine overexpression, gene knock-out and knock-in, simultaneous targeting of multiple antigens, precise control of CAR expression and signaling to increase their effectiveness [29]. The suppression or improper presentation of tumor antigens, which occur frequently in brain tumors due to abnormalities in components of human leukocyte antigen (HLA) class I antigen processing machinery has no adverse effects on this form of therapy because recognition and effector mechanisms of CAR T cells are HLA class I independent. In addition, CARs supersede T cells in that, besides small peptide sequences, they can also recognize tumor antigens in the form of carbohydrates, glycolipids, and proteins [30].
Design considerations
The optimization of CAR design is highly dependent upon antigen and tumor-specific properties. At the first step of designing CAR T cells, patient T cells are collected by leukapheresis and a specific subset of T cells are collected. T cells are activated by engaging their T cell receptors, and co-stimulatory receptors (mostly anti-CD3 and anti-CD28) and/or adhesion molecule(s) (Fig. 1). Stimulated T cells are genetically modified by viral transduction to intensify the expression of CAR. The engineered CAR T cells are expanded ex vivo in media supplemented with common γ-chain cytokine cocktails e.g. IL-2, IL-15, IL-7. And IL-21. Superior CAR T cell products are generated by a less differentiated memory phenotype with increased mitochondrial fitness [31]. Ex vivo cultures of about 2 weeks are capable of producing millions of engineered CAR T cells with therapeutic potential to be infused back into the patient. Certain factors, such as the effect of intrinsic T cell product variability on efficacy, consistency over time, and the extent of T cells reaching the CNS, need to be taken into consideration when using CAR T cells to treat brain tumors.
CAR T cells have been designed to secrete proinflammatory cytokines to aid in their function and proliferation while serving as a defense against immunosuppressive cytokines. In solid tumor models, CAR T cells secreting IL-12, and IL-18 have been evinced to exert longer-lasting tumor responses preclinically [32, 33]. Enhanced anti-tumor efficacy of CAR T cells with constitutive IL-7 and IL-15 signaling has also been reported, as well as with the inducible release of an IL-15 super-agonist complex by T cells upon interaction with the cognate antigen [34, 35]. Secreted pro-inflammatory molecules, in addition, exert some paracrine effects like reshaping the tumor microenvironment to be antitumorigenic, and regression of solid tumors [36, 37]. Co-expression of dominant-negative transforming growth factor β (TGFβ) receptor II (TGFβRII) can block TGFβ signaling within CAR T cells leading to reduced exhaustion, and improved anti-tumor efficacy [38]. As a measure of imparting resistance to the immunosuppressive tumor microenvironment, engineered T cells can be made to recognize soluble ligands so that the immunosuppressive cytokine signal can potentially be converted into an immune-stimulatory one [39, 40]. CAR T cells engineered to secrete a programmed cell death ligand 1 (PD-L1) antibody, CAR T cells with programmed cell death 1 (PD-1) and lymphocyte activation gene 3 (Lag3) genes knocked out using clustered regularly interspaced short palindromic repeats (CRISPR)/ CRISPR-associated protein 9 (Cas9) technology, and CAR T cells designed with a PD-1 ectodomain linked to the transmembrane, and cytoplasmic domains of CD28 in order to convert an immunosuppressive signal into a co-stimulatory one have all been explored exhibiting encouraging outcomes [41,42,43].
Tumor antigen expression on normal tissues often hinders the usage of CAR T cells due to the concern of unexpected side effects and toxicities in healthy tissues. To overcome this challenge CARs designed with ScFvs with varying affinity allow for differential recognition of the targeted tumor antigen [44]. Despite several developments, recurring tumors might still circumvent monovalent CAR T cells by downregulating the targeted antigen or by creating antigen-negative clones [45]. Encompassing bispecific target domains within CAR design can minimize antigen escape by rendering recognition of either of two targeted antigens for activation of T cells (Figs. 2a and b). Well-characterized antigens remain the focus for designing bispecific ‘OR-gated’ CARs against brain tumors (Fig. 2a). This strategy can be highly useful in the case of recurrent tumors where malignant cells devoid of the single targeted antigen have been expressed as a result of earlier CAR T treatment. In a CAR-based study, bispecific targeting of IL-13 receptor α2 (IL13Rα2) and human epidermal growth factor receptor 2 (HER2) was able to control glioblastoma for nearly a month whereas single-targeted CAR therapy was ineffective due to antigen escape [46]. Regarding design considerations of bispecific CARs, the ‘tandem’ design sequentially arranges the heavy and light chains of each ScFv while the ‘loop’ pattern resembles the bivalent antibody structure (Fig. 2b). In a study with CD19/CD22 bispecific CARs, the ‘loop’ design proved overwhelmingly superior to the ‘tandem’ design stressing the importance of bivalent designs, linker lengths, and order of ScFv for CAR performance [47]. The incorporation of a third targeting domain (trispecific CARS) can further intensify this approach (Fig. 2c) [48]. Another exciting approach involves the use of EGFRvIII-specific CAR T cells engineered to secrete bispecific T cell engagers (BiTE) [49]. These bispecific monoclonal antibodies can link T cells to wild-type EGFR, overcoming the resistance of EGFRvIII heterogenous glioblastoma to EGFRvIII-specific CAR T cells. Alternatively, multi-antigen targeting can be addressed by expressing two CARs on the same T cell, and/or by mixing different single-targeted CAR T cells (Fig. 2d) [50, 51]. Interestingly, monitoring of safety and efficacy of mixed CAR T cells might be easier from previous experience with single-targeted CAR T cells. On the contrary, bispecific CAR T cells seem to be more effective experimentally compared to a pool of single-targeted CAR T cells, probably because of the effects of local competition [46, 52]. The future of CAR T cell engineering lies in overcoming reliance on selective tumor antigen(s) and allowing them the flexibility to target several antigens, while at the same time imparting in them the capacity to be turned on or off in a timely and effective manner to avoid toxicities. Thus, when designing new approaches, safety switches such as suicide genes, and using inducible and/or controllable CAR systems are attractive approaches to address the issue of CAR T cell-induced toxicity. SynNotch system utilizes ‘AND’ and ‘NOT’ gated CARs i.e. CAR expression can only be induced upon recognition of a second tumor-associated antigen [53]. The limitation of such an approach is that both antigens must be expressed at considerably high levels by tumor cells. This requirement poses a potential problem for anti-glioma CAR T cells due to high heterogeneity in antigen expression levels. Universal CARs have been designed in order to fight tumor heterogeneity by utilizing adapters to connect CAR with targeted antigens thereby permitting antigen switch without re-engineering T cells [54, 55]. Additional approaches to enhance CAR T cell anti-tumor activity include the incorporation of the hypoxia transcription factor-1 alpha (HIF-1α) subdomain in a CAR construct. This results in the activation of CAR T cells only under hypoxic conditions such as within a tumor microenvironment [56]. This strategy may attribute an on-target effect with minimized off-tumor toxicity.

Multi-antigen targeting CAR T cells that prevent tumor antigen escape to intensify therapeutic attributes. a Bispecific/bivalent ‘OR-gated’ CAR T cells. Each CAR exhibits a comprehensive signal domain that, when present with either alike antigen, intensifies the antitumor activity of CAR T cells. b Bispecific ‘tandem’ CAR T cells. One CAR simultaneously expresses two different antigen-binding domains. c Trivalent ‘OR-gated’ CAR T cells. Three CARs expressing T cells intensify the antitumor effects by recognition of one, two, or three tumor-specific (targeted/validated) antigens. d A mixture of different single-targeted CAR T cells also ensures multi-antigen targeting
Delivery/trafficking
Brain tumors present unique challenges to host T cells, or so to say, any immune cells owing to the highly selective nature of the BBB and the blood CSF barrier (BCSFB). These barriers control immunological entrance into the brain and CSF and restrict immune extravasation through post-capillary vesicles. CAR T cells can be used as delivery vehicles when combined with constitutively or inducibly expressed cytokines, chemokines, antibody fragments, or other biomolecules. Circulating T lymphocytes must first attach to the vascular endothelium via integrins, adhesion molecules, and chemokines. In the absence of inflammation, activated T cells can be recruited beyond the BBB, however, CD8 T cells still require the presentation of the major MHC class I antigens on luminal endothelial cells.
Systemic administration of CAR T cells through the intravenous route is most common in cases of hematological and solid tumors. Experimentally, CD19-CAR T cells were shown to reach CNS and combat malignancy [27]. The adoptively transferred cells were activated in the periphery due to the presence of disease. The fact that activated T cells are more likely to travel to the CNS may have a considerable impact on the effectiveness of CNS trafficking. Locoregional routes including intratumoral, and intraventricular routes of administration bypass most of the barriers to reaching brain parenchyma. Comparative studies revealed that local administration of CAR T cells beats systemic delivery for glioblastoma, and breast cancer brain metastases, with improved targeting of multifocal disease, thus illustrating the surveillance of CAR T cells throughout the CNS [57,58,59]. From a safety point of view, locoregional distribution is anticipated to reduce off-tumor targeting of other systemic tissues and intravenous first-pass pulmonary damage [60].
CAR T cells can be engineered to express chemokine receptors to enhance intra-tumoral T cell trafficking. A clinical trial has revealed that CD19-CAR T cells can fight CNS leukemia by trafficking into the CNS [27]. Interestingly, the forced expression of C-C chemokine receptor type 4 (CCR4) could enhance CAR T cell accumulation and therapeutic response in Hodgkin’s lymphoma [61]. Detection of chemokine (C-C motif) ligand (CCL)2, CCL4, CCL5, CCL17, and CCL21 which have all been linked to glioma also, ignite promise regarding the efficacy of them against glioma. However, CCR4 may be a flexible tactic for brain cancer [62, 63]. Tumor tropism of adoptively transplanted T lymphocytes by controlling tumor chemokine release to glioblastoma expressing CCL2, (a ligand for CCR4) has been demonstrated [64, 65]. In line with the findings, the incorporation of CCR2, a receptor for CCL2 in CAR T cells has enhanced efficacy against neuroblastoma [66]. Chemokine receptor-engineered T cells should be designed for the chemokine signaling specific to each tumor type to improve desired localization. Additional targeting of endothelial adhesion molecules or vascular cytokines (upregulated in brain tumors) could also be a viable strategy for increasing CAR T cell accumulation at the tumor site [67, 68].
Using imaging techniques to track the migration of CAR T cells in real time can offer significant evidence for on- and off-target localization, compared to the sampling of blood, CSF, or invasive biopsies, which provide more generalized information about cells in circulation or in the biopsied tissue, respectively. Nuclear imaging techniques (single-photon emission computerized tomography, positron emission tomography, etc.), coupled with anatomical detailing (computerized tomography scan, magnetic resonance imaging, etc.) can enable sensitive, non-invasive tracking of perfusion in brain tumors [69, 70]. Several reporter systems using human genes as non-immunogenic alternatives are being tested with the aim to study the trafficking of T cells while simultaneously minimizing the risk of immunological rejection. Though this strategy yields signals based on the extent of the presence of metabolically active cells, it is also influenced by background uptake of the nucleoside, and the BBB permeability and tissue penetrability of the probe [71, 72]. Cell-antigen-specific visualization with positron emission tomography has been applied to track T cells engineered with antigen tags. Metal chelator-based strategies are also displaying promise, especially in tracking CARs with ScFvs [73]. In summary, pre-labelling techniques should be employed when sensitivity and early trafficking are crucial, while injected antibodies and reporter genes perform better over repeated imaging sessions and the signal is connected with the number of living cells.
Clinical considerations
Brain cancer is possibly the current forerunner among different tumor types to undergo clinical trials with CAR T cell-based treatments. Multiple potential targets for CAR T cell-based therapies have been identified through immuno-histochemical investigations of brain malignancies. In the majority of cases, malignant brain tumor patients in advanced stages are enrolled after verification of expression of targeted tumor antigen(s). The intended dose is divided into multiple weekly infusions. Splitting of doses minimizes the safety risks compared to single bolus administration [74]. In addition, repeated administration allows increased overall dosing of functional CAR T cells over a longer duration of time, thus lengthening the therapeutic window. Positron emission tomography and magnetic resonance imaging are undertaken to evaluate the response of CAR T cells in volunteers/patients. Immunologic correlative studies are conducted over the course of treatment to assess the persistence of CAR T cells in peripheral blood and CSF. Continuous monitoring for activation of the endogenous immune system indicated by changes in cytokine levels in blood and CSF is also undertaken simultaneously. Alterations in antigen expressions, and variations in cytokine levels in the tumor milieu are also monitored.
Targets attributed clinical promise
Targeting a small number of antigens, the initial clinical trials with CAR T cells for brain tumors have started with most patients with recurrent or resistant glioblastoma. Lead tumor antigen candidates were chosen based on evidence of negligible expression in the normal brain, and the history of overexpression on malignant cells (Fig. 3). This is particularly important to reduce off-target toxicities. Target-based evaluation of CAR T cell therapy for brain cancer management is uncovering salient insights regarding the safety and bioactivity of the same, steering toward future clinical translations.

A glance at CAR T cell-based therapeutic prospect in brain cancer management. CAR T cells recognize some brain tumor-specific antigens that are targeted with an ambition to eliminate brain cancer cells. Some malignant growths in the brain can damage the BBB and help CAR T cells to cross the BBB to reach the tumor site. In addition, some delivery strategies allow delivering CAR T cells to the tumor site enabling the achievement of better therapeutic efficacy in brain tumor management
IL13Rα2
IL13Rα2, a high-affinity IL-13 receptor that is typically overexpressed on glioblastoma cells and barely expressed on healthy brain tissue, was one of the first targets for CAR T cell therapy of brain tumors. IL-13 can bind to the decoy receptor IL13Rα2, which lacks a functional cytoplasmic domain, thus unable to trigger an intracellular signaling pathway [75]. Expression of IL13Rα2 increases with advancing stages of the disease. The mesenchymal subclass of glioblastoma is also associated with IL13Rα2 related gene signatures [76]. Importantly, both glioma stem-like cells and differentiated tumor cells express IL13Rα2, thus maximizing the circumference of IL13Rα2-targeted CAR T cell therapy.
