
Abstract
The nuclear speckle-type pox virus and zinc finger (POZ) protein (SPOP), a representative substrate-recognition subunit of the cullin-RING E3 ligase, has been characterized to play a dual role in tumorigenesis and cancer progression. Numerous studies have determined that SPOP suppresses tumorigenesis in a variety of human malignancies such as prostate, lung, colon, gastric, and liver cancers. However, several studies revealed that SPOP exhibited oncogenic function in kidney cancer, suggesting that SPOP could exert its biological function in a cancer type-specific manner. The role of SPOP in thyroid, cervical, ovarian, bone and neurologic cancers has yet to be determined. In this review article, we describe the structure and regulation of SPOP in human cancer. Moreover, we highlight the critical role of SPOP in tumorigenesis based on three major categories: physiological evidence (animal models), pathological evidence (human cancer specimens) and biochemical evidence (downstream ubiquitin substrates). Furthermore, we note that SPOP could be a promising therapeutic target for cancer treatment.
Similar content being viewed by others


Introduction
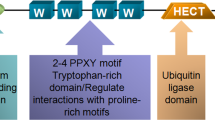
Protein degradation is critical for maintaining cellular homeostasis, and abnormal accumulation of proteins may lead to various diseases including human cancers [1]. There are two major proteolytic pathways that are conserved in eukaryotes: lysosomal-mediated proteolysis and proteasome-mediated degradation [2]. By promoting protein ubiquitination and degradation, the ubiquitin proteasome system (UPS) is responsible for the destruction of approximately 80% of intracellular proteins, thereby regulating an array of biological processes including cell proliferation, apoptosis, invasion and metastasis [3,4,5]. It is well accepted that UPS-mediated protein degradation is composed of two discrete steps: (1) a substrate protein is labeled by a single ubiquitin protein (monoubiquitination) or multiple ubiquitin molecules (polyubiquitination); (2) the ubiquitinated substrate is subsequently degraded by the 26S proteasome complex [6]. Biochemically, the first step of protein ubiquitination is achieved through three unique and consecutive enzymatic reactions, which are catalyzed by a ubiquitin-activating E1 enzyme, a ubiquitin-conjugating E2 enzyme and a ubiquitin-protein E3 ligase [7]. Mechanistically, ubiquitin is activated by the E1 enzyme in an ATP-dependent fashion. Then, the active form of ubiquitin is conjugated to the E2 enzyme, after which it is transferred to the specific target substrate depending on the E3 ligase involved [7]. Hence, E3 ligases are crucial for determining the substrate specificity for degradation (Fig. 1) [8].

UPS-mediated protein degradation. Ubiquitin is activated by the E1 enzyme in an ATP-dependent fashion. Then, the active form of ubiquitin is conjugated to the E2 enzyme, after which it is transferred to a specific target substrate depending on the E3 ligase involved. E3 ligases are crucial for determining the substrate specificity for degradation
The human genome encodes more than 600 putative E3 ligases [9], among which the cullin-RING E3 ligase (CRL) complex family is the largest and consists of eight members including CRL1, CRL2, CRL3, CRL4A, CRL4B, CRL5, CRL7 and CRL9 [10, 11]. Generally, the CRL E3 ligases are composed of a cullin (Cul) protein as the scaffold protein, a RING-box protein (RBX1 or RBX2) that is essential for recruiting the E2 enzyme, a substrate-recognition subunit (SRS) and an adaptor protein (SKP1, elongin B/C or DDB1) that links the SRS to the complex [12]. Interestingly, CRL3 contains only three primary components including the Cul-3 protein, the RBX1 protein and a Bric-a-brac-Tramtrack/Broad (BTB) protein, and the BTB protein serves as both the SRS and the adaptor protein for substrate binding within this complex (Fig. 2) [13, 14]. The nuclear speckle-type pox virus and zinc finger protein (SPOP), a representative SRS of CRL3, has been greatly explored for its dual functions in tumorigenesis [15, 16]. Therefore, in this review, we will mainly describe the structure and regulation of SPOP and discuss the role of SPOP in tumorigenesis on the basis of three major categories: physiological evidence (animal models), pathological evidence (human cancer specimens) and biochemical evidence (downstream ubiquitin substrates). We will also note that SPOP is a promising therapeutic target for cancer treatments.

Cul-3 structure is illustrated. CRL3 contains three primary components including a Cul-3 protein as the scaffold protein, a RING-box protein (RBX1) that is essential for recruiting the E2 enzyme, and a Bric-a-brac-Tramtrack/Broad (BTB) protein; the BTB protein serves as both a substrate-recognition subunit (SRS) and the adaptor protein for substrate binding within this complex
Structure of SPOP
SPOP was first discovered in 1997 by Nagai and his colleagues and harbors a typical POZ domain [17]. After that, accumulating evidence has elucidated the structure of SPOP. Structurally, the SPOP protein comprises an N-terminal MATH domain, an internal BTB/POZ domain, a BACK domain, a 3-box domain and a C-terminal nuclear localization sequence (NLS) (Fig. 3) [18, 19]. The MATH domain of SPOP plays a central role in selectively recognizing and recruiting substrates, and the majority of mutations are located in this domain [18]. This domain specifically recognized short linear motifs of substrates that serve as the specific SPOP-binding (SB) motifs. In turn, the substrate proteins require the existence of a prerequisite SB 5-residue motif φ-π-S-S/T-S/T (φ-nonpolar; π-polar amino acid), termed the SPOP-binding consensus (SBC) [18]. Moreover, the BTB domain is primarily involved in SPOP dimerization as well as Cul-3 binding. Specifically, an α3-β4 loop consisting of 10 amino acid residues in the BTB domain is crucial for this binding [20,21,22]. The BACK domain serves as a second place to mediate dimerization [19]. Furthermore, the 3-box domain, a pair of α-helices stretching beyond the BTB domain, has also been suggested to enhance the SPOP-Cul-3 interaction [23]. The dimerization interface in the BTB and BACK domains and the C-terminus work independently and form higher-order SPOP oligomers [24], which augments the E3 ligase activity by increasing the substrate avidity and the effective concentration of the E2 enzyme [23].

SPOP structure is illustrated. The SPOP protein comprises an N-terminal MATH domain, an internal BTB/POZ domain, a BACK domain, a 3-box domain and a C-terminal nuclear localization sequence (NLS). The MATH domain of SPOP selectively recognizes and recruits substrates and includes key amino acid residues such as Y87, F102, Y123, W131 and F133
Tumor suppressive role of SPOP in cancers
Multiple studies have determined that SPOP could suppress tumorigenesis in several types of human malignancies, including prostate, lung, gastric, liver, colon and endometrial cancers (Fig. 4, Table 1).

The tumor suppresssive role of SPOP in human cancers. SPOP suppresses tumorigenesis and progression via regulation of cell growth, apoptosis, migration, invasion and drug resistance by targeting its downstream substrates in several types of human malignancies, including prostate, lung, gastric, liver, colon and endometrial cancers
Prostate cancer (PrCa)
PrCa is a common diagnosed cancer among men worldwide [80]. SPOP was reported as a frequently mutated gene in PrCa by Kan et al. in 2010 [52]. After that, whole-genome as well as exome sequencing analyses have revealed SPOP mutations in primary prostate neoplastic tumors but not in matched normal prostate tissues [53, 54]. SPOP mutations (such as Y87, F102, S119, Y123, F125, K129, W131, F133 and K134) often occur in the MATH domain of SPOP (Fig. 3) [54], and these mutations have been identified as an early event in the development of genomic instability and tumorigenesis in PrCa [55,56,57,58]. Notably, a number of animal models have been generated to explore the physiological role of SPOP in the development of PrCa and prostate tumorigenesis. For instance, homozygous deletion of SPOP (SPOP−/−) in zebrafish showed impaired brain, eye and body development that was largely rescued via microinjection of SPOP mRNA [58]. Furthermore, SPOP−/− mice died between embryonic day 18.5 and postnatal day 1 [59]. Blattner et al. constructed a prostate-specific SPOP-F133 V mutation-carrying transgenic mouse and found that PrCa was developed in part due to the activation of the PI3K/mTOR and AR signaling pathways as well as the loss of PTEN [60]. Additionally, the PrCa-derived SPOP-F133 V mutation selectively damaged the homology-directed repair function mediated by wild-type SPOP (wt-SPOP) [58]. Clinical data showed that SPOP mutations and downregulation were detected in human PrCa tissues, and these mutations were also tightly correlated with a worse prognosis in patients with PrCa [61]. More importantly, extensive biochemical evidence has further indicated that SPOP functions as a tumor suppressor by promoting the degradation of oncogenic substrates in PrCa, including SRC3 [62], AR [63], TRIM24 [64], c-Myc [65], DEK [66], SENP7 [67], EglN2 [68], ATF2 [69], Cdc20 [70], ERG [71, 81], BRD4 [72,73,74], PD-L1 [75] and cyclin E1 [76]. Due to the many publications and space limitations, we will not describe the tumor suppressive role that SPOP plays by promoting the ubiquitination and degradation of its substrates in PrCa in detail. Therefore, we sincerely apologize to some researchers for not citing their important and meaningful papers.
Lung cancer (LC)
LC is one of the leading causes of cancer-related death in the world [82]. Downregulation of SPOP has been observed in non-small cell LC (NSCLC) tissues compared with normal tissues at both the transcriptional and translational levels [83]. Furthermore, the level of SPOP was confirmed to be associated with several clinicopathologic parameters, and a decrease in SPOP was considered a predictor of poor prognosis in patients with NSCLC, suggesting that SPOP could be a potential tumor suppressor in LC [83]. The sirtuin (SIRT) family of NAD-dependent protein lysine deacylases has been reported to participate in multiple biological processes such as transcription regulation, metabolism and DNA repair [84,85,86]. Notably, one group showed that SPOP promoted the proteasomal degradation of SIRT2 by binding to it, thus suppressing the growth of NSCLC cells [47]. Moreover, this ability was inhibited by mutation of SPOP in NSCLC cells. Furthermore, compared with the normal cells, NSCLC cell lines had elevated SIRT2 and reduced SPOP levels [47]. Fas-associated protein with death domain (FADD) is the key adaptor protein that transmits extrinsic apoptotic cell death signals by recruiting complexes of caspase 8 to death receptors [87, 88]. Emerging evidence has also shown that FADD expression is involved in tumorigenesis and cancer progression. For example, overexpression of FADD might serve as a biomarker in head and neck squamous cell carcinoma [89]. Furthermore, a high level of FADD protein has also been reported to be associated with poor outcome in LC, suggesting that it could become a potent prognostic biomarker in LC patients [48, 90]. Luo et al. found that SPOP directly bound to FADD and promoted its ubiquitination and degradation, blocking the development of NSCLC [48]. Therefore, SPOP exerts anticancer effects by targeting FADD in LC. Interestingly, an oncogenic role of SPOP in LC has also been indicated recently [91]. SPOP was found to be widely expressed in different LC cell lines. Conversely, knockdown of SPOP by shRNA in LC cells led to DNA damage repair defects, increased cell apoptosis and sensitization to irradiation under DNA damage conditions [91]. Therefore, in-depth investigation is essential to determine the role of SPOP in LC.
Gastric cancer (GC)
GC is one of the leading causes of cancer-related death worldwide and has a poor response to current chemotherapy [80]. The sonic hedgehog (Shh) signaling pathway is crucial for growth control and patterning during embryonic development and adult homeostasis [92]. Glioma-associated oncogenic (Gli) proteins are the main effectors of the Shh pathway, including Gli1, Gli2 and Gli3 [93, 94]. Among the three Gli proteins, Gli2 is the principal transcriptional activator that regulates Shh signaling in skin development and tumorigenesis. One study revealed that Gli2−/− mice displayed hair morphogenesis defects similar to those of Shh−/− mice [95]. Furthermore, a high level and increased activity of Gli2 in the epidermis is sufficient to promote the formation of basal cell carcinoma and maintain tumor growth [96, 97]. Moreover, upregulation of Gli2 protein was detected in GC specimens and Gli2 was also correlated with lymphovascular invasion in GC [98]. One group showed that the expression of SPOP was much lower in GC tissues than in adjacent normal tissues, while a high level of SPOP was negatively associated with poor clinicopathologic outcome [37]. Additionally, overexpression of SPOP significantly inhibited growth, metastasis, and colony formation in vitro. Mechanistically, using immunofluorescent staining, it was observed that elevation of SPOP accelerated the degradation of Gli2 without affecting its synthesis via intracellular interactions in GC cells [37]. However, whether SPOP functions as a tumor suppressor in GC should be further validated with more studies in the future.