An E13Y site-directed mutation was introduced in IL-13 based on work on cytotoxin-conjugated IL-13 that defined mutations increasing the specificity of IL-13 for IL13Rα2 over the more ubiquitously expressed IL13R1/IL4R complex [4, 77]. The engineered CAR T cells acted specifically upon glioma cells consistent with the expectations.
Second-generation CARs designed with an additional 4-1BB co-stimulatory domain and optimized spacer domain, presented a nearly 10-fold increase in anti-tumor potential compared to first-generation IL-13 zetakines [58, 78]. During the initial days, two clinical trials were undertaken to evaluate the feasibility and safety of locoregional intracranial tumoral delivery of autologous [79], and allogenic [80] CAR T cells (first-generation IL-13 zetakine CD8+ T cell clones) for resectable, and non-resectable glioblastoma, respectively. While short-lived anti-glioma activity was observed among a few patient volunteers, dose-limiting toxicity was completely absent at a dose of 1 × 108 CAR T cells. The first-generation IL-13 zetakine CAR T cells demonstrated limited persistence, in line with the transiency of the anti-glioma effect observed and led to initiatives to improve CAR design for the betterment of therapeutic efficacy [57, 81]. Second-generation IL13Rα2-targeted, 4-1BB co-stimulatory CAR T cells underwent clinical trial [82] whereby CAR T cells were introduced in r/r IL13Rα2+ malignant glioma patients through either intratumoral, intraventricular or dual intratumoral/intraventricular routes. With the phase I study still ongoing, safety and tolerability with regard to the absence of dose-limiting toxicity are already reported [57]. One of the patients has experienced dramatic upgradation regarding the quality of life, including discontinuation of glucocorticoids on receiving intraventricular CAR T cell administrations that lasted for about 7.5 months. Increases in endogenous immune cells and inflammatory cytokines detected following each intraventricular infusion of CAR T cells may be due to the activation of the host immune system. Thus, a complete response was generated inspite of non-uniform expressions of IL13Rα2 on respondent tumors. Recurrent tumors appeared outside the brain with lowered expression of IL13Rα2 highlighting antigen escape as a mode of tumor resistance to immunotherapy. Another trial [83] is still assessing the effect of IL13Rα2 CAR T cells on medulloblastoma, glioblastoma, and ependymoma. CAR T cells after lymphodepletion for the treatment of IL13Rα2 positive recurrent or refractory brain tumors in children are under clinical evaluation [84].
EGFR
EGFRvIII, a deletion mutant of endogenous EGFR, frequently exhibits genetic amplification and/or mutation during glioblastoma, it is an attractive target for CAR T treatment. EGFRvIII presents multiple advantages viz. tumor-restricted expression of mutation, and an immunogenic epitope in the form of a truncated extracellular domain responsible for activation of signaling cascades for the onset and progression of glioblastoma. The tumor-restricted expression profile of EGFRvIII makes it an appealing target. However, this receptor’s expression is unstable over the course of the disease, raising the possibility of antigen-negative escape variants under targeted therapy. Targeting wild-type EGFR could not solve the issue completely as the expression of wild-type EGFR is not restricted to tumor cells only, thus triggering the likelihood of off-target toxicity [85].
In a clinical trial [86], recurrent glioblastoma patients were treated with third-generation, ScFv-based EGFRvIII-targeted CAR containing co-stimulatory domains for CD28 and 4-1BB. Intravenous administration of CAR started with doses as low as 1 × 107 cells followed by systemic delivery of IL-2. At low doses of 1 × 107 to 1 × 109 cells, no evidence of off-tumor toxicity was observed. However, higher doses of ≥1 × 1010 cells, pulmonary toxicities became prevalent. At a dose of 3 × 107 cells, one patient encountered hypoxia and dyspnea as serious adverse effects. One death was recorded at a dose of 6 × 1010 cells whereby pulmonary edema was evidenced on post-mortem examination [87]. These unwanted effects might be attributed to activated T cells congesting the pulmonary vasculature in a dose-dependent manner, which is probably a limitation of the route of administration used. It is worth mentioning that EGFRs are expressed in pulmonary cells too. Overexpression of EGFRvIII on lung carcinomas has been utilized for CAR targeting whereby EGFRvIII-targeted CAR vectors have demonstrated promising anti-cancer activity against non-small cell lung carcinoma both in vivo and in vitro [88]. In the clinical trial under discussion [86]. CAR T cell persistence was on the lower side resulting in median survival of 6.9 months. Despite the fact that the findings primarily emphasize the difficulties in using CARs to treat brain tumors, one patient gave rise to some optimism by managing 12.5 months of progression-free survival. A humanized EGFRvIII-specific CAR exhibited promising activity and safety profile against glioblastoma in preclinical studies [89]. Thus, it was advanced to a phase I clinical evaluation [90]. Patient volunteers received a single intravenous infusion of the engineered CAR vectors followed by surgical resection. Significant movement of CAR T cells to the desired site triggered optimistic promise. Downregulated EGFRvIII in recurrent tumors indicated initial antigen-specific targeting by CARs. Due to the inceptive heterogeneous expression of EGFRvIII on glioblastoma, the designed CARs could only target a fraction of malignant cells. The immunosuppressive tumor microenvironment was exacerbated by CAR T cell administration, including increases in indoleamine 2,3-dioxygenase (IDO1), programmed death-ligand 1 (PDL1), and IL-10. Non-CAR T cells, primarily polyclonal regulatory T cells, also contributed to the immunosuppression by virtue of their expression of CD4, CD25, and forkhead box P3 (FoxP3), which are markers for regulatory T cells [91]. The immunosuppressive response to CAR T treatment raises the possibility that protective measures like immune checkpoint inhibition can complement EGFRvIII-CAR T therapy, thus igniting possibilities of combination therapy. Further digging into this idea, another clinical trial [92] evaluated the synergistic response of EGFRvIII-CAR T cells with the anti-PD-1 antibody pembrolizumab, which has been discussed later on. Clearly, low affinity to endogenous EGFR and enhanced binding to tumor-specific EGFRvIII binding is necessary for the high efficacy of CARs. Antibodies such as mAb806 have been designed to target activated EGFRvIII and amplified EGFR. A clinical study with an indium-EGFR806 antibody has displayed nearly zero uptakes by normal tissues [93]. Clinical trials are on the way [94, 95] at Seattle Children’s Hospital. USA with EGFR806-CAR to assess on target-off tumor behavior on pediatric recurrent and/or refractory solid tumors, especially within the CNS.
HER2
HER2 is a highly alluring target antigen owing to its high abundance in brain tumors, its role in tumor progression, and the capacity of HER2-specific CAR T cells to eradicate both differentiated cells and cancer-initiating cells [96]. However, in the clinical setup, the first patient treated with HER2-targeted CAR T therapy succumbed to death, raising safety concerns [97]. Intravenous infusion of a dose of as high as 1 × 1010 cells led to death led to off-target toxicity on lung epithelial tissue, triggering cytokine storm, respiratory distress, and pulmonary edema as a result of the accumulation of HER2-CAR T cells in the lung and abdominal/mediastinal lymph nodes. Failure of the ScFv from high-affinity trastuzumab antibody, and CAR co-stimulatory domains consisting of both CD28 and 4-1BB mandated scientists to restructure the design. A newer optimized CAR containing ScFv from lower affinity trastuzumab, redesigned co-stimulatory domains lower cytokine release, and use of lower affinity HER2-monoclonal antibody FRP5 for improved tumor targeting advanced to investigative stages [98]. Initially, these HER2-CAR T cells demonstrated a satisfactory safety profile with low persistence in sarcoma patients [99]. The CARs were engineered into virus-specific T cells in an attempt to enhance their persistence assuming co-stimulation would result from the engagement of T cells with latent virus antigens on antigen-presenting cells. In a study on 16 patients [100] with progressive glioblastoma, the safety of autologous HER2-CAR virus-specific T cells was instituted. Though these CAR vectors established safety, their efficacy was below par highlighting the requirement for improvements regarding function, persistence, and expansion. In glioblastoma patients, up to 1 × 108 HER2-specific CAR T cells with a CD28ζ endodomain were successfully administered without dose-limiting toxic effects [101]. Inspired by preclinical outcomes of low cytokine production of good anti-tumor efficacy of 4-1BB versus CD28 co-stimulation, two phase I clinical trials [102, 103] have started with optimized HER2-CARs among patients with HER2-positive malignant glioma and brain metastasis of HER2-positive breast cancer, respectively [59]. Interestingly, HER2-amplified breast cancer depicts much higher expression of HER2 compared to glioma [104]. Thus, clinical trials under discussion might also shed light on the effect of locoregional intracranial ventricular delivery to increase specificity. Another two clinical trials [105, 106] are evaluating locoregional delivery of HER2-specific CAR T cells on HER2-positive, recurrent and/or refractory pediatric CNS tumors. Another interventional trial [107] has initiated in 2022 with HER2-specific CAR T vectors against child ependymoma.
Others
The reported success of CAR T therapy so far, emphasizes the necessity for an appropriate CAR target to be highly expressed across tumor(s) and intratumoral cellular subsets. Since brain tumor cells express multiple markers that are also shared by parts of the normal brain, it has been difficult to identify suitable antigens in brain tumors. Repercussions of off-tumor targeting are far less admissible within the CNS compared to other systems. Moreover, resistance to CAR T cells led by the heterogeneity of the tumor antigen(s) in brain tumors, rationalizes the necessity for a larger library of targetable antigens.
B7-homolog 3 (B7-H3) alias CD276, a member of the B7 family of immune checkpoint inhibitors, is expressed in cases of hematological cancers and solid tumors including higher grades of glioma. In most of the normal tissues, the immune-histochemical analysis does not detect B7-H3 since normal tissues lack the ability to translate B7-H3 mRNA due to hindrance caused by microRNAs (miRs). Apart from the expression on the tumor itself, negatively regulating T cell activation, B7-H3 is also expressed on tumor-associated angiogenic vessels and fibroblasts [108]. Thus, targeting of B7-H3 by CAR T cells can act by not only direct targeting but also by suppression of angiogenesis and by the disruption of the stroma. Gaining inspiration from the preclinical promise exhibited by Majzner et al. [109] and Tang et al. [110], a phase I/II clinical trial [111] has been initiated in 2022 to assess the effect of B7-H3 CAR T cell therapy in between two successive cycles of temozolomide in the treatment of recurrent and/or refractory glioblastoma. Another trial [112] is assessing B7-H3-Specific CAR T cell locoregional immunotherapy for diffuse intrinsic pontine glioma/diffuse midline glioma and recurrent or refractory pediatric CNS tumors.
CD147, also known as extracellular matrix metalloproteinase (MMP) inducer is a type I transmembrane adhesion molecule pertaining to the superfamily immunoglobulin. CD147 prompts the production of MMPs 1, 2, 3, 9, 14, and 15 from fibroblasts to degrade the extracellular matrix and aid in tumor progression. Interestingly, the extent of expression of CD147 on glioma cells inversely correlates with disease prognosis, thereby making it a potential CAR target during the early stages of the disease [113]. An early phase I trial [114] has attempted to shed light on the safety, tolerability, and efficacy of CD147-specific CAR T cells against recurrent glioblastoma.
The appearance of ganglioside GD2 in different tumor samples makes it a sought-after tumor antigen for CAR T-mediated targeting [115]. The report claimed GD2 acts as a cancer-initiating cell marker in the case of breast cancer, which further boosts its attraction as a target tumor antigen among researchers [116]. Orthotopic xenograft models of patient-derived diffuse midline glioma are efficiently eliminated by GD2-specific CAR T cells [117]. GD2-specific CAR T cells have been administered safely to neuroblastoma patients in earlier studies [118, 119]. In a clinical trial [120] with anti-GD2 CAR T cells against neuroblastoma, no dose-limiting toxicity was observed with 1 × 107 cells/m2, along with undetectable CAR T cell levels in peripheral blood. Expansion of engineered vectors was observed at 1 × 108 cells/m2 while disease progression restarted at day 45 when CAR T cells were no longer detectable. An interventional trial [121] is assessing the safety and efficacy of C7R (an IL-7 cytokine receptor) expressing GD2 CAR T cells in advanced stages of glioma. An interventional trial [122] is assessing anti-GD2 CAR T cells against GD2-positive, pediatric neuroblastoma. GD2 CAR T cells are also being assessed against diffuse pontine glioma and diffuse midline glioma, clinically [123].
Tumor-targeting peptides can be used as a tumor-targeting domain of CARs as an alternative to designs based on antibodies. Chlorotoxin (CLTX) is a small, nature-derived peptide able to bind to primary brain tumors while exhibiting negligible affinity for normal tissues [124]. On incorporation into CARs, CLTX reroutes T cells towards targeted tumor detection with little detectable off-target consequences. Earlier studies have also reported no dose-limiting toxicity in patients receiving CLTX-bioconjugates, thus adding fuel to the idea of utilizing it as a CAR target [125]. Moreover, CLTX-specific CAR T cells depicted promising anti-tumor effects in glioblastoma orthotropic xenograft models [126]. Interestingly, expressions of MMP2, chloride voltage-gated channel 3 (CLCN3), and annexin A3 (ANXA3) seem to be indispensable for the binding of CLTX to glioblastoma cells [96]. The bioactivity of CLTX CAR T cells significantly has been shown to be decreased when MMP2 was knocked out, demonstrating the requirement of MMP2 expression for efficient targeting. Gaining inspiration from the previous outcomes, a phase I clinical study [127] is presently evaluating the therapeutic effect of CLTX CAR T cells on patients with recurrent or progressive MMP2-positive glioblastoma.