Hepatocellular carcinoma (HCC)
HCC arises in patients as a consequence of long-standing preexisting liver illness, including viral hepatitis, alcohol abuse, and metabolic disease [99]. A previous study revealed using exome sequencing that SPOP was heterozygously mutated in one hepatoblastoma case and that it may normally play a tumor suppressive role in hepatoblastoma [40]. In addition, Huang et al. found a reduction in SPOP expression in HCC tissues and a low level of SPOP was associated with a high grade and intrahepatic metastasis in patients with HCC [41]. Moreover, in vitro experiments revealed that SPOP suppressed the growth and migration of HCC cells in part through blockade of ZEB2 expression [41]. Furthermore, one group also confirmed that the expression of SPOP was downregulated in HCC specimens and suggested SPOP as a predictor of poor prognosis for HCC patients [42]. Mechanistic studies implied that SPOP promoted the ubiquitin-mediated degradation and proteolysis of SENP7 by recognizing and binding to it, which eventually decreased the level of vimentin and attenuated the metastasis of HCC cells [42]. In addition, experiments on subcutaneous tumor mouse model also confirmed the inhibitory effect of SPOP on liver and lung metastases in HCC [42]. Overall, these studies indicate that SPOP might be a potential tumor suppressor in HCC.
Colorectal cancer (CRC)
CRC is the third most frequent cancer in the Western hemisphere and the incidence increases with increasing age [100]. Numerous studies have explored the role of SPOP in tumorigenesis and progression in CRC. Frequent downregulation of SPOP was detected in CRC tissues compared with paired adjacent normal tissues at both the mRNA and protein levels, and this downregulation was also significantly related to clinicopathologic parameters such as poor differentiation, distant metastasis, and high TNM stage [29]. Furthermore, a decrease in SPOP expression might serve as a potent predictive factor of poor prognosis for patients with CRC according to Kaplan-Meier survival analysis. In an in vitro study, overexpression of SPOP dramatically inhibited the proliferation and migration of CRC cells via upregulation of E-cadherin and downregulation of vimentin, MMP2, and MMP7, while this process was reversed by silencing of SPOP [29]. Moreover, one study also found that SPOP ablated MMP2 expression in CRC cells by suppressing the SP1 phosphorylation and nuclear translocation that was involved in the PI3K/Akt signaling pathway [101]. Histone deacetylase 6 (HDAC6) belongs to the HDACs family and is prominently involved in carcinogenesis and cancer progression [102, 103]. Numerous studies have found that high expression of HDAC6 exists in several human cancers and is associated with a significantly poor prognosis in diseases such as breast cancer [104] and PrCa [105]. HDAC6 contains two functional catalytic domains and deacetylates many nonhistone oncogenic proteins [103]. For example, HDAC6-mediated deacetylation leads to activation of HSP90 and promotes the binding of client proteins to HSP90 including AR, which enhances the activity of AR and suppresses its degradation [106, 107]. Additionally, the elevation of HDAC6 induced by P62 (also known as sequestosome-1, SQSTM1) promotes the epithelial-mesenchymal transition (EMT) process and impairs autophagic flux, facilitating the growth, migration and invasion of prostate cancer cells [108]. EMT is a reprogramming process in which epithelial cells take on mesenchymal phenotype after stimulation with EMT inducers [109]. The cells will lose the features of polarized immotile epithelial cells and obtain motile mesenchymal cell characteristics, leading to enhancement of migration and invasiveness [109]. Moreover, the expression of E-cadherin is decreased, while the levels of several mesenchymal markers including vimentin, Twist, ZEB1, ZEB2, and Slug are upregulated [109]. More importantly, multiple agents have been confirmed to exert their anticancer effects by inactivating and downregulating HDAC6 in PrCa cells [110,111,112,113]. It has been noted that SPOP specifically interacts with HDAC6 and promotes its polyubiquitination and degradation in human 293 T cells [30]. Notably, the growth and migration of SPOP-depleted colon cancer cells was partly reversed through knockdown of HDAC6 [30]. In line with the studies above describing the tumor suppressive function of SPOP in CRC, Zhi et al. showed that SPOP accelerated the ubiquitination and degradation of Gli2 by directly binding and interacting with it in CRC cells, enhancing the apoptotic signals of cells via a decrease in Bcl-2 and blocking the progression of CRC [31]. These results indicate that SPOP is a tumor suppressor in CRC, and it accomplishes this role by promoting the ubiquitination-mediated degradation of HDAC6 and Gli2.
Endometrial cancer (EC)
EC is the sixth most commonly diagnosed cancer in women worldwide [80], and multiple studies have been conducted to explore the function of SPOP in the tumorigenesis of EC. Hence, we will mainly discuss how SPOP plays a role in EC in this section. Recently, a SPOP−/− mouse model was generated and exhibited an infertility phenotype with a reduction in progesterone receptors (PRs) in the uterus [114]. In addition, based on artificially induced decidualization and steroid hormone-processing mouse models, researchers have demonstrated that SPOP is required for embryonic implantation and for endometrial decidualization [115]. Collectively, these findings support a crucial role for SPOP in regulating normal uterine function. However, several genomic analyses have provided evidence that SPOP is frequently mutated in human EC [32,33,34,35,36], indicating that wild-type SPOP may act as a tumor suppressor in this disease. Zhang et al. discovered that estrogen receptor-α (ERα) is a specific substrate for SPOP, and ERα is also considered a main promoter of EC and facilitates the tumorigenesis of EC [36]. The SPOP protein accelerated the ubiquitin-mediated degradation of ERα by recognizing specific domains that contained abundant Ser/Thr (S/T)-rich degrons, and this action was reversed via the knockdown of SPOP by siRNAs in EC cells [36]. However, intriguingly, a recent study suggested that three BETs (BRD2, BRD3 and BRD4) were preferentially degraded by EC-related SPOP mutation and enhanced the sensitivity to BET inhibitors in EC cells [73]. Therefore, the definitive role of SPOP in the carcinogenesis of EC remains to be further elucidated in the future.
Oncogenic role of SPOP in kidney cancer (KC)
In contrast to the tumor-suppressive role of the SPOP protein in many human cancers described above, the oncogenic function of SPOP has been confirmed in KC. Studies have shown that SPOP is significantly upregulated in renal cell carcinoma (RCC) tissues at both the transcriptional and translational levels [43,44,45], and this upregulation was positively associated with cancer metastasis in patients with RCC [45]. In contrast, the malignant behaviors of RCC A498 and ACHN cells were reversed after SPOP knockdown using siRNA, as manifested by apoptosis induction, migration inhibition and increased sensitivity to sorafenib [116]. The SPOP protein has been identified as a nuclear protein in human normal embryonic 293 (HEK293) cells, whereas it has shown to be predominately transferred and accumulated in the cytoplasm of RCC cells under hypoxia [46]. Furthermore, Li et al. constructed stable polyclonal HEK293 cells transfected with cytoplasmic SPOP (cyto-SPOP) and found that 80% of nude mice injected with HEK293-cyto-SPOP developed tumor xenografts subcutaneously whereas no tumor growth was produced in the control groups [46]. Biochemically, cyto-SPOP promoted the ubiquitination and degradation of several tumor suppressors (including PTEN, DUSP7, Daxx and Gli2) in the cytoplasm, facilitating proliferation and inhibiting apoptosis in RCC cells [46]. Taken together, these findings suggest that SPOP plays an oncogenic role in KC cells via its cytoplasmic accumulation, resulting in degradation of tumor suppressive substrates of SPOP (Fig. 5, Table 1).

The oncogenic role of SPOP in kidney cancer. SPOP serves as an oncoprotein in kidney cancer by promotting the ubiquitination and degradation of PTEN, DUSP7, Daxx and Gli2, thus facilitating proliferation and inhibiting apoptosis in RCC cells
SPOP functions to be determined in tumorigenesis
Breast cancer (BC)
BC is one of the most common malignancies and is the second cause of death among women in the world [117]. One group reported that SPOP mediated the ubiquitination and degradation of c-Myc in triple-negative BC (TNBC) both in vitro and in vivo, while this process could be prevented by the lncRNA LINC01638, which interacted with c-Myc and subsequently promoted the expression of metadherin (MTDH) and Twist1 [25]. Moreover, Li et al. demonstrated that SPOP destabilized SRC-3 by hastening its polyubiquitination and proteasomal degradation in a phosphorylation-dependent manner in BC cells, thereby significantly reducing the levels of some molecules involved in SRC-3-mediated oncogenic signaling such as IGF-1 and MMP-2 [26]. In addition, knockdown of SPOP by shRNA promoted the proliferation and invasion of BC cells and elevated the cancer growth rate in a tumor xenograft mouse model, and these results were greatly influenced by upregulation of the SRC-3 oncoprotein [26]. For the majority of women with BC, tumor tissues show biological expression of receptors for estrogen and progesterone, hormones that are known to promote the growth and proliferation of cancer cells [118, 119]. PRs are member of the sex steroid receptor family, which modulates the function of sexual organs in a ligand-dependent manner, and PR has two coexpressed isoforms (PRA and PRB) [120]. There is compelling evidence suggesting that progesterone and PR play a crucial role in the development of BC [121, 122]. Gao et al. revealed that SPOP targeted PR for ubiquitin-mediated proteolysis and inhibited the PR transactivation induced by progesterone, S phase entry, and ERK1/2 activation, indicating that PR could be a bona fide substrate of SPOP in human BC [27]. Hence, these studies recommend that SPOP serves as a tumor suppressor by regulating the degradation of its substrates in BC. Breast cancer metastasis suppressor 1 (BRMS1) is a member of a subclass of the metastasis suppressor family, and its repressive effects on distant metastasis have been observed in several human cancers including BC [123, 124]. Intriguingly, one group suggested that BRMS1 may be a promising substrate that could be ubiquitinated and degraded by interacting with SPOP in BC cells [125]. Furthermore, deletion of SPOP dramatically promoted the expression of BRMS1 and decreased the level of OPN and uPA targeted and inhibited by BRMS1 in BC cells, suggesting that SPOP can also exert an oncogenic role in BC tumorigenesis and progression (Fig. 6) [125].

The dual roles of SPOP in breast cancer. SPOP might serve as a tumor suppressor or oncoprotein in breast cancer by regulating the degradation of its various substrates, but this role requires further validation
Cervical cancer (CC) and ovarian cancer (OC)
Although the prevalence of CC and OC survivors is on the rise due to improved outcomes after therapy, these diseases have also represented a serious threat to women’s health [126]. However, studies focusing on the role of SPOP in the tumorigenesis of CC and OC are still lacking. Only one group indicated that overexpression of the SPOP protein was essential for the apoptosis of HeLa cells, while this proapoptotic function was countered by ablation of SPOP [28]. Furthermore, one study used a tissue microarray to reveal a deletion mutant of SPOP in the majority of OC tissues that was not found in normal ovarian tissues, and this deletion mutant was correlated with high histological type and grade in OC [49]. Similarly, Jiang et al. identified several novel mutations in OC cell lines and tissues through wholeexome sequencing, including mutations in SPOP [50]. Thus, more studies should be carried out to clarify the function of SPOP in these two cancers and identify several potential substrates of SPOP during the carcinogenesis of CC and OC.