Glioblastoma stem-like cells play a role in mediating resistance to radiation and chemotherapy. Thus, elimination of glioblastoma stem-like cells is a prerequisite for lowering tumor recurrence. In a novel approach to specifically target glioblastoma stem-like cells, CAR T vectors have been fabricated against CD133, one of the surface markers of glioblastoma stem-like cells. CD133 CAR T cells have displayed cytotoxic potential on patient-derived glioblastoma stem-like cells [128]; however, the safety aspect still persists as CD133 is also expressed on neuronal stem cells. CD171 is a neuronal cell adhesion molecule playing a vital role in adhesion, migration, and differentiation. It is also prominently expressed in treatment-resistant cancers. A phase I trial [129] is evaluating the safety and feasibility of CD171-specific CAR T cells against recurrent/refractory neuroblastoma.
It has been revealed that glioblastoma cells overexpress the erythropoietin-producing hepatocellular carcinoma A2 (EphA2) that plays an essential role in carcinogenesis and cell migration [130]. A clinical trial [131] had been initiated to examine the efficacy of EphA2-CAR T cells on glioblastoma patients but has been discontinued.
Combination therapy
Combination therapy may be a prospective approach to address the unfavorable glioma environment and improve CAR T cell activity and durability. Combining CAR T cell therapy with a checkpoint inhibitor would aid in overcoming the barriers to T cell invasion and functionality. Two clinical studies have shown that neoadjuvant anti-PD1 immunotherapy can improve survival and modify the tumor microenvironment in glioma patients indicating blocking of PD-1 could alter the tumor microenvironment and act in conjunction with CAR T treatment to lengthen the survival of glioma patients [132, 133]. Alternative tactics might include genetically deleting PD1 from CAR T cell products or designing CAR T cells to produce a blocking antibody against PD1/PDL1. A combination trial of EGFRvIII-CAR T cells with an anti-PD-1 antibody (pembrolizumab) has been undertaken [92]. Another trial is evaluating the combination of IL13Rα2-CAR T cells with Nivolumab [134]. However, the safety, feasibility, and potential anti-tumor activity of CAR T cells in combination with checkpoint inhibitors are awaited from these trials. Another interventional trial [135] is assessing anti-GD2 CAR T cells along with IL-15, against GD2-positive, pediatric neuroblastoma. A schematic overview of checkpoint inhibition in combination with CAR T cell therapy has been depicted in Fig. 4a.

An overview of combination therapy with CAR T cells. a Combination of CAR T cells with checkpoint inhibitors. b Combination of CAR T cells with immunosuppressive drugs. c Natural killer group 2D (NKG2D) ligand-based targeting by natural killer cells coupled with CAR T cells highlights a potential for combinatorial treatment. Extrinsic checkpoint blockade rests on programmed cell death 1 (PD-1) receptor/ligand hindering antibodies. Genetic engineering enables intrinsic PD-1 checkpoint blockage to express proteins or nucleic acid that disrupt PD-1/PD-L1 signaling. The PD-1 dominant negative receptor, which competes with native PD-1 receptor and inhibits inhibitory signaling through native wild-type receptors, lacks the intracellular signaling domain in its creation. PD-1/PD-L1 inhibiting single-chain variable fragments (ScFvs), which provide local antibody inhibition of PD-1 receptor/ligand interaction, can also be ensured by CAR T cells. Furthermore, gene-editing techniques can eliminate PD-1 expression by editing the programmed cell death protein 1 gene locus. CAR T cell therapy coupled with immunosuppressants attenuates non-specific toxicities by suppressing inflammation. The usefulness of combination therapy in the treatment of cancer is reinforced by the fact that NKG2D ligand-based targeting of natural killer cells in conjunction with CAR T cells enhances the anticancer effect of CAR T cells
Drugs such as temozolomide, dexamethasone, and bevacizumab, which are often used in combination to manage and treat brain tumors, may have an effect on activation of CAR T cells, calling for careful monitoring. Although it is conceivable that lymphodepletion will enhance CAR T cells’ clinical responses against brain tumors, principally for intravenous administration, but its therapeutic efficacy in the case of brain tumors requires more clinical validation. The reported EGFRvIII-CAR clinical trials that included lymphodepletion preconditioning [86] did not show noticeably superior CAR T cell persistence, expansion, or patient outcomes when compared to a study that did not use lymphodepletion [87, 90, 91]. Though this could be due to a variety of factors, including CAR design, target selection, and patient enrollment, it emphasizes the importance of further clinical evaluation of the role of lymphodepletion in the application of CAR T cells for brain tumors, specifically for locoregional delivery. A dual-intensified regimen of temozolomide has been used as a preconditioning lymphodepleting regimen in an EGFRvIII CAR T [136] trial against grade IV glioma. As lymphodepletion has been proven to be crucial for the successful anti-tumor activity of CAR T cells regarding T cell receptors and tumor-initiating lymphocytes when the therapeutic cells are administered intravenously, the dual use of temozolomide for the treatment of glioma and for lymphodepleting preconditioning is an appealing method. On the other hand, dexamethasone. Frequently used to alleviate tumor-associated swelling of the brain, imparts toxicity to immune cells including T cells, and thus potentially apprehends CAR T cell activity. Dexamethasone has been demonstrated to impart deleterious effects in vaccine trials for glioblastoma at very low concentrations and is known to impede dendritic cell priming of the T cell immune response [137]. CAR persistence and effectiveness have been demonstrated to be disrupted by long-term systemic corticosteroid treatment in trials [74]. Given the significance of dexamethasone in the management of glioma symptoms, a successful CAR T cell trial design must identify its allowable concentrations. At a dexamethasone level of 4–6 mg/day, a clinically relevant response has been achieved with CAR T cells [57]. Bevacizumab, a non-steroid anti-inflammatory drug, has the potential to be an effective alternative to immunosuppressive corticosteroids like dexamethasone for treating cerebral edema symptoms [138]. Bevacizumab may also improve CAR T cell therapy by promoting tumor lymphocyte trafficking and counteracting the immune-suppressive effects of VEGF. Clinical validation of the benefits of combinatorial treatment of brain cancer with CAR T cells and bevacizumab is awaited, following promising preclinical outcomes [139]. An overview of the mechanistic impacts of different immunosuppressive drugs in combination with CAR T cells in the efficacious management of brain cancers has been depicted in Fig. 4b.
Stressed cells typically express natural killer group 2D (NKG2D) ligands, such as MHC class I-related chains and UL16-binding proteins, to help immune effector cells detect and eliminate them [140]. Glioblastoma cells, and glioblastoma stem-like cells both represent overexpressed NKG2D ligands. NKG2D ligand expression on glioblastoma cells can be increased by chemotherapy or radiotherapy, highlighting the potential for combinatorial treatment approaches [141]. Considering the expression of NKG2D ligands on normal tissues under stress, harm in humans cannot be completely ruled out. A phase I study [142] evaluating the clinical response of NKG2D-based CAR T vectors on solid tumors including glioblastoma, has been withdrawn citing administrative reasons. The mechanistic insight has been hypothesized in Fig. 4c.
Preclinical advances
Resistance to CAR T cells is occurred due to the heterogeneity of the antigens presented by brain tumors, stressing the need for a larger library of targeted antigens. The effectiveness of CD19-CAR T cells highlights the requirement that a suitable CAR target is highly expressed across a variety of tumor types and intratumoral cellular subsets. The use of EGFRvIII-specific CAR T cells with disrupted PD-1 signaling via a CRISPR-Cas9 approach resulted in significantly longer survival of orthotopically engrafted mice [143]. Potential therapeutic use of B7-H3-CAR T cells against specific types of brain malignancies was shown by preclinical anti-tumor activity against a variety of pediatric tumors, including medulloblastoma [109]. In line with the finding, Tang et al. [110] also concluded that B7-H3 is commonly overexpressed in glioblastoma, and could be used as a therapeutic target in CAR T therapy.
Carbonic anhydrase (CA) IX is induced by hypoxia and is thus frequently overexpressed in many solid tumors, including glioblastoma. The extent of hypoxia in the tumor microenvironment regulates the level of CAIX expression in cancer cells, which is likely to lead to significant alterations in its expression in cancer cells [144]. CAIX-specific CAR T cells have been tested against glioblastoma cells in vitro and in vivo-xenografted mouse model after direct intratumoral injection [145]. Glioblastoma cells were successfully removed by CAIX-specific CAR T cells, thereby also enhancing the longevity of mice carrying tumors. However, CAIX expression in a patient might exhibit significant intra- and inter-tumor heterogeneity owing to the variability in the expression of CAIX on cancer cells depending on the degree of hypoxia. This expression pattern is likely to aid in the generation of cancer cell escape variants, which could pose a significant barrier to the successful application of CAIX-specific CAR T cell-based therapy.
CD70 is a transmembrane protein expressed on certain hematological and solid tumors besides being expressed on activated lymphocytes and matured dendritic cells. Human and mouse CD70-specific CAR T cells were able to recognize and eliminate CD70-positive glioblastoma tumors in vitro, as well as in xenograft and syngeneic models, without imparting significant toxicity [146]. CXCR1 and CXCR2-modified CD70-specific CAR T cells have been demonstrated to improve T cell trafficking and antitumor efficacy via IL-8-mediated chemotaxis in an in vivo glioblastoma model [147]. The higher CAR T cell antitumor activity resulted in improved tumor regression and survival of mice compared to those treated with unmodified CD70-specific CAR T cells. A major advantage of this tumor antigen is its confined expression mostly to malignant tumors [148]. Targeting CD70-positive T cells and dendritic cells may be of concern, but CD70-specific CAR T cells do not appear to have an impact on these activated immune cells [149]. In glioblastoma, preclinical investigations using CD70-specific CAR T cells have come up with promising findings, motivating researchers to move with this tumor antigen for possible clinical translations [150].
In gliomas, the expression of chondroitin sulfate proteoglycan 4 (CSPG4, also known as NG2) is inversely linked with patient survival [151]. Convincingly disproving the notion that CSPG4 expression on normal tissues might pose a problem when targeting this molecule as a tumor antigen, is the inability of CSPG4-specific CAR T cells to identify and lyse normal cells in vitro [152]. Further, in glioblastoma tissue and tumor-associated vasculature, CSPG4 is highly expressed with minimal heterogeneity but is not detected in the normal brain parenchyma [153]. In orthotopic a glioblastoma neurosphere xenograft model, intracranial injection of CSPG4 CAR T cells has been demonstrated to halt tumor progression [153]. In addition, the expression of CSPG4 on cancer-initiating cells is expected to allow CAR T cells to recognize and eliminate this particularly hostile cell subpopulation.
EphA2 is overexpressed in glioblastoma cells and glioblastoma cancer-initiating cells, without a detectable presence in normal tissues [154]. In vitro, EphA2 targeting CAR T cells effectively eliminates differentiated glioblastoma cells and glioblastoma cancer stem-like cells, significantly prolonging the survival of orthotopic xenograft mice [155]. A subsequent study utilized a short spacer region to fabricate an optimized version of the anti-EphA2 CAR construct [156]. Since the survival of glioma-bearing mice was prolonged to a similar amount utilizing a 20-fold lower dose, this CAR demonstrated stronger anti-glioma action compared to the previous CAR vector targeting EphA2.
Trophoblast cell surface antigen 2 (TROP2) is regarded as a stem cell marker and is expressed by solid tumor cells. High levels of this transmembrane glycoprotein have been demonstrated on surgically removed patient glioblastoma cells with minimal expression on normal brain tissues [157]. Since TROP2 is involved in the formation of new blood vessels in glioblastoma patients via upregulation of VEGF, targeting TROP2 might also aid to inhibit cancer growth by preventing neoangiogenesis. Encouraging outcomes with TROP2-specific CAR T cells against solid tumor cells viz. breast, pancreas, and prostate cancer cells is inspiring the scientific community to investigate similar vectors against glioblastoma cells [158].
Trispecific CAR T cells that target HER2, IL13Ra2, and EphA2 offer more thorough coverage of tumor antigens and have been demonstrated to considerably extend the longevity of mice, bearing glioblastoma patient-derived xenografts [48]. BiTE-armored CAR T cells successfully eradicated cancer cells and extended the survival of mice orthotopically grafted with either glioblastoma cells or patient-derived glioma neurospheres [49]. Engineering the CAR construct to induce or constitutively release active cytokines in order to boost CAR T cell activity and persistence is an approach to improve antitumor efficacy. In the orthotopic glioma xenograft mouse model, IL13Rα2 CAR T cells that were also designed to express IL-15 exhibited elevated anti-glioma activity, enhanced persistence, and considerably longer survival than the control [159].
Beyond membrane-associated proteins, the pool of brain tumor antigens that CAR T cells can target is anticipated to grow. Chheda et al. [160] revealed a shared neoantigen among patients with diffuse intrinsic pontine glioma as a result of a mutation in the H3.3K27 gene. When the altered peptide was used to stimulate HLA-A2+, and CD8+ T cells, the result was the creation of a TCR clone that, when expressed on T cells, caused cytotoxicity against tumor cells carrying the same mutation [160]. Additionally, CARs have also been created against soluble proteins, suggesting the possibility to target secreted components unique to brain tumors [40, 161].
Current challenges with adoptive T cell therapy for brain cancer
CAR T treatment has not yet achieved the same level of resounding success against solid tumors as it has, in treating hematological cancers. Insufficient trafficking of CAR T cells to the tumor site, defective recognition of the targeted tumor antigen and expression of the targeted antigen on normal tissues result in off-tumor toxicity. Off-tumor toxicity may also be impacted by factors such as the limited durability and low proliferation of effector immune cells in the tumor microenvironment, and the uncontrolled expression and timing of effector activities that lead to deleterious effects and provide many escape routes. Several CAR T designing strategies have been undertaken to get rid of these challanges, which have been discussed in the earlier sections. With rare exceptions, CARs have the drawback of necessitating the expression of the target antigen on the cell surface, while T cell receptors recognize mostly intracellular moieties which are transported to the cell membrane by MHC class I antigens. Failure of CAR T cell treatments in glioblastoma may be caused by intratumoral heterogeneity, antigen/epitope loss after therapy, and selectivity. The ability of differentiated cancer cells to proliferate and endure, and the capacity of cancer-initiating cells to evade detection by CAR T cells owing to their lack of expression of the targeted antigen are thought to be the causes of the resistance. These cells also have the ability to advance the disease with a modified phenotype [162]. Thus, it is essential to select a tumor antigen with high homogenous expression and high-expression stability to inhibit this cancer escape mechanism. An ideal target is assumed to be expressed uniformly on all differentiated and cancer-initiating cells. Another alternative can be to develop strategies to improve the capacity of CAR T cells to destroy cancer cells that do not express the targeted antigen, may be by formulating bispecific or trispecific CAR T vectors. Cancer-initiating cells have been suggested to be partially responsible for treatment failure and disease recurrence due to their self-renewal ability and treatment resistance. CD133-specific CAR T cells have been shown to successfully eliminate glioblastoma cancer-initiating cells in an orthotopic in vivo model [128]. CAR T cells targeting B7-H3, CSPG4 and HER2 have also been shown to eliminate both differentiated glioblastoma and glioblastoma-initiating cells [153, 163].