Thyroid cancer (TC)
TC is the most common endocrine malignancy worldwide [127], consisting of papillary (75–85%), follicular (10–20%), medullary (~ 5%) and anaplastic carcinomas (< 5%) [128]. Among the four types of TCs, only follicular thyroid carcinoma (FTC) has a benign counterpart named follicular thyroid adenoma (FTA). In fact, it has been widely accepted that FTA precedes FTC and has a favorable prognosis compared to FTC [129]. In this scenario, FTA originating from the thyroid follicle penetrates the tumor capsule and finally leads to the formation of FTC [130]. A few studies have investigated the genetic alterations responsible for the progression of FTA to FTC in recent years. Yoo et al. first identified the somatic mutation of SPOP-P94R in both minimally invasive FTC and FTA, and the mutations were confirmed by polymerase chain reaction (PCR) and Sanger sequencing assays [77]. Moreover, one group also found SPOP mutations in both FTC and FTA by conducting whole-exome sequencing and copy number profiling [78]. In addition, the results of evolutionary age analyses further showed that FTA genomes were as old as FTC genomes, implying the stability of the follicular thyroid tumor genomes during the transition from FTA to FTC [78]. Interestingly, one recent study found that adenomatoid nodules shared few overlapping gene mutations and expression patterns with coincidental papillary thyroid carcinoma (PTC) and the mutation of SPOP only existed in adenomatoid nodules [79]. Further studies should be performed in the future to explore several potential substrates of SPOP involved in the tumorigenesis.
Glioma and osteosarcoma (OS)
The function of SPOP has also been reported in glioma and osteosarcoma. Glioma is one of the most common primary brain tumors and has a high mortality worldwide [131]. A previous genome-wide linkage study revealed a series of single nucleotide polymorphisms (SNPs) in SPOP located on 17q12–21.32, and these SNPs may play an important role in gliomagenesis and cancer progression [38]. Furthermore, molecular studies found that SPOP was markedly downregulated in glioma samples compared to normal brain tissues, and low expression of SPOP displayed a potential for indicating poor prognosis in patients with glioma [39]. Conversely, overexpression of SPOP significantly suppressed glioma cell migration and invasion in vitro [39]. OS is a primary bone malignancy that predominantly occurs in children and adolescents [132]. Chen et al. showed that the expression of SPOP was decreased in both OS cell lines and clinical tissues [51]. Moreover, the migratory and invasive abilities of OS cells were dramatically enhanced after silencing of SPOP, whereas the anticancer effect was rescued by restoring the expression of SPOP, which negatively modulates the PI3K/Akt/NF-κB signaling pathway [51].
Regulation of SPOP
The function of SPOP is regulated at different levels, including genetic alteration, and transcriptional, translational and post-transcriptional modifications (PTMs). Genetic alterations of SPOP, including mutation and aberrant expression, have been extensively observed in various human cancers and are discussed in the above text [133]. In addition, studies with regard to the PTMs of SPOP are still lacking. Therefore, in this section, we will mainly focus on transcriptional and translational regulation as well as other emerging mechanisms that modulate the biological functions of SPOP in physiological and tumorigenic processes. Reportedly, SPOP is a direct transcriptional target of hypoxic stress and hypoxia-inducible factors (HIFs) in clear cell renal carcinoma (ccRCC) [46]. In ccRCC cells, SPOP mRNA and protein levels are elevated under hypoxic conditions. Additionally, hypoxia is sufficient to induce the cytoplasmic accumulation of SPOP, which promotes the ubiquitin-mediated degradation of several tumor suppressors (Daxx, PTEN, DUSP7, and Gli2) and leads to tumorigenesis in kidney cancer [46]. Methylation, a common modification of DNA catalyzed by a family of DNA methyltransferase enzymes, directly regulates the transcription of SPOP by affecting its promoter [31, 134]. Zhi et al. indicated that hypermethylation of the specific CpG sites within the SPOP gene promoter region decreased the transcriptional activities of SPOP, thereby causing the progression and metastasis of CRC [31]. Several miRNAs have also been identified to be involved in the regulation of SPOP in human cancers. For example, miR-145 and miR-543 reduced endogenous SPOP levels in human CRC, BC, CC, HCC and GC cells by directly targeting a conserved putative binding site in the 3′-untranslated regions (3′-UTRs) of SPOP transcripts, promoting the invasion and migration of cancer cells [135, 136]. Additionally, one group independently showed that SPOP might be a new target of miR-372 and miR-373. Specifically, miR-372 and miR-373 enhanced the stemness of CRC cells and induced a poor differentiation status in CRC by inhibiting SPOP expression [137]. More recently, Zhang et al. also reported that miR-373 promoted the proliferation, invasion, and migration of oral squamous cell carcinoma (OSCC) cells by negatively regulating the expression of SPOP [138].
Liquid-liquid phase separation (LLPS) of proteins is an emerging and important mechanism for regulating the function of SPOP [139, 140]. During this process, multiple membraneless compartments are formed, through which enzymes and substrates are concentrated and protein degradation is enhanced [139]. Studies have shown that the SPOP protein exists in various nuclear bodies including nuclear speckles, promyelocytic leukemia (PML) bodies, DNA damage loci, and other substrate-containing bodies [17, 19]. Recently, Bouchard et al. found that localization of SPOP-substrate complexes in membraneless organelles triggered by LLPS was essential for SPOP-mediated ubiquitination and the subsequent degradation of specific substrates in cells [140]. Conversely, tumor-associated SPOP mutations disrupted LLPS and SPOP-substrate accumulation, thus inhibiting ubiquitin-dependent proteolysis of downstream proteins.
SPOP as a therapeutic target
Since evidence has indicated oncogenic or suppressive roles of the SPOP protein depending on the specific cancer types, the SPOP protein could function as a novel therapeutic target for treating human cancers. Structurally, the SPOP protein can selectively bind to specific substrates by recognizing their SPOP-binding consensus (SBC) motif [18, 141]. On this basis, Quo et al. performed a computational screening and identified 109 small molecule inhibitors that disrupted the interactions between SPOP and its substrates [142]. Among these molecules, compound 6a showed the potential for competing with the puc_SBC1 peptide to bind to SPOP, while compound 6b was found to exert a strong inhibitory activity in disrupting the SPOP-PTEN and SPOP-DUSP7 interaction and blocking the downstream PTEN/Akt pathway in vitro, inhibiting the proliferation of A498 ccRCC cells [142]. Subsequent cellular thermal shift assay (CETSA) analysis further verified that compound 6b stabilized the substrates of SPOP in ccRCC cells [142]. Because the SPOP protein has context-dependent functions in different cancer types, future studies should focus on designing tissue- or cell-specific cancer drugs.