Adhesion molecules on endothelial cells, as well as specific antigen expression by antigen-presenting cells, are thought to be required for the recruitment of antigen-specific CD8+ T cells across the BBB. T cell recruitment, on the other hand, is frequently reduced across BBB during CNS malignancies [164]. This decrease may give cancer cells an immune escape mechanism. Glioblastoma multiforme was earlier believed to uniformly damage the BBB; thus, drug, antibody, and immune cell permeability should not be an issue [165]. However, it has been demonstrated subsequently that, even in the presence of significant tumor burden, the BBB can remain intact [166]. To address this limitation, direct administration of CAR T cells to the tumor site has been investigated as a method of delivery, eliminating the need for cells to migrate across the BBB. Locoregional infusion of CAR T cells has been demonstrated to enhance T cell tumor infiltration and tumor control in brain malignancies as compared to intravenous delivery [58, 167, 168]. However, some concerns still persist regarding cytokine syndrome and neurotoxicity by the direct delivery of CAR T cells into the brain [169]. The incidence of high-grade neurotoxicity occurs in approximately 30% of patients treated with CD19-CARs [170, 171]. The gamut of these neurotoxicities is referred to as CAR T-related encephalopathy syndrome. CAR-related neurotoxicity is associated with disease burden, high CAR T dose, and cytokine release syndrome. CAR T cell-mediated CAR T-related encephalopathy syndrome was mimicked in a humanized mouse model with a high leukemia burden [172]. Since human monocytes were the primary source of IL-1 and IL-6 during CAR T-related encephalopathy syndrome, monocyte depletion alleviates toxicities. IL-1 blocking demonstrates a reduction of neurotoxicities; while, IL-6 receptor blockade can only inhibit the symptoms. The use of CAR T cells with suicide genes or switchable signaling components, multiple dosing of lower numbers of CAR T cells in a single infusion, choosing optimal CAR T cells with respect to binding avidity, co-administration of a steroid and/or a monoclonal antibody targeting proinflammatory cytokine receptors are the potential strategies that all have been investigated to reduce CAR T toxicities.
Perspectives and critical analyses
Advancements in immunotherapy are igniting hope to treat solid tumors. As living T cell ‘micropharmacies’, CAR T cells can be used to deliver immunomodulatory molecules to the tumor microenvironment. Despite the majority of clinical trials are still in their early phases (Table 2), the results supported the safety of targeting certain tumor antigens in the CNS. Clinical trials using CAR T therapy [86, 90] have demonstrated progression-free survival for over a year; however, additional research is required to interpret the influence of underlying variations in tumor biology and the immunological microenvironment on the improved responsiveness of the patients. The inability of conventional preclinical models to accurately forecast the risk of toxicity in humans has been regarded as a significant challenge. In order to address these issues, various cutting-edge platforms that are currently available, such as suicide switches, RNA CARs, logic-gated CARs, etc. could be exploited.
Tumor tissue analyses demonstrated that antigen loss is a therapeutic escape mechanism [173]. Newer-generation CAR vectors came ahead with a prospect of overcoming antigen loss. The interactions between the host immune system and engineered T cells are also a matter of concern. According to clinical data, the tumor microenvironment responds to IL13Rα2 CAR T therapy by boosting CD3+, CD14+, and CD15+ immune cells as well as inflammatory cytokines after locoregional CAR infusion. Since CAR T therapy requires a highly expressed target throughout tumors and heterogeneity of tumor antigens leads to probable resistance towards CAR T therapy, the recognition of a broader pool of antigens is an urgent need.
The specificity of a few of the novel tumor-specific antigens under study raises significant questions. A potential concern regarding B7-H3 CAR T cells remains in the possibility of modulation of the inhibition of translation of B7-H3 mRNA by microRNAs during inflammation. This could potentially result in the expression of B7-H3 on healthy tissues, which would cause off-target toxicity [177]. While CD147 is overexpressed on malignant cells, it is still expressed at a low level on various normal tissues such as epithelial and endothelial cells of the brain and heart tissues [178]. Thus, there is a concern that CD147-specific CAR T cells may cause ‘on-target off-tumor’ side effects. Clinical outcomes are yet to reach a decisive verdict. Similarly, GD2 is expressed in normal brain tissues. Substantial CNS toxicity has been reported along with T cell infiltration into brain regions representing GD2-positive normal cells when GD2 CAR T cells were tested to treat neuroblastoma [179]. The future of CAR T cell engineering lies in abrogating their dependence on particular tumor antigen(s) and incorporating freedom to target a range of antigens, as well as giving them the ability to be turned on/off efficaciously. In order to address the challenges with CAR T cell-induced toxicity, inducible and/or programmable CAR systems and safety switches like suicide genes are appealing options when building novel strategies [180]. As discussed earlier, combination therapies may be a promising strategy to endorse CAR T cell activity and persistence. The clinical and molecular endpoints and correlating data that are gathered before, during, and after therapy can have an impact on future combination trials in order to comprehend the mechanisms of response and resistance to CAR-mediated treatment of brain cancer.
Several small molecule inhibitors have been discovered that can inhibit tumor growth, survival, angiogenesis, and metastasis by interfering with different transcription factors. Tyrosine kinase inhibitors have only had a modest therapeutic impact when used as monotherapy in clinical trials for glioblastoma [181]. However, tyrosine kinase inhibitors in combination with CAR T cells have demonstrated synergistic results in the treatment of other forms of solid tumors, making them a compelling candidate for glioblastoma testing [182, 183]. LB-100, a protein phosphatase 2A inhibitor, might augment the efficacy of CAIX-specific CAR T cells in the treatment of glioblastoma [184].
In many clinical studies, CAR T cells used as monotherapy have been found not to be sufficient to induce sustained clinical responses. However, one might still argue that the lack of clinical response to the treatment with CAR T cells alone is partially due to the recruitment of patients at advanced stages of the disease with a high tumor burden. An alternative therapeutic strategy with CAR T cells is the incorporation of a CAR construct into other types of effector immune cells such as NK cells. Oncolytic viruses can accelerate tumor lysis by selectively attacking cancer cells while sparing healthy cells, which can send out alarm signals and boost the immune systems [185]. In addition, oncolytic viruses may work in conjunction with CAR T therapy provided they are genetically transformed to express therapeutic transgenes endorsing a suppressive tumor microenvironment. Other promising combinatorial approaches include antagonistic antibodies specific to the 4-1BB co-stimulatory receptor, which can rapidly activate CAR T cells [186]. Vaccines containing glioma-associated antigens or dendritic cells loaded with mRNA or tumor lysate have been used to treat primary brain tumors, and they may work in tandem with CAR T therapy to overcome tumor heterogeneity and induce an endogenous immune response [187]. A combination of anti-podoplanin CAR T cells with oncolytic herpes virus G47D successfully reduced ‘on-target off tumor’ toxicity acting against patient-derived glioma stem cells while sparing normal cells [188].
Tumor irradiation acts somewhat as an in situ vaccine as it endorses the release of tumor-associated antigens that prompt antigen-presenting cells to migrate to draining lymph nodes, where they prime cytotoxic CD8+ T cells to generate an adaptive immune response including increased expression of T cell receptors [189]. Given that radiation plays a part in the recruitment of the adaptive immune response that is stimulated by the stimulator of the interferon gene (STING), it may also help with the trafficking and persistence issues of CAR T cells. Since the expansion of T cell receptors and activation of dendritic cells might result in ‘epitope spreading’ and immunologic memory against several tumor antigens, radiation and concurrent STING activation may also address post-CAR T antigen escape [190].
Patients with glioma have seen relatively modest success from CAR T cells, despite the fact that they have demonstrated exceptional effectiveness in the treatment of hematological malignancies [26]. Clearly, the ability of CAR T cell therapy to treat glioma is largely dependent on how the design of the cell constructs themselves are altered. CAR T cells can be significantly modified using contemporary genetic technologies like CRISPR/Cas9 in order to increase their therapeutic efficacy, persistence, and safety. Majority of glioma models employ immunosuppressed mice, which do not accurately replicate the tumor microenvironment to detect CAR T cell responses [191]. For the effective development of manipulation tactics, a deeper comprehension of the CNS environment in which CAR T cells function is important. Given how powerful immunosuppressive myeloid cells are and how they are present within the CNS, gene modifications intended to offset their effects may be supportive.
For the treatment of brain cancer, significant progress has been made with the identification of tumor-specific antigens and the development of cutting-edge CAR designs. Even though combinatorial targeting has shown promise in addressing tumor heterogeneity, research is still going on to find the ideal combination of targeted antigens. Combinatorial regimens that combine CAR therapeutics with immunomodulators and immune checkpoint inhibitors are likely to improve the effectiveness with which CAR T cell therapies can cause “epitope spreading” and so address the issue of antigen loss or antigen low escape. The majority of the confusion is caused by uncertainty regarding the dynamic changes tumors go through after an immune assault. Therefore, identifying the changes in intra-tumor subpopulations at the single-cell level following CAR therapy might direct CAR T cell therapy to properly address tumor heterogeneity. Overall, several strategies, including the insertion of cytokine transgenes, gene knock-out and knock-in, control of CAR production and activity, as well as multi-antigen targeting, show good promise in the field of anti-glioma CAR T cell treatment. The predicted advancements depend on continued development in removing the main obstacles to CAR T cell therapy of brain tumors i.e. tumor heterogeneity, T cell exhaustion, suppressive microenvironment, antigen escape, and insufficient T cell trafficking. An enhanced focus on the identification of ideal target antigens is an important area of future research that is required to achieve the potential of CAR T cell therapy for any form of solid tumors. The tumor surfaceome can now be completely cataloged to find a suitable set of differentially expressed cell surface antigens that can be incorporated into multi-specific CAR T cells. Efforts to boost tumor sensitivity to CAR therapy by antigen modulation may also improve outcomes in the long run, but they must be carefully assessed for potential off-target effects. Exhaustion-resistant CAR T cells need to be designed to produce a powerful, long-lasting cytotoxic response. Developing ways to make T cells exhaustion-resistant will significantly improve the efficacy of adoptive cell treatments. Measures to minimize exhaustion are also likely to limit the impact of inhibitory signals produced by the tumor microenvironment.
Conclusion and outlooks
CAR T cells could be a promising therapeutic option for brain cancer since so many potential target tumor antigens have been identified so far. However, one of the biggest hurdles in this field is choosing a suitable antigen to target, especially one that is expressed in brain cancer and brain cancer-initiating cells and destroys them without harming normal brain cells. Understanding the optimal trafficking of CAR T cells to brain tumors is still to be clearly interpreted. Though most clinical trials have demonstrated that CAR T cells as a monotherapy are often ineffective in cases of solid tumors due to their immune escape mechanisms, the efficacy and scope of CAR T cell therapies are continually being expanded, and innovative approaches are being explored to improve safety as well as efficacy. These innovative approaches would lead to the wider applications of this technology for the efficient management of brain cancer through the optimization of CAR T cell biology and others. However, concerns with CAR T cells also spin around their cost and scale-up, which also require substantial attention.
Abbreviations
- CAR T cells:
-
Chimeric antigen receptor T cells
- CNS:
-
Central nervous system
- CARs:
-
Chimeric antigen receptors
- MHC:
-
Major histocompatibility complex
- BBB:
-
Blood-brain barrier
- BBTB:
-
Blood-brain tumor barrier
- TCR:
-
T cell receptor
- TRAC:
-
T cell receptor α constant
- FDA:
-
Food and drug administration
References
Wang SS, Bandopadhayay P, Jenkins MR. Towards immunotherapy for pediatric brain tumors. Trends Immunol. 2019;40(8):748–61. https://doi.org/10.1016/j.it.2019.05.009.
Wu S, Yang W, Zhang H, Ren Y, Fang Z, Yuan C, et al. The prognostic landscape of tumor-infiltrating immune cells and immune checkpoints in glioblastoma. Technol Cancer Res Treat. 2019a;18:1533033819869949. https://doi.org/10.1177/1533033819869949.
Yip A, Webster RM. The market for chimeric antigen receptor T cell therapies. Nat Rev Drug Discov. 2018;17(3):161–2. https://doi.org/10.1038/nrd.2017.266.
Akhavan D, Alizadeh D, Wang D, Weist MR, Shepphird JK, Brown CE. CAR T cells for brain tumors: lessons learned and road ahead. Immunol Rev. 2019;290(1):60–84. https://doi.org/10.1111/imr.12773.
McGrail DJ, Pilié PG, Rashid NU, Voorwerk L, Slagter M, Kok M, et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann Oncol. 2021;32(5):661–72. https://doi.org/10.1016/j.annonc.2021.02.006.
Kheirollahi M, Dashti S, Khalaj Z, Nazemroaia F, Mahzouni P. Brain tumors: Special characters for research and banking. Adv Biomed Res. 2015;4:4. https://doi.org/10.4103/2277-9175.148261.
Marinelli JP, Beeler CJ, Carlson ML, Caye-Thomasen P, Spear SA, Erbele ID. Global incidence of sporadic vestibular schwannoma: a systematic review. Otolaryngol Head Neck Surg. 2022;167(2):209–14. https://doi.org/10.1177/01945998211042006.