Conclusions and perspectives
In conclusion, SPOP plays a dual role in the development and progression of human cancer by targeting its various substrates. Due to the oncogenic role of SPOP in kidney cancer, SPOP inhibitors are necessary for the suppression of SPOP to treat kidney cancer. To take advantage of the tumor suppressive role of SPOP in prostate cancer and other cancers, one alternative approach is to modulate the upstream effectors of SPOP, leading to the upregulation of SPOP. For example, downregulation of miR-145 and miR-543 could increase the SPOP level, and inhibit the migration and invasion of cancer cells [135, 136]. Downregulation of miR-372 and miR-373 could lead to upregulation of SPOP and subsequent inhibition of stemness in CRC cells [137].
Since most studies focus on the function and molecular mechanisms of SPOP in prostate cancer, the detailed role of SPOP in other tumors must be explored. Conditional engineered mouse models are important for determining the function of SPOP and the mechanism by which SPOP contributes to tumorigenesis and progression. Systematic approaches are needed to screen the substrates of SPOP in human cancer. It is also important to determine the reason why SPOP exhibits a dual role in certain tissues. Overall, further exploration is required to discover a rationale for designing therapeutic strategies using SPOP inhibitors or promoters for human cancer patients.
Availability of data and materials
Not applicable.
Abbreviations
- AR:
-
Androgen receptor
- ATF2:
-
Activating Transcription Factor 2
- BRMS1:
-
Breast cancer metastasis suppressor 1
- BTB:
-
Bric-a-brac-Tramtrack/Broad
- ccRCC:
-
Clear cell renal carcinoma
- cdc20:
-
Cell division cycle 20
- CETSA:
-
Cellular thermal shift assay
- CRC:
-
Colorectal cancer
- CRL:
-
Cullin-RING E3 ligase
- EC:
-
Endometrial cancer
- EMT:
-
Epithelial-mesenchymal transition
- ERα:
-
Estrogen receptor-α
- FADD:
-
Fas-associated protein with death domain
- FTA:
-
Follicular thyroid adenoma
- FTC:
-
Follicular thyroid carcinoma
- Gli:
-
Glioma-associated oncogenic proteins
- HCC:
-
Hepatocellular carcinoma
- HDAC6:
-
Histone deacetylase 6
- LIPS:
-
Liquid-liquid phase separation
- MMP:
-
Matrix metalloproteinase
- mTORC1:
-
Mammalian target of rapamycin
- NF-κB:
-
Nuclear factor-kappa B
- NLS:
-
Nuclear localization sequence
- NSCLC:
-
Non-small cell lung cancer
- OSCC:
-
Oral squamous cell carcinoma
- PCR:
-
Polymerase chain reaction
- PD-L1:
-
Programmed death ligand 1
- PI3K:
-
Phosphatidylinositol 3-kinase
- PML:
-
Promyelocytic leukemia
- PRs:
-
Progesterone receptors
- PTC:
-
Papillary thyroid carcinoma
- PTM:
-
Post-transcriptional modification
- RCC:
-
Renal cell carcinoma
- SBC:
-
SPOP-binding consensus
- SENP7:
-
SUMO specific peptidase 7
- SHH:
-
Sonic hedgehog
- SNPs:
-
Single nucleotide polymorphisms
- SPOP:
-
Speckle-type pox virus and zinc finger protein
- SRS:
-
Substrate-recognition subunit
- TNBC:
-
Triple-negative breast cancer
- TRIM24:
-
Tripartite motif containing 24
- uPA:
-
Urokinase-type plasminogen activator
- UPS:
-
Ubiquitin proteasome system
- UTR:
-
Untranslated regions
References
Christianson JC, Ye Y. Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat Struct Mol Biol. 2014;21:325–35.
Ciechanover A. Intracellular protein degradation: from a vague idea through the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Bioorg Med Chem. 2013;21:3400–10.
Wang Z, Liu P, Inuzuka H, Wei W. Roles of F-box proteins in cancer. Nat Rev Cancer. 2014;14:233–47.
Chen HY, Chen RH. Cullin 3 ubiquitin ligases in Cancer biology: functions and therapeutic implications. Front Oncol. 2016;6:113.
Skaar JR, Pagan JK, Pagano M. SCF ubiquitin ligase-targeted therapies. Nat Rev Drug Discov. 2014;13:889–903.
Ravid T, Hochstrasser M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat Rev Mol Cell Biol. 2008;9:679–90.
Bedford L, Lowe J, Dick LR, Mayer RJ, Brownell JE. Ubiquitin-like protein conjugation and the ubiquitin-proteasome system as drug targets. Nat Rev Drug Discov. 2011;10:29–46.
Weissman AM, Shabek N, Ciechanover A. The predator becomes the prey: regulating the ubiquitin system by ubiquitylation and degradation. Nat Rev Mol Cell Biol. 2011;12:605–20.
Li W, Bengtson MH, Ulbrich A, Matsuda A, Reddy VA, Orth A, Chanda SK, Batalov S, Joazeiro CA. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle's dynamics and signaling. PLoS One. 2008;3:e1487.
Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20.
Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434.
Buetow L, Huang DT. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat Rev Mol Cell Biol. 2016;17:626–42.
Genschik P, Sumara I, Lechner E. The emerging family of CULLIN3-RING ubiquitin ligases (CRL3s): cellular functions and disease implications. EMBO J. 2013;32:2307–20.
Schumacher FR, Siew K, Zhang J, Johnson C, Wood N, Cleary SE, Al Maskari RS, Ferryman JT, Hardege I, Yasmin, et al. Characterisation of the Cullin-3 mutation that causes a severe form of familial hypertension and hyperkalaemia. EMBO Mol Med. 2015;7:1285–306.
Liu J, Ghanim M, Xue L, Brown CD, Iossifov I, Angeletti C, Hua S, Negre N, Ludwig M, Stricker T, et al. Analysis of Drosophila segmentation network identifies a JNK pathway factor overexpressed in kidney cancer. Science. 2009;323:1218–22.
Brenner JC, Chinnaiyan AM. Disruptive events in the life of prostate cancer. Cancer Cell. 2011;19:301–3.