Chakraborty P, Das SS, Dey A, Chakraborty A, Bhattacharyya C, Kandimalla R, et al. Quantum dots: The cutting-edge nanotheranostics in brain cancer management. J Control Release. 2022;350:698–715. https://doi.org/10.1016/j.jconrel.2022.08.047.
Qi X, Jha SK, Jha NK, Dewanjee S, Dey A, Deka R, et al. Antioxidants in brain tumors: current therapeutic significance and future prospects. Mol Cancer. 2022;21(1):204. https://doi.org/10.1186/s12943-022-01668-9.
Dewanjee S, Dua TK, Bhattacharjee N, Das A, Gangopadhyay M, Khanra R, et al. Natural Products as Alternative Choices for P-Glycoprotein (P-gp) Inhibition. Molecules. 2017;22(6):871. https://doi.org/10.3390/molecules22060871.
Zhao XQ, He LM, Mao KY, Chen DM, Jiang HB, Liu ZP. The research status of immune checkpoint blockade by anti-CTLA4 and a nti-PD1/PD-l1 antibodies in tumor immunotherapy in China: a bibliometrics study. Medicine. 2018;97(15):e0276. https://doi.org/10.1097/MD.0000000000010276.
Sampson JH, Gunn MD, Fecci PE, Ashley DM. Brain immunology and immunotherapy in brain tumours. Nat Rev Cancer. 2020;20(1):12–25. https://doi.org/10.1038/s41568-019-0224-7.
Haist C, Poschinski Z, Bister A, Hoffmann MJ, Grunewald CM, Hamacher A, et al. Engineering a single-chain variable fragment of cetuximab for CAR T-cell therapy against head and neck squamous cell carcinomas. Oral Oncol. 2022;129:105867. https://doi.org/10.1016/j.oraloncology.2022.105867.
Rafiq S, Hackett CS, Brentjens RJ. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat Rev Clin Oncol. 2020;17(3):147–67. https://doi.org/10.1038/s41571-019-0297-y.
Umut Ö, Gottschlich A, Endres S, Kobold S. CAR T cell therapy in solid tumors: a short review. Memo. 2021;14(2):143–9. https://doi.org/10.1007/s12254-021-00703-7.
Holzinger A, Abken H. Treatment with Living Drugs: Pharmaceutical Aspects of CAR T Cells. Pharmacology. 2022;107(9–10):446–63. https://doi.org/10.1159/000525052.
Schaap FG, French PJ, Bovée JV. Mutations in the isocitrate dehydrogenase genes IDH1 and IDH2 in tumors. Adv Anat Pathol. 2013;20(1):32–8. https://doi.org/10.1097/PAP.0b013e31827b654d.
Berghoff AS, Kiesel B, Widhalm G, Wilhelm D, Rajky O, Kurscheid S, et al. Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro Oncol. 2017;19(11):1460–8. https://doi.org/10.1093/neuonc/nox054.
Valdebenito S, D'Amico D, Eugenin E. Novel approaches for glioblastoma treatment: Focus on tumor heterogeneity, treatment resistance, and computational tools. Cancer Rep (Hoboken). 2019;2(6):e1220. https://doi.org/10.1002/cnr2.1220.
Ocasio JK, Babcock B, Malawsky D, Weir SJ, Loo L, Simon JM, et al. scRNA-seq in medulloblastoma shows cellular heterogeneity and lineage expansion support resistance to SHH inhibitor therapy. Nat Commun. 2019;10(1):5829. https://doi.org/10.1038/s41467-019-13657-6 Erratum in: Nat Commun. 2022;13(1):3048.
Han D, Xu Z, Zhuang Y, Ye Z, Qian Q. Current Progress in CAR-T Cell Therapy for Hematological Malignancies. J Cancer. 2021;12(2):326–34. https://doi.org/10.7150/jca.48976.
Borogovac A, Keruakous A, Bycko M, Holter Chakrabarty J, Ibrahimi S, Khawandanah M, et al. Safety and feasibility of outpatient chimeric antigen receptor (CAR) T-cell therapy: experience from a tertiary care center. Bone Marrow Transplant. 2022;57(6):1025–7. https://doi.org/10.1038/s41409-022-01664-z.
Fu W, Lei C, Liu S, Cui Y, Wang C, Qian K, et al. CAR exosomes derived from effector CAR-T cells have potent antitumour effects and low toxicity. Nat Commun. 2019;10(1):4355. https://doi.org/10.1038/s41467-019-12321-3.
Avanzi MP, Yeku O, Li X, Wijewarnasuriya DP, van Leeuwen DG, Cheung K, et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and through Activation of the Endogenous Immune System. Cell Rep. 2018;23(7):2130–41. https://doi.org/10.1016/j.celrep.2018.04.051.
Wagner J, Wickman E, DeRenzo C, Gottschalk S. CAR T Cell Therapy for Solid Tumors: Bright Future or Dark Reality? Mol Ther. 2020;28(11):2320–39. https://doi.org/10.1016/j.ymthe.2020.09.015.
Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer J. 2021;11(4):69. https://doi.org/10.1038/s41408-021-00459-7.
Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385(9967):517–28. https://doi.org/10.1016/S0140-6736(14)61403-3.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377(26):2531–44. https://doi.org/10.1056/NEJMoa1707447.
Hou AJ, Chen LC, Chen YY. Navigating CAR-T cells through the solid-tumour microenvironment. Nat Rev Drug Discov. 2021;20(7):531–50. https://doi.org/10.1038/s41573-021-00189-2.
Chandran SS, Klebanoff CA. T cell receptor-based cancer immunotherapy: emerging efficacy and pathways of resistance. Immunol Rev. 2019;290(1):127–47. https://doi.org/10.1111/imr.12772.
Rostamian H, Khakpoor-Koosheh M, Fallah-Mehrjardi K, Mirzaei HR, Brown CE. Mitochondria as playmakers of CAR T-cell fate and longevity. Cancer Immunol Res. 2021;9(8):856–61. https://doi.org/10.1158/2326-6066.CIR-21-0110.
Koneru M, Purdon TJ, Spriggs D, Koneru S, Brentjens RJ. IL-12 secreting tumor-targeted chimeric antigen receptor T cells eradicate ovarian tumors in vivo. Oncoimmunology. 2015;4(3):e994446. https://doi.org/10.4161/2162402X.2014.994446.
Hu B, Ren J, Luo Y, et al. Augmentation of antitumor immu- nity by human and mouse CAR T cells secreting IL-18. Cell Rep. 2017;20(13):3025–33. https://doi.org/10.1016/j.celrep.2017.09.002.
Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, et al. Constitutive signaling from an engineered IL7 receptor promotes durable tumor elimination by tumor-redirected T cells. Cancer Discov. 2017;7(11):1238–47. https://doi.org/10.1158/2159-8290.CD-17-0538.
Tang L, Zheng Y, Melo MB, Mabardi L, Castaño AP, Xie YQ, et al. Enhancing T cell therapy through TCR-signaling-responsive nanoparticle drug delivery. Nat Biotechnol. 2018;36(8):707–16. https://doi.org/10.1038/nbt.4181.
Chinnasamy D, Yu Z, Kerkar SP, Zhang L, Morgan RA, Restifo NP, et al. Local delivery of interleukin-12 using T cells targeting VEGF receptor-2 eradicates multiple vascularized tumors in mice. Clin Cancer Res. 2012;18(6):1672–83. https://doi.org/10.1158/1078-0432.CCR-11-3050.
Koneru M, O'Cearbhaill R, Pendharkar S, Spriggs DR, Brentjens RJ. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16(ecto) directed chimeric antigen receptors for recurrent ovarian cancer. J Transl Med. 2015;13:102. https://doi.org/10.1186/s12967-015-0460-x.
Kloss CC, Lee J, Zhang A, Chen F, Melenhorst JJ, Lacey SF, et al. Dominant-negative TGF-β receptor enhances PSMA-targeted human CAR T cell proliferation and augments prostate cancer eradication. Mol Ther. 2018;26(7):1855–66. https://doi.org/10.1016/j.ymthe.2018.05.003.
Mohammed S, Sukumaran S, Bajgain P, Watanabe N, Heslop HE, Rooney CM, et al. Improving chimeric antigen receptor-modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Mol Ther. 2017;25(1):249–58. https://doi.org/10.1016/j.ymthe.2016.10.016.
Chang ZL, Lorenzini MH, Chen X, Tran U, Bangayan NJ, Chen YY. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat Chem Biol. 2018;14(3):317–24. https://doi.org/10.1038/nchembio.2565.
Liu X, Ranganathan R, Jiang S, Fang C, Sun J, Kim S, et al. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res. 2016;76(6):1578–90. https://doi.org/10.1158/0008-5472.CAN-15-2524.
Suarez ER, Chang de K, Sun J, Sui J, Freeman GJ, Signoretti S, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget. 2016;7(23):34341–55. https://doi.org/10.18632/oncotarget.9114.
Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. 2017;23:2255–66. https://doi.org/10.1158/1078-0432.CCR-16-1300.
Ponterio E, De Maria R, Haas TL. Identification of targets to redirect CAR T cells in glioblastoma and colorectal cancer: an arduous venture. Front Immunol. 2020;11:565631. https://doi.org/10.3389/fimmu.2020.565631.
Walsh Z, Ross S, Fry TJ. Multi-specific CAR targeting to prevent antigen escape. Curr Hematol Malig Rep. 2019;14(5):451–9. https://doi.org/10.1007/s11899-019-00537-5.
Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J Clin Invest. 2016;126(8):3036–52. https://doi.org/10.1172/JCI83416.
Qin H, Ramakrishna S, Nguyen S, Fountaine TJ, Ponduri A, Stetler-Stevenson M, et al. Preclinical development of bivalent chimeric antigen receptors targeting both CD19 and CD22. Mol Ther Oncolytics. 2018;11:127–37. https://doi.org/10.1016/j.omto.2018.10.006.
Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro-Oncology. 2018;20(4):506–18. https://doi.org/10.1093/neuonc/nox182.
Choi BD, Yu X, Castano AP, Bouffard AA, Curry WT, Carter BS, et al. Abstract LB-066: BiTE-armored CARs overcome antigen escape in EGFRvIII-targeted therapy for glioblastoma. Cancer Res. 2019;79(13_Supplement):LB–066. https://doi.org/10.1158/1538-7445.AM2019-LB-066.
Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. 2016;16(9):566–81. https://doi.org/10.1038/nrc.2016.97.
Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest. 2016;126(10):3814–26. https://doi.org/10.1172/JCI87366.
Zah E, Lin MY, Silva-Benedict A, Jensen MC, Chen YY. T cells expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res. 2016;4(6):498–508. https://doi.org/10.1158/2326-6066.CIR-15-0231.
Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell. 2016;164(4):770–9. https://doi.org/10.1016/j.cell.2016.01.011.
Rodgers DT, Mazagova M, Hampton EN, Cao Y, Ramadoss NS, Hardy IR, et al. Switch-mediated activation and retargeting of CART cells for B-cell malignancies. Proc Natl Acad Sci U S A. 2016;113(4):E459–68. https://doi.org/10.1073/pnas.1524155113.
Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell. 2018;173(6):1426–1438.e11. https://doi.org/10.1016/j.cell.2018.03.038.
Juillerat A, Marechal A, Filhol JM, Valogne Y, Valton J, Duclert A, et al. An oxygen sensitive self-decision making engineered CAR T-cell. Sci Rep. 2017;7:39833. https://doi.org/10.1038/srep39833.
Brown CE, Alizadeh D, Starr R, Weng L, Wagner JR, Naranjo A, et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N Engl J Med. 2016;375(26):2561–9. https://doi.org/10.1056/NEJMoa1610497.
Brown CE, Aguilar B, Starr R, Yang X, Chang WC, Weng L, et al. Optimization of IL13Rα2-targeted chimeric antigen receptor T cells for improved anti-tumor efficacy against glioblastoma. Mol Ther. 2018;26(1):31–44. https://doi.org/10.1016/j.ymthe.2017.10.002.
Priceman SJ, Tilakawardane D, Jeang B, Aguilar B, Murad JP, Park AK, et al. Regional delivery of chimeric antigen receptor-engineered T cells effectively targets HER2 breast cancer metastasis to the brain. Clin Cancer Res. 2018;24(1):95–105. https://doi.org/10.1158/1078-0432.CCR-17-2041.
Turk OM, Woodall RC, Gutova M, Brown CE, Rockne RC, Munson JM. Delivery strategies for cell-based therapies in the brain: overcoming multiple barriers. Drug Deliv Transl Res. 2021;11(6):2448–67. https://doi.org/10.1007/s13346-021-01079-1.
Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113(25):6392–402. https://doi.org/10.1182/blood-2009-03-209650.
Jacquelot N, Duong CPM, Belz GT, Zitvogel L. Targeting chemokines and chemokine receptors in melanoma and other cancers. Front Immunol. 2018;9:2480. https://doi.org/10.3389/fimmu.2018.02480.
Foeng J, Comerford I, McColl SR. Harnessing the chemokine system to home CAR-T cells into solid tumors. Cell Rep Med. 2022;3(3):100543. https://doi.org/10.1016/j.xcrm.2022.100543.
Brown CE, Vishwanath RP, Aguilar B, Starr R, Najbauer J, Aboody KS, et al. Tumor-derived chemokine MCP-1/CCL2 is sufficient for mediating tumor tropism of adoptively transferred T cells. J Immunol. 2007;179(5):3332–41. https://doi.org/10.4049/jimmunol.179.5.3332.
Vitanza NA, Johnson AJ, Wilson AL, Brown C, Yokoyama JK, Künkele A, et al. Locoregional infusion of HER2-specific CAR T cells in children and young adults with recurrent or refractory CNS tumors: an interim analysis. Nat Med. 2021;27(9):1544–52. https://doi.org/10.1038/s41591-021-01404-8.
Richards RM, Sotillo E, Majzner RG. CAR T cell therapy for neuroblastoma. Front Immunol. 2018;9:2380. https://doi.org/10.3389/fimmu.2018.02380.