Nagai Y, Kojima T, Muro Y, Hachiya T, Nishizawa Y, Wakabayashi T, Hagiwara M. Identification of a novel nuclear speckle-type protein, SPOP. FEBS Lett. 1997;418:23–6.
Zhuang M, Calabrese MF, Liu J, Waddell MB, Nourse A, Hammel M, Miller DJ, Walden H, Duda DM, Seyedin SN, et al. Structures of SPOP-substrate complexes: insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol Cell. 2009;36:39–50.
Marzahn MR, Marada S, Lee J, Nourse A, Kenrick S, Zhao H, Ben-Nissan G, Kolaitis RM, Peters JL, Pounds S, et al. Higher-order oligomerization promotes localization of SPOP to liquid nuclear speckles. EMBO J. 2016;35:1254–75.
Xu L, Wei Y, Reboul J, Vaglio P, Shin TH, Vidal M, Elledge SJ, Harper JW. BTB proteins are substrate-specific adaptors in an SCF-like modular ubiquitin ligase containing CUL-3. Nature. 2003;425:316–21.
Furukawa M, He YJ, Borchers C, Xiong Y. Targeting of protein ubiquitination by BTB-Cullin 3-Roc1 ubiquitin ligases. Nat Cell Biol. 2003;5:1001–7.
Pintard L, Willis JH, Willems A, Johnson JL, Srayko M, Kurz T, Glaser S, Mains PE, Tyers M, Bowerman B, Peter M. The BTB protein MEL-26 is a substrate-specific adaptor of the CUL-3 ubiquitin-ligase. Nature. 2003;425:311–6.
Errington WJ, Khan MQ, Bueler SA, Rubinstein JL, Chakrabartty A, Prive GG. Adaptor protein self-assembly drives the control of a cullin-RING ubiquitin ligase. Structure. 2012;20:1141–53.
van Geersdaele LK, Stead MA, Harrison CM, Carr SB, Close HJ, Rosbrook GO, Connell SD, Wright SC. Structural basis of high-order oligomerization of the cullin-3 adaptor SPOP. Acta Crystallogr D Biol Crystallogr. 2013;69:1677–84.
Luo L, Tang H, Ling L, Li N, Jia X, Zhang Z, Wang X, Shi L, Yin J, Qiu N, et al. LINC01638 lncRNA activates MTDH-Twist1 signaling by preventing SPOP-mediated c-Myc degradation in triple-negative breast cancer. Oncogene. 2018;37:6166–79.
Li C, Ao J, Fu J, Lee DF, Xu J, Lonard D, O'Malley BW. Tumor-suppressor role for the SPOP ubiquitin ligase in signal-dependent proteolysis of the oncogenic co-activator SRC-3/AIB1. Oncogene. 2011;30:4350–64.
Gao K, Jin X, Tang Y, Ma J, Peng J, Yu L, Zhang P, Wang C. Tumor suppressor SPOP mediates the proteasomal degradation of progesterone receptors (PRs) in breast cancer cells. Am J Cancer Res. 2015;5:3210–20.
Byun B, Tak H, Joe CO. BTB/POZ domain of speckle-type POZ protein (SPOP) confers proapoptotic function in HeLa cells. Biofactors. 2007;31:165–9.
Xu J, Wang F, Jiang H, Jiang Y, Chen J, Qin J. Properties and clinical relevance of speckle-type POZ protein in human colorectal Cancer. J Gastrointest Surg. 2015;19:1484–96.
Tan Y, Ci Y, Dai X, Wu F, Guo J, Liu D, North BJ, Huo J, Zhang J. Cullin 3SPOP ubiquitin E3 ligase promotes the poly-ubiquitination and degradation of HDAC6. Oncotarget. 2017;8:47890–901.
Zhi X, Tao J, Zhang L, Tao R, Ma L, Qin J. Silencing speckle-type POZ protein by promoter hypermethylation decreases cell apoptosis through upregulating hedgehog signaling pathway in colorectal cancer. Cell Death Dis. 2016;7:e2569.
DeLair DF, Burke KA, Selenica P, Lim RS, Scott SN, Middha S, Mohanty AS, Cheng DT, Berger MF, Soslow RA, Weigelt B. The genetic landscape of endometrial clear cell carcinomas. J Pathol. 2017;243:230–41.
Le Gallo M, Rudd ML, Urick ME, Hansen NF, Zhang S, Program NCS, Lozy F, Sgroi DC, Vidal Bel A, Matias-Guiu X, et al. Somatic mutation profiles of clear cell endometrial tumors revealed by whole exome and targeted gene sequencing. Cancer. 2017;123:3261–8.
Le Gallo M, O'Hara AJ, Rudd ML, Urick ME, Hansen NF, O'Neil NJ, Price JC, Zhang S, England BM, Godwin AK, et al. Exome sequencing of serous endometrial tumors identifies recurrent somatic mutations in chromatin-remodeling and ubiquitin ligase complex genes. Nat Genet. 2012;44:1310–5.
Le Gallo M, Bell DW. The emerging genomic landscape of endometrial cancer. Clin Chem. 2014;60:98–110.
Zhang P, Gao K, Jin X, Ma J, Peng J, Wumaier R, Tang Y, Zhang Y, An J, Yan Q, et al. Endometrial cancer-associated mutants of SPOP are defective in regulating estrogen receptor-alpha protein turnover. Cell Death Dis. 2015;6:e1687.
Zeng C, Wang Y, Lu Q, Chen J, Zhang J, Liu T, Lv N, Luo S. SPOP suppresses tumorigenesis by regulating hedgehog/Gli2 signaling pathway in gastric cancer. J Exp Clin Cancer Res. 2014;33:75.
Liu Y, Melin BS, Rajaraman P, Wang Z, Linet M, Shete S, Amos CI, Lau CC, Scheurer ME, Tsavachidis S, et al. Insight in glioma susceptibility through an analysis of 6p22.3, 12p13.33-12.1, 17q22-23.2 and 18q23 SNP genotypes in familial and non-familial glioma. Hum Genet. 2012;131:1507–17.
Ding D, Song T, Jun W, Tan Z, Fang J. Decreased expression of the SPOP gene is associated with poor prognosis in glioma. Int J Oncol. 2015;46:333–41.