Lanitis E, Irving M, Coukos G. Targeting the tumor vasculature to enhance T cell activity. Curr Opin Immunol. 2015;33:55–63. https://doi.org/10.1016/j.coi.2015.01.011.
Liu G, Rui W, Zhao X, Lin X. Enhancing CAR-T cell efficacy in solid tumors by targeting the tumor microenvironment. Cell Mol Immunol. 2021;18(5):1085–95. https://doi.org/10.1038/s41423-021-00655-2.
Yaghoubi SS, Jensen MC, Satyamurthy N, Budhiraja S, Paik D, Czernin J, et al. Noninvasive detection of therapeutic cytolytic T cells with 18F-FHBG PET in a patient with glioma. Nat Clin Pract Oncol. 2009;6(1):53–8. https://doi.org/10.1038/ncponc1278.
Keu KV, Witney TH, Yaghoubi S, Rosenberg J, Kurien A, Magnusson R, et al. Reporter gene imaging of targeted T cell immunotherapy in recurrent glioma. Sci Transl Med. 2017;9(373):eaag2196. https://doi.org/10.1126/scitranslmed.aag2196.
Miyagawa T, Gogiberidze G, Serganova I, Cai S, Balatoni JA, Thaler HT, et al. Imaging of HSV-tk reporter gene expression: comparison between [18F] FEAU,[18F] FFEAU, and other imaging probes. J Nucl Med. 2008;49(4):637–48. https://doi.org/10.2967/jnumed.107.046227.
Moroz MA, Zhang H, Lee J, Moroz E, Zurita J, Shenker L, et al. Comparative analysis of T cell imaging with human nuclear reporter genes. J Nucl Med. 2015;56(7):1055–60. https://doi.org/10.2967/jnumed.115.159855.
Gosmann D, Russelli L, Weber WA, Schwaiger M, Krackhardt AM, D'Alessandria C. Promise and challenges of clinical non-invasive T-cell tracking in the era of cancer immunotherapy. EJNMMI Res. 2022;12(1):5. https://doi.org/10.1186/s13550-022-00877-z.
Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(224):224ra25. https://doi.org/10.1126/scitranslmed.3008226.
Stern LA, Gholamin S, Moraga I, Yang X, Saravanakumar S, Cohen JR, et al. Engineered IL13 variants direct specificity of IL13Rα2-targeted CAR T cell therapy. Proc Natl Acad Sci U S A. 2022;119(33):e2112006119. https://doi.org/10.1073/pnas.2112006119.
Brown CE, Warden CD, Starr R, Deng X, Badie B, Yuan YC, et al. Glioma IL13Rα2 is associated with mesenchymal signature gene expression and poor patient prognosis. PLoS One. 2013;8(10):e77769. https://doi.org/10.1371/journal.pone.0077769 Erratum in: PLoS One. 2018;;13(9):e0204463.
Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004;64(24):9160–6. https://doi.org/10.1158/0008-5472.CAN-04-0454.
Jonnalagadda M, Mardiros A, Urak R, Wang X, Hoffman LJ, Bernanke A, et al. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid fc receptor binding and improve T cell persistence and antitumor efficacy. Mol Ther. 2015;23(4):757–68. https://doi.org/10.1038/mt.2014.208.
U.S. National Institutes of Health n.d.-m. ClinicalTrials.gov Cellular Adoptive Immunotherapy Using Genetically Modified T-Lymphocytes in Treating Patients With Recurrent or Refractory High-Grade Malignant Glioma. Official Title: Pilot Feasibility and Safety Study of Cellular Immunotherapy for Recurrent/Refractory Malignant Glioma Using Genetically-Modified Autologous CD8+ T Cell Clones. Identifier: NCT00730613. Available online: https://clinicaltrials.gov/ct2/show/NCT00730613. Accessed 17 Oct 2022.
U.S. National Institutes of Health n.d.-ad. ClinicalTrials.gov Phase I Study of Cellular Immunotherapy for Recurrent/Refractory Malignant Glioma Using Intratumoral Infusions of GRm13Z40–2, An Allogeneic CD8+ Cytolitic T-Cell Line Genetically Modified to Express the IL 13-Zetakine and HyTK and to be Resistant to Glucocorticoids, in Combination With Interleukin-2. Official Title: Phase I Study of Cellular Immunotherapy for Recurrent/Refractory Malignant Glioma Using Intratumoral Infusions of GRm13Z40–2, An Allogeneic CD8+ Cytolitic T-Cell Line Genetically Modified to Express the IL 13-Zetakine and HyTK and to be Resistant to Glucocorticoids, in Combination With Interleukin-2. Identifier: NCT01082926. Available online: https://www.clinicaltrials.gov/ct2/show/NCT01082926. Accessed 17 Oct 2022.
Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC, et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin Cancer Res. 2015;21(18):4062–72. https://doi.org/10.1158/1078-0432.CCR-15-0428.
U.S. National Institutes of Health n.d.-u. ClinicalTrials.gov Genetically Modified T-cells in Treating Patients With Recurrent or Refractory Malignant Glioma. Official Title: Phase I Study of Cellular ImmunoTx Using Memory Enriched T Cells Lentivirally Transduced to Express an IL13Rα2-Specific, Hinge-Optimized, 41BB-Costimulatory Chimeric Receptor and a Truncated CD19 for Pts With Rec/Ref MaligGlioma. Identifier: NCT02208362 Available online: https://clinicaltrials.gov/ct2/show/NCT02208362. Accessed 17 Oct 2022.
U.S. National Institutes of Health n.d.-f. ClinicalTrials.gov Brain Tumor-Specific Immune Cells (IL13Ralpha2-CAR T Cells) for the Treatment of Leptomeningeal Glioblastoma, Ependymoma, or Medulloblastoma. Official Title: A Phase 1 Study to Evaluate IL13Rα2-Targeted Chimeric Antigen Receptor (CAR) T Cells for Adult Patients With Leptomeningeal Glioblastoma, Ependymoma or Medulloblastoma. Identifier: NCT04661384 Available online: https://clinicaltrials.gov/ct2/show/NCT04661384. Accessed 20 Oct 2022.
U.S. National Institutes of Health n.d.-i. ClinicalTrials.gov CAR T Cells After Lymphodepletion for the Treatment of IL13Rα2 Positi ve Recurrent or Refractory Brain Tumors in Children. Official Title: Phase I Study of Cellular Immunotherapy Using Memory Enric hed T Cells Lentivirally Transduced to Express an IL13Rα2-Targeting, Hinge-Optimized, 41BB-Costimulatory Chimeric Receptor a nd a Truncated CD19 for Children With Recurrent/Refractory Malignant Brain Tumors. Identifier: NCT04510051 Available online: https://clinicaltrials.gov/ct2/show/NCT04510051. Accessed 20 Oct 2022.
Allen GM, Lim WA. Rethinking cancer targeting strategies in the era of smart cell therapeutics. Nat Rev Cancer. 2022. https://doi.org/10.1038/s41568-022-00505-x.
U.S. National Institutes of Health n.d.-h. ClinicalTrials.gov CAR T Cell Receptor Immunotherapy Targeting EGFRvIII for Patients With Malignant Gliomas Expressing EGFRvIII. Official Title: A Phase I/II Study of the Safety and Feasibility of Administering T Cells Expressing Anti-EGFRvIII Chimeric Antigen Receptor to Patients With Malignant Gliomas Expressing EGFRvIII. Identifier: NCT01454596 Available online: https://clinicaltrials.gov/ct2/show/NCT01454596. Accessed 18 Oct 2022.
Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot trial of adoptive transfer of chimeric antigen receptortransduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother. 2019;42(4):126–35. https://doi.org/10.1097/CJI.0000000000000260.
Zhang Z, Jiang J, Wu X, Zhang M, Luo D, Zhang R, et al. Chimeric antigen receptor T cell targeting EGFRvIII for metastatic lung cancer therapy. Front Med. 2019;13(1):57–68. https://doi.org/10.1007/s11684-019-0683-y.
Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. 2015;7(275):275ra22. https://doi.org/10.1126/scitranslmed.aaa4963.
U.S. National Institutes of Health n.d.-d. ClinicalTrials.gov Autologous T Cells Redirected to EGFRVIII-With a Chimeric Antigen Receptor in Patients With EGFRVIII+ Glioblastoma. Official Title: Pilot Study of Autologous T Cells Redirected to EGFRVIII-With a Chimeric Antigen Receptor in Patients With EGFRVIII+ Glioblastoma. Identifier: NCT02209376 Available online: https://clinicaltrials.gov/ct2/show/NCT02209376. Accessed 18 Oct 2022.
O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017;9(399):eaaa0984. https://doi.org/10.1126/scitranslmed.aaa0984.
U.S. National Institutes of Health n.d.-k. ClinicalTrials.gov CART-EGFRvIII + Pembrolizumab in GBM. Official Title: Phase 1 Study of EGFRvIII-Directed CAR T Cells Combined With PD-1 Inhibition in Patients With Newly Diagnosed, MGMT-Unmethylated Glioblastoma Identifier: NCT03726515 Available online: https://clinicaltrials.gov/ct2/show/NCT03726515. Accessed 17 Oct 2022.
Scott AM, Lee FT, Tebbutt N, Herbertson R, Gill SS, Liu Z, et al. A phase I clinical trial with monoclonal antibody ch806 targeting transitional state and mutant epidermal growth factor receptors. Proc Natl Acad Sci U S A. 2007;104(10):4071–6. https://doi.org/10.1073/pnas.0611693104.
U.S. National Institutes of Health (n.d.-a). ClinicalTrials.gov EGFR806-specific CAR T Cell Locoregional Immunotherapy for EGFR-positive Recurrent or Refractory Pediatric CNS Tumors. Official Title: Phase 1 Study of EGFR806-specific CAR T Cell Locoregional Immunotherapy for EGFR-positive Recurrent or Refractory Pediatric Central Nervous System Tumors. Identifier: NCT03638167 Available online: https://clinicaltrials.gov/ct2/show/NCT03638167. Accessed 17 Oct 2022.
U.S. National Institutes of Health n.d.-p. ClinicalTrials.gov EGFR806 CAR T Cell Immunotherapy for Recurrent/Refractory Solid Tumors in Children and Young Adults. Official Title: Phase I Study of EGFR806 CAR T Cell Immunotherapy for Recurrent/Refractory Solid Tumors in Children and Young Adults. Identifier: NCT03618381 Available online: https://clinicaltrials.gov/ct2/show/NCT03618381. Accessed 17 Oct 2022.
Maggs L, Cattaneo G, Dal AE, Moghaddam AS, Ferrone S. CAR T cell-based immunotherapy for the treatment of glioblastoma. Front Neurosci. 2021;15:662064. https://doi.org/10.3389/fnins.2021.662064.
Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18(4):843–51. https://doi.org/10.1038/mt.2010.24.
Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. 2010;16(2):474–85. https://doi.org/10.1158/1078-0432.CCR-09-1322.
Ahmed N, Brawley VS, Hegde M, Robertson C, Ghazi A, Gerken C, et al. Human epidermal growth factor receptor 2 (HER2) –specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol. 2015;33(15):1688–96. https://doi.org/10.1200/JCO.2014.58.0225.
U.S. National Institutes of Health n.d.-o. ClinicalTrials.gov CMV-specific Cytotoxic T Lymphocytes Expressing CAR Targeting HER2 in Patients With GBM (HERT-GBM). Official Title: Administration of HER2 Chimeric Antigen Receptor Expressing CMV-Specific Cytotoxic T Cells Ins Patients With Glioblastoma Multiforme (HERT-GBM). Identifier: NCT01109095 Available online: https://clinicaltrials.gov/ct2/show/NCT01109095. Accessed 17 Oct 2022.
Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. HER2-specific c AQ10 himeric antigen receptor-modified virus-specific T cells for progressive glioblastoma: a phase 1 dose-escalation trial. JAMA Oncol. 2017;3(8):1094–101. https://doi.org/10.1001/jamaoncol.2017.0184.
U.S. National Institutes of Health n.d.-aa. ClinicalTrials.gov Memory-Enriched T Cells in Treating Patients With Recurrent or Refractory Grade III-IV Glioma. Official Title: Phase I Study of Cellular Immunotherapy Using Memory-Enriched T Cells Lentivirally Transduced to Express a HER2-Specific, Hinge-Optimized, 41BB-Costimulatory Chimeric Receptor and a Truncated CD19 for Patients With Recurrent/Refractory Malignant Glioma. Identifier: NCT03389230 Available online: https://clinicaltrials.gov/ct2/show/NCT03389230. Accessed 17 Oct 2022.
U.S. National Institutes of Health n.d.-v. ClinicalTrials.gov HER2-CAR T Cells in Treating Patients With Recurrent Brain or Leptomeningeal Metastases. Official Title: A Phase 1 Cellular Immunotherapy Study of Intraventricularly Administered Autologous HER2-Targeted Chimeric Antigen Receptor (HER2-CAR) T Cells in Patients With Brain and/or Leptomeningeal Metastases From HER2 Positive Cancers. Identifier: NCT03696030 Available online: https://clinicaltrials.gov/ct2/show/NCT03696030. Accessed 17 Oct 2022.
Koirala N, Dey N, Aske J, De P. Targeting cell cycle progression in HER2+ breast cancer: an emerging treatment opportunity. Int J Mol Sci. 2022;23(12):6547. https://doi.org/10.3390/ijms23126547.
U.S. National Institutes of Health n.d.-w. ClinicalTrials.gov HER2-specific CAR T Cell Locoregional Immunotherapy for HER2-positive Recurrent/Refractory Pediatric CNS Tumors. Official Title: Phase 1 Study of HER2-Specific CAR T Cell Locoregional Immunotherapy for HER2 Positive Recurrent/Refractory Pediatric Central Nervous System Tumors. Identifier: NCT03500991 Available online: https://clinicaltrials.gov/ct2/show/NCT03500991. Accessed 19 Oct 2022.
U.S. National Institutes of Health n.d.-ag. ClinicalTrials.gov T Cells Expressing HER2-specific Chimeric Antigen Receptors (CAR) for Patients With HER2-Positive CNS Tumors (iCAR). Official Title: Phase I Study of Intracranial Injection of T Cells Expressing HER2-specific Chimeric Antigen Receptors (CAR) in Subjects With HER2-Positive Tumors of the Central Nervous System (iCAR). Identifier: NCT02442297 Available online: https://clinicaltrials.gov/ct2/show/NCT02442297. Accessed 17 Oct 2022.
U.S. National Institutes of Health n.d.-x. ClinicalTrials.gov HER2-specific Chimeric Antigen Receptor (CAR) T Cells for Children With Ependymoma. Official Title: Phase 1 Trial of Autologous HER2-specific CAR T Cells in Pediatric Patients With Refractory or Recurrent Ependymoma. Identifier: NCT04903080 Available online: https://clinicaltrials.gov/ct2/show/NCT04903080. Accessed 19 Oct 2022.
Yang K, Wu Z, Zhang H, Zhang N, Wu W, Wang Z, et al. Glioma targeted therapy: insight into future of molecular approaches. Mol Cancer. 2022;21(1):39. https://doi.org/10.1186/s12943-022-01513-z.
Majzner RG, Theruvath JL, Nellan A, Heitzeneder S, Cui Y, Mount CW, et al. CAR T cells targeting B7-H3, a Pan-Cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin Cancer Res. 2019;25(8):2560–74. https://doi.org/10.1158/1078-0432.CCR-18-0432.
Tang X, Zhao S, Zhang Y, Wang Y, Zhang Z, Yang M, et al. B7-H3 as a novel CAR-T therapeutic target for glioblastoma. Mol Ther Oncolytics. 2019;14:279–87. https://doi.org/10.1016/j.omto.2019.07.002.
U.S. National Institutes of Health n.d.-e. ClinicalTrials.gov B7-H3 CAR-T for Recurrent or Refractory Glioblastoma. Official Title: B7-H3-Targeted Chimeric Antigen Receptor (CAR) T Cells in Treating Patients With Recurrent or Refractory Glioblastoma. Identifier: NCT04077866 Available online: https://clinicaltrials.gov/ct2/show/NCT04077866. Accessed 17 Oct 2022.
U.S. National Institutes of Health n.d.-af. ClinicalTrials.gov Study of B7-H3-Specific CAR T Cell Locoregional Immunotherapy for Diffuse Intrinsic Pontine Glioma/Diffuse Midline Glioma and Recurrent or Refractory Pediatric Central Nervous System Tumors. Official Title: Phase 1 Study of B7-H3-Specific CAR T Cell Locoregional Immunotherapy for Diffuse Intrinsic Pontine Glioma/Diffuse Midline Glioma and Recurrent or Refractory Pediatric Central Nervous System Tumors. Identifier: NCT04185038 Available online: https://clinicaltrials.gov/ct2/show/NCT04185038. Accessed 19 Oct 2022.
Xin X, Zeng X, Gu H, Li M, Tan H, Jin Z, et al. CD147/EMMPRIN overexpression and prognosis in cancer: a systematic review and meta-analysis. Sci Rep. 2016;6:32804. https://doi.org/10.1038/srep32804.
U.S. National Institutes of Health n.d.-l. ClinicalTrials.gov CD147-CART Cells in Patients With Recurrent Malignant Glioma. Official Title: A Clinical Study to Investigate the Safety, Tolerance and Efficacy Evaluation of Single-centre, Open-label of Local Treatment of CD147-CART in Recurrent Glioblastoma. Identifier: NCT04045847 Available online: https://clinicaltrials.gov/ct2/show/NCT04045847. Accessed 18 Oct 2022.
Golinelli G, Grisendi G, Prapa M, Bestagno M, Spano C, Rossignoli F, et al. Targeting GD2-positive glioblastoma by chimeric antigen receptor empowered mesenchymal progenitors. Cancer Gene Ther. 2020;27(7–8):558–70. https://doi.org/10.1038/s41417-018-0062-x.
Battula VL, Shi Y, Evans KW, Wang RY, Spaeth EL, Jacamo RO, et al. Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J Clin Invest. 2012;122(6):2066–78. https://doi.org/10.1172/JCI59735.
Mount CW, Majzner RG, Sundaresh S, Arnold EP, Kadapakkam M, Haile S, et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M diffuse midline gliomas. Nat Med. 2018;24(5):572–9. https://doi.org/10.1038/s41591-018-0006-x.
Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptorpositive T cells in patients with neuroblastoma. Blood. 2011;118:6050–6. https://doi.org/10.1182/BLOOD-2011-05-354449.
Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, et al. CAR T cells administered in combination with Lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol Ther. 2017;25(9):2214–24. https://doi.org/10.1016/j.ymthe.2017.05.012.
U.S. National Institutes of Health n.d.-b. ClinicalTrials.gov A Phase I Trial of Anti-GD2 T-cells (1RG-CART). Official Title: A Cancer Research UK Phase I Trial of Anti-GD2 Chimeric Antigen Receptor (CAR) Transduced T-cells (1RG-CART) in Patients With Relapsed or Refractory Neuroblastoma Identifier: NCT02761915 Available online: https://clinicaltrials.gov/ct2/show/NCT02761915. Accessed 19 Oct 2022.
U.S. National Institutes of Health n.d.-g. ClinicalTrials.gov C7R-GD2.CAR T Cells for Patients With GD2-expressing Brain Tumors (GAIL-B). Official Title: Phase I Study of Autologous T Lymphocytes Expressing GD2-specific Chimeric Antigen and Constitutively Active IL-7 Receptors for the Treatment of Patients With GD2-expressing Brain Tumors (GAIL-B). Identifier: NCT04099797 Available online: https://clinicaltrials.gov/ct2/show/NCT04099797. Accessed 18 Oct 2022.
U.S. National Institutes of Health n.d.-c. ClinicalTrials.gov Anti-GD2 CAR T Cells in Pediatric Patients Affected by High Risk and/or Relapsed/Refractory Neuroblastoma or Other GD2-positive Solid Tumors. Official Title: Phase I/II Study of Anti-GD2 Chimeric Antigen Receptor-Expressing T Cells in Pediatric Patients Affected by High Risk and/or Relapsed/Refractory Neuroblastoma or Other GD2-positive Solid Tumors. Identifier: NCT03373097 Available online: https://clinicaltrials.gov/ct2/show/NCT03373097. Accessed 19 Oct 2022.
U.S. National Institutes of Health n.d.-s. ClinicalTrials.gov GD2 CAR T Cells in Diffuse Intrinsic Pontine Gliomas (DIPG) & Spinal Diffuse Midline Glioma (DMG). Official Title: Phase 1 Clinical Trial of Autologous GD2 Chimeric Antigen Receptor (CAR) T Cells (GD2CART) for Diffuse Intrinsic Pontine Gliomas (DIPG) and Spinal Diffuse Midline Glioma (DMG). Identifier: NCT04196413 Available online: https://clinicaltrials.gov/ct2/show/NCT04196413. Accessed 19 Oct 2022.
Delinois LJ, Peón H, Villalobos-Santos JC, Ramírez-Paz J, Miller J, Griebenow KH, et al. A cytochrome c-Chlorotoxin hybrid protein as a possible Antiglioma drug. ChemMedChem. 2020;15(22):2185–92. https://doi.org/10.1002/cmdc.202000373.
Cohen G, Burks SR, Frank JA. Chlorotoxin-a multimodal imaging platform for targeting glioma tumors. Toxins (Basel). 2018;10(12):496. https://doi.org/10.3390/toxins10120496.
Wang D, Starr R, Chang WC, Aguilar B, Alizadeh D, Wright SL, et al. Chlorotoxin-directed CAR T cells for specific and effective targeting of glioblastoma. Sci Transl Med. 2020;12(533):eaaw2672. https://doi.org/10.1126/scitranslmed.aaw2672.
U.S. National Institutes of Health n.d.-n. ClinicalTrials.gov Chimeric Antigen Receptor (CAR) T Cells With a Chlorotoxin Tumor-Targeting Domain for the Treatment of MMP2+ Recurrent or Progressive Glioblastoma. Official Title: A Phase 1 Study to Evaluate Chimeric Antigen Receptor (CAR) T Cells With a Chlorotoxin Tumor-Targeting Domain for Patients With MMP2+ Recurrent or Progressive Glioblastoma. Identifier: NCT04214392 Available online: https://clinicaltrials.gov/ct2/show/NCT04214392. Accessed 18 Oct 2022.
Zhu X, Prasad S, Gaedicke S, Hettich M, Firat E, Niedermann G. Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget. 2015;6(1):171–84. https://doi.org/10.18632/oncotarget.2767.
U.S. National Institutes of Health n.d.-r. ClinicalTrials.gov Engineered Neuroblastoma Cellular Immunotherapy (ENCIT)-01. Official Title: A Phase 1 Feasibility and Safety Study of Cellular Immunotherapy for Recurrent/Refractory Neuroblastoma Using Autologous T-cells Lentivirally Transduced to Express CD171-specific Chimeric Antigen Receptors. Identifier: NCT02311621 Available online: https://clinicaltrials.gov/ct2/show/NCT02311621. Accessed 19 Oct 2022.
Wu N, Zhao X, Liu M, Liu H, Yao W, Zhang Y, et al. Role of microRNA-26b in glioma development and its mediated regulation on EphA2. PLoS One. 2011;6(1):e16264. https://doi.org/10.1371/journal.pone.0016264.
U.S. National Institutes of Health n.d.-j. ClinicalTrials.gov CAR-T Cell Immunotherapy for EphA2 Positive Malignant Glioma Patients. Official Title: Chimeric Antigen Receptor-Modified T Cells for EphA2 Positive Recurrent and Metastatic Malignant Glioma. Identifier: NCT02575261 Available online: https://clinicaltrials.gov/ct2/show/NCT02575261. Accessed 18 Oct 2022.
Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25(3):477–86. https://doi.org/10.1038/s41591-018-0337-7.
Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, López-Janeiro A, Porciuncula A, Idoate MA, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med. 2019;25(3):470–6. https://doi.org/10.1038/s41591-018-0339-5.
U.S. National Institutes of Health n.d.-y. ClinicalTrials.gov IL13Ra2-CAR T Cells With or Without Nivolumab and Ipilimumab in Treating Patients With GBM. Official Title: A Phase 1 Study to Evaluate IL13Rα2-Targeted Chimeric Antigen Receptor (CAR) T Cells Combined With Checkpoint Inhibition for Patients With Resectable Recurrent Glioblastoma. Identifier: NCT04003649 Available online: https://clinicaltrials.gov/ct2/show/NCT04003649. Accessed 18 Oct 2022.
U.S. National Institutes of Health n.d.-t. ClinicalTrials.gov GD2 Specific CAR and Interleukin-15 Expressing Autologous NKT Cells to Treat Children With Neuroblastoma (GINAKIT2). Official Title: GD2 Specific Chimeric Antigen Receptor (CAR) and Interleukin-15 Expressing Autologous Natural Killer T-cells to Treat Children With Neuroblastoma Identifier: NCT03294954 Available online: https://clinicaltrials.gov/ct2/show/NCT03294954. Accessed 19 Oct 2022.
U.S. National Institutes of Health n.d.-q. ClinicalTrials.gov EGFRvIII CAR T Cells for Newly-Diagnosed WHO Grade IV Malignant Glioma (ExCeL). Official Title: EGFRvIII Chimeric Antigen Receptor (CAR) Gene-modified T Cells for Patients With Newly-Diagnosed GBM During Lymphopenia. Identifier: NCT02664363 Available online: https://clinicaltrials.gov/ct2/show/NCT02664363. Accessed 18 Oct 2022.
Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565(7738):234–9. https://doi.org/10.1038/s41586-018-0792-9.
Jessurun CAC, Hulsbergen AFC, de Wit AE, Tewarie IA, Snijders TJ, Verhoeff JJC, et al. The combined use of steroids and immune checkpoint inhibitors in brain metastasis patients: a systematic review and meta-analysis. Neuro-Oncology. 2021;23(8):1261–72. https://doi.org/10.1093/neuonc/noab046.
Bocca P, Di Carlo E, Caruana I, Emionite L, Cilli M, De Angelis B, et al. Bevacizumab-mediated tumor vasculature remodelling improves tumor infiltration and antitumor efficacy of GD2-CAR T cells in a human neuroblastoma preclinical model. Oncoimmunology. 2017;7(1):e1378843. https://doi.org/10.1080/2162402X.2017.1378843.
Jeon S, Kim HK, Kwon JY, Baek SH, Ri HS, Choi HJ, et al. Role of sevoflurane on natural killer group 2, member D-mediated immune response in non-small-cell lung cancer: an in vitro study. Med Sci Monit. 2020;26:e926395. https://doi.org/10.12659/MSM.926395.
Mangani D, Weller M, Roth P. The network of immunosuppressive pathways in glioblastoma. Biochem Pharmacol. 2017;130:1–9. https://doi.org/10.1016/j.bcp.2016.12.011.
U.S. National Institutes of Health n.d.-ab. ClinicalTrials.gov NKG2D-based CAR T-cells Immunotherapy for Patient With r/r NKG2 DL+ Solid Tumors. Official Title: A Phase I Clinical Trial of NKG2D-based CAR T-cells Injection for Subjects With Relapsed/Refractory NKG2DL+ Solid Tumors. Identifier: NCT04270461 Available online: https://clinicaltrials.gov/ct2/show/NCT04270461. Accessed 18 Oct 2022.
Choi BD, Yu X, Castano AP, Darr H, Henderson DB, Bouffard AA, et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J Immunother Cancer. 2019a;7(1):304. https://doi.org/10.1186/s40425-019-0806-7.
Huang BR, Liu YS, Lai SW, Lin HJ, Shen CK, Yang LY, et al. CAIX regulates GBM motility and TAM adhesion and polarization through EGFR/STAT3 under hypoxic conditions. Int J Mol Sci. 2020;21(16):5838. https://doi.org/10.3390/ijms21165838.
Cui J, Zhang Q, Song Q, Wang H, Dmitriev P, Sun MY, et al. Targeting hypoxia downstream signaling protein, CAIX, for CAR T-cell therapy against glioblastoma. Neuro-Oncology. 2019;21(11):1436–46. https://doi.org/10.1093/neuonc/noz117.
Jin L, Ge H, Long Y, Yang C, Chang YE, Mu L, et al. CD70, a novel target of CAR T-cell therapy for gliomas. Neuro-Oncology. 2018;20(1):55–65. https://doi.org/10.1093/neuonc/nox116.
Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun. 2019;10(1):4016. https://doi.org/10.1038/s41467-019-11869-4.
Harper T, Sharma A, Kaliyaperumal S, Fajardo F, Hsu K, Liu L, et al. Characterization of an anti-CD70 half-life extended bispecific T-cell engager (HLE-BiTE) and associated on-target toxicity in Cynomolgus monkeys. Toxicol Sci. 2022;189(1):32–50. https://doi.org/10.1093/toxsci/kfac052.
Sauer T, Parikh K, Sharma S, Omer B, Sedloev D, Chen Q, et al. CD70-specific CAR T cells have potent activity against acute myeloid leukemia without HSC toxicity. Blood. 2021;138(4):318–30. https://doi.org/10.1182/blood.2020008221.
Park YP, Jin L, Bennett KB, Wang D, Fredenburg KM, Tseng JE, et al. CD70 as a target for chimeric antigen receptor T cells in head and neck squamous cell carcinoma. Oral Oncol. 2018;78:145–50. https://doi.org/10.1016/j.oraloncology.2018.01.024.
Tsidulko AY, Kazanskaya GM, Kostromskaya DV, Aidagulova SV, Kiselev RS, Volkov AM, et al. Prognostic relevance of NG2/CSPG4, CD44 and Ki-67 in patients with glioblastoma. Tumour Biol. 2017;39(9):1010428317724282. https://doi.org/10.1177/1010428317724282.
Geldres C, Savoldo B, Hoyos V, Caruana I, Zhang M, Yvon E, et al. T lymphocytes redirected against the chondroitin sulfate proteoglycan-4 control the growth of multiple solid tumors both in vitro and in vivo. Clin Cancer Res. 2014;20(4):962–71. https://doi.org/10.1158/1078-0432.CCR-13-2218.
Pellegatta S, Savoldo B, Di Ianni N, Corbetta C, Chen Y, Patané M, et al. Constitutive and TNFα-inducible expression of chondroitin sulfate proteoglycan 4 in glioblastoma and neurospheres: implications for CAR-T cell therapy. Sci Transl Med. 2018;10(430):eaao2731. https://doi.org/10.1126/scitranslmed.aao2731 Erratum in: Sci Transl Med. 2018;10 (435).
Hamaoka Y, Negishi M, Katoh H. EphA2 is a key effector of the MEK/ERK/RSK pathway regulating glioblastoma cell proliferation. Cell Signal. 2016;8:937–45. https://doi.org/10.1016/j.cellsig.2016.04.009.
Chow KK, Naik S, Kakarla S, Brawley VS, Shaffer DR, Yi Z, et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther. 2013;21(3):629–37. https://doi.org/10.1038/mt.2012.210.
Yi Z, Prinzing BL, Cao F, Gottschalk S, Krenciute G. Optimizing EphA2-CAR T cells for the adoptive immunotherapy of glioma. Mol Ther Methods Clin Dev. 2018;9:70–80. https://doi.org/10.1016/j.omtm.2018.01.009.
Hou B, Tang Y, Li W, Zeng Q, Chang D. Efficiency of CAR-T therapy for treatment of solid tumor in clinical trials: a meta-analysis. Dis Markers. 2019;2019:3425291. https://doi.org/10.1155/2019/3425291.
Bedoya DM, King T, Posey AD. Generation of CART cells targeting oncogenic TROP2 for the elimination of epithelial malignancies. Cytotherapy. 2019;21(5):S11–2. https://doi.org/10.1016/j.jcyt.2019.03.570.
Krenciute G, Prinzing BL, Yi Z, Wu MF, Liu H, Dotti G, et al. Transgenic expression of IL15 improves Antiglioma activity of IL13Rα2-CAR T cells but results in antigen loss variants. Cancer Immunol Res. 2017;7:571–81. https://doi.org/10.1158/2326-6066.CIR-16-0376.
Chheda ZS, Kohanbash G, Okada K, Jahan N, Sidney J, Pecoraro M, et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J Exp Med. 2018;215(1):141–57. https://doi.org/10.1084/jem.20171046.
Hou AJ, Chang ZL, Lorenzini MH, Zah E, Chen YY. TGF-β-responsive CAR-T cells promote anti-tumor immune function. Bioeng Transl Med. 2018;3(2):75–86. https://doi.org/10.1002/btm2.10097.
Larson RC, Maus MV. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat Rev Cancer. 2021;21(3):145–61. https://doi.org/10.1038/s41568-020-00323-z.
Nehama D, Di Ianni N, Musio S, Du H, Patané M, Pollo B, et al. B7-H3-redirected chimeric antigen receptor T cells target glioblastoma and neurospheres. EBioMedicine. 2019;47:33–43. https://doi.org/10.1016/j.ebiom.2019.08.030.
Cordell EC, Alghamri MS, Castro MG, Gutmann DH. T lymphocytes as dynamic regulators of glioma pathobiology. Neuro-Oncology. 2022;24(10):1647–57. https://doi.org/10.1093/neuonc/noac055.
Nduom EK, Yang C, Merrill MJ, Zhuang Z, Lonser RR. Characterization of the blood-brain barrier of metastatic and primary malignant neoplasms. J Neurosurg. 2013;119(2):427–33. https://doi.org/10.3171/2013.3.JNS122226.
Sarkaria JN, Hu LS, Parney IF, Pafundi DH, Brinkmann DH, Laack NN, et al. Is the blood-brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncology. 2018;20(2):184–91. https://doi.org/10.1093/neuonc/nox175.
Mulazzani M, Fräßle SP, von Mücke-Heim I, Langer S, Zhou X, Ishikawa-Ankerhold H, et al. Long-term in vivo microscopy of CAR T cell dynamics during eradication of CNS lymphoma in mice. Proc Natl Acad Sci U S A. 2019;116(48):24275–84. https://doi.org/10.1073/pnas.1903854116.
Theruvath J, Sotillo E, Mount CW, Graef CM, Delaidelli A, Heitzeneder S, et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat Med. 2020;26(5):712–9. https://doi.org/10.1038/s41591-020-0821-8.
Neill L, Rees J, Roddie C. Neurotoxicity-CAR T-cell therapy: what the neurologist needs to know. Pract Neurol. 2020;20(4):285–93. https://doi.org/10.1136/practneurol-2020-002550.
Gust J, Hay KA, Hanafi LA, Li D, Myerson D, Gonzalez-Cuyar LF, et al. Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. 2017;7(12):1404–19. https://doi.org/10.1158/2159-8290.CD-17-0698.
Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma. N Engl J Med. 2019;380(1):45–56. https://doi.org/10.1056/NEJMoa1804980.
Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24(6):739–48. https://doi.org/10.1038/s41591-018-0036-4.
Petersen CT, Krenciute G. Next generation CAR T cells for the immunotherapy of high-grade glioma. Front Oncol. 2019;9:69. https://doi.org/10.3389/fonc.2019.00069.
U.S. National Institutes of Health n.d.-z. ClinicalTrials.gov Intracerebral EGFR-vIII CAR-T Cells for Recurrent GBM (INTERCEPT). Official Title: INTERCEPT: INTracerebral EGFR-vIII Chimeric Antigen Receptor Gene-Modified T CElls for PaTients With Recurrent GBM. Identifier: NCT03283631 Available online: https://clinicaltrials.gov/ct2/show/NCT03283631. Accessed 25 Oct 2022.
U.S. National Institutes of Health n.d.-ae. ClinicalTrials.gov Pilot Study of B7-H3 CAR-T in Treating Patients With Recurrent and Refractory Glioblastoma. Official Title: A Pilot Study of Chimeric Antigen Receptor (CAR) T Cells Targeting B7-H3 Antigen in Treating Patients With Recurrent and Refractory Glioblastoma. Identifier: NCT04385173 Available online: https://clinicaltrials.gov/ct2/show/NCT04385173. Accessed 25 Oct 2022.
U.S. National Institutes of Health n.d.-ac. ClinicalTrials.gov Personalized Chimeric Antigen Receptor T Cell Immunotherapy for Patients With Recurrent Malignant Gliomas. Official Title: A Pilot Study to Evaluate the Safety and Efficacy of Personalized ChimericAntigen Receptor T Cell Immunotherapy for Patients With Recurrent Malignant Gliomas Based on the Expression of Tumor Specific/Associated Antigens. Identifier: NCT03423992 Available online: https://clinicaltrials.gov/ct2/show/NCT03423992. Accessed 25 Oct 2022.
Liu C, Zhang G, Xiang K, Kim Y, Lavoie RR, Lucien F, et al. Targeting the immune checkpoint B7-H3 for next-generation cancer immunotherapy. Cancer Immunol Immunother. 2022;71(7):1549–67. https://doi.org/10.1007/s00262-021-03097-x.
Masre SF, Jufri NF, Ibrahim FW, Abdul Raub SH. Classical and alternative receptors for SARS-CoV-2 therapeutic strategy. Rev Med Virol. 2021;31(5):1–9. https://doi.org/10.1002/rmv.2207.
Richman SA, Nunez-Cruz S, Moghimi B, Li LZ, Gershenson ZT, Mourelatos Z, et al. High-affinity GD2-specific CAR T cells induce fatal encephalitis in a preclinical neuroblastoma model. Cancer Immunol Res. 2018;6(1):36–46. https://doi.org/10.1158/2326-6066.CIR-17-0211.
Gargett T, Brown MP. The inducible caspase-9 suicide gene system as a "safety switch" to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Front Pharmacol. 2014;5:235. https://doi.org/10.3389/fphar.2014.00235.
Kim G, Ko YT. Small molecule tyrosine kinase inhibitors in glioblastoma. Arch Pharm Res. 2020;43(4):385–4. https://doi.org/10.1007/s12272-020-01232-3.
Wu X, Luo H, Shi B, Di S, Sun R, Su J, et al. Combined antitumor effects of Sorafenib and GPC3-CAR T cells in mouse models of hepatocellular carcinoma. Mol Ther. 2019b;27(8):1483–94. https://doi.org/10.1016/j.ymthe.2019.04.020.
Li H, Ding J, Lu M, Liu H, Miao Y, Li L, et al. CAIX-specific CAR-T cells and Sunitinib show synergistic effects against metastatic renal cancer models. J Immunother. 2020;43(1):16–28. https://doi.org/10.1097/CJI.0000000000000301.
Cui J, Wang H, Medina R, Zhang Q, Xu C, Indig IH, et al. Inhibition of PP2A with LB-100 enhances efficacy of CAR-T cell therapy against glioblastoma. Cancers (Basel). 2020;12(1):139. https://doi.org/10.3390/cancers12010139.
Xu B, Tian L, Chen J, Wang J, Ma R, Dong W, et al. An oncolytic virus expressing a full-length antibody enhances antitumor innate immune response to glioblastoma. Nat Commun. 2021;12(1):5908. https://doi.org/10.1038/s41467-021-26003-6.
Guedan S, Alemany R. CAR-T cells and oncolytic viruses: joining forces to overcome the solid tumor challenge. Front Immunol. 2018;9:2460. https://doi.org/10.3389/fimmu.2018.02460.
Sampson JH, Maus MV, June CH. Immunotherapy for brain tumors. J Clin Oncol. 2017;35(21):2450–6. https://doi.org/10.1200/JCO.2017.72.8089.
Chalise L, Kato A, Ohno M, Maeda S, Yamamichi A, Kuramitsu S, et al. Efficacy of cancer-specific anti-podoplanin CAR-T cells and oncolytic herpes virus G47Δ combination therapy against glioblastoma. Mol Ther Oncolytics. 2022;20(26):265–74. https://doi.org/10.1016/j.omto.2022.07.006.
Lhuillier C, Rudqvist NP, Elemento O, Formenti SC, Demaria S. Radiation therapy and anti-tumor immunity: exposing immunogenic mutations to the immune system. Genome Med. 2019;11(1):40. https://doi.org/10.1186/s13073-019-0653-7.
Lai J, Mardiana S, House IG, Sek K, Henderson MA, Giuffrida L, et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat Immunol. 2020;21(8):914–26. https://doi.org/10.1038/s41590-020-0676-7.
Hetze S, Sure U, Schedlowski M, Hadamitzky M, Barthel L. Rodent models to analyze the glioma microenvironment. ASN Neuro. 2021;13:17590914211005074. https://doi.org/10.1177/17590914211005074.
Acknowledgements
National Natural Science Foundation of China is acknowledged for awarding Regional Science Foundation Project (no: 72164001). Jadavpur University, India is acknowledged for awarding a state government research fellowship to PC (Ref no: R-11/13/20 dated 22.01.2020). The authors are thankful to Gannan Medical University, China, Jadavpur University, India, Sharda University, India, Adamas University, India, and Presidency University, India for providing the necessary facilities during the formulation of this manuscript.
Funding
This is review article; and there is no financial support for this work.
Author information
Authors and Affiliations
Contributions
Conceptualization, SKJ, ZH, SD, and NKJ; data accumulation, writing, and original draft preparation: ZH, PC, SD, XYC and JW; editing: MG, AD, and JW; supervision: SD, NKJ, JW and SKJ; All authors have read and approved the submitted version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval
This is review article; data is not related to Ethics approval.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Huang, Z., Dewanjee, S., Chakraborty, P. et al. CAR T cells: engineered immune cells to treat brain cancers and beyond. Mol Cancer 22, 22 (2023). https://doi.org/10.1186/s12943-022-01712-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-022-01712-8