Jia D, Dong R, Jing Y, Xu D, Wang Q, Chen L, Li Q, Huang Y, Zhang Y, Zhang Z, et al. Exome sequencing of hepatoblastoma reveals novel mutations and cancer genes in the Wnt pathway and ubiquitin ligase complex. Hepatology. 2014;60:1686–96.
Huang Y, Tan N, Jia D, Jing Y, Wang Q, Li Z, Zhang J, Liu L, Li J, Chen Z, He X. Speckle-type POZ protein is negatively associated with malignancies and inhibits cell proliferation and migration in liver cancer. Tumour Biol. 2015;36:9753–61.
Ji P, Liang S, Li P, Xie C, Li J, Zhang K, Zheng X, Feng M, Li Q, Jiao H, et al. Speckle-type POZ protein suppresses hepatocellular carcinoma cell migration and invasion via ubiquitin-dependent proteolysis of SUMO1/sentrin specific peptidase 7. Biochem Biophys Res Commun. 2018;502:30–42.
Harb OA, Elfeky MA, El Shafaay BS, Taha HF, Osman G, Harera IS, Gertallah LM, Abdelmonem DM, Embaby A. SPOP, ZEB-1 and E-cadherin expression in clear cell renal cell carcinoma (cc-RCC): Clinicopathological and prognostic significance. Pathophysiology. 2018;25:335–45.
Chauhan A, Bhattacharyya S, Ojha R, Mandal AK, Singh SK. Speckle-type POZ protein as a diagnostic biomarker in renal cell carcinoma. J Cancer Res Ther. 2018;14:977–82.
Zhao W, Zhou J, Deng Z, Gao Y, Cheng Y. SPOP promotes tumor progression via activation of beta-catenin/TCF4 complex in clear cell renal cell carcinoma. Int J Oncol. 2016;49:1001–8.
Li G, Ci W, Karmakar S, Chen K, Dhar R, Fan Z, Guo Z, Zhang J, Ke Y, Wang L, et al. SPOP promotes tumorigenesis by acting as a key regulatory hub in kidney cancer. Cancer Cell. 2014;25:455–68.
Luo J, Bao YC, Ji XX, Chen B, Deng QF, Zhou SW. SPOP promotes SIRT2 degradation and suppresses non-small cell lung cancer cell growth. Biochem Biophys Res Commun. 2017;483:880–4.
Luo J, Chen B, Gao CX, Xie HK, Han CN, Zhou CC. SPOP promotes FADD degradation and inhibits NF-kappaB activity in non-small cell lung cancer. Biochem Biophys Res Commun. 2018;504:289–94.
Hu X, Yang Z, Zeng M, Liu YI, Yang X, Li Y, Li XU, Yu Q. Speckle-type POZ (pox virus and zinc finger protein) protein gene deletion in ovarian cancer: fluorescence in situ hybridization analysis of a tissue microarray. Oncol Lett. 2016;12:658–62.
Jiang W, Ye S, Xiang L, Yang W, He T, Pei X, Guo L, Yang H. Establishment and molecular characterization of a human ovarian clear cell carcinoma cell line (FDOV1). J Ovarian Res. 2018;11:58.
Chen L, Pei H, Lu SJ, Liu ZJ, Yan L, Zhao XM, Hu B, Lu HG. SPOP suppresses osteosarcoma invasion via PI3K/AKT/NF-kappaB signaling pathway. Eur Rev Med Pharmacol Sci. 2018;22:609–15.
Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, Yue P, Haverty PM, Bourgon R, Zheng J, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–73.
Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C, et al. The genomic complexity of primary human prostate cancer. Nature. 2011;470:214–20.
Barbieri CE, Baca SC, Lawrence MS, Demichelis F, Blattner M, Theurillat JP, White TA, Stojanov P, Van Allen E, Stransky N, et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet. 2012;44:685–9.
Romanel A, Garritano S, Stringa B, Blattner M, Dalfovo D, Chakravarty D, Soong D, Cotter KA, Petris G, Dhingra P, et al. Inherited determinants of early recurrent somatic mutations in prostate cancer. Nat Commun. 2017;8:48.
Jung SH, Shin S, Kim MS, Baek IP, Lee JY, Lee SH, Kim TM, Lee SH, Chung YJ. Genetic progression of high grade prostatic intraepithelial Neoplasia to prostate Cancer. Eur Urol. 2016;69:823–30.
Hjorth-Jensen K, Maya-Mendoza A, Dalgaard N, Sigurethsson JO, Bartek J, Iglesias-Gato D, Olsen JV, Flores-Morales A. SPOP promotes transcriptional expression of DNA repair and replication factors to prevent replication stress and genomic instability. Nucleic Acids Res. 2018;46:9484–95.
Boysen G, Barbieri CE, Prandi D, Blattner M, Chae SS, Dahija A, Nataraj S, Huang D, Marotz C, Xu L, et al. SPOP mutation leads to genomic instability in prostate cancer. Elife. 2015;4:e09207.
Claiborn KC, Sachdeva MM, Cannon CE, Groff DN, Singer JD, Stoffers DA. Pcif1 modulates Pdx1 protein stability and pancreatic beta cell function and survival in mice. J Clin Invest. 2010;120:3713–21.
Blattner M, Liu D, Robinson BD, Huang D, Poliakov A, Gao D, Nataraj S, Deonarine LD, Augello MA, Sailer V, et al. SPOP mutation drives prostate tumorigenesis in vivo through coordinate regulation of PI3K/mTOR and AR signaling. Cancer Cell. 2017;31:436–51.
Garcia-Flores M, Casanova-Salas I, Rubio-Briones J, Calatrava A, Dominguez-Escrig J, Rubio L, Ramirez-Backhaus M, Fernandez-Serra A, Garcia-Casado Z, Lopez-Guerrero JA. Clinico-pathological significance of the molecular alterations of the SPOP gene in prostate cancer. Eur J Cancer. 2014;50:2994–3002.
Geng C, He B, Xu L, Barbieri CE, Eedunuri VK, Chew SA, Zimmermann M, Bond R, Shou J, Li C, et al. Prostate cancer-associated mutations in speckle-type POZ protein (SPOP) regulate steroid receptor coactivator 3 protein turnover. Proc Natl Acad Sci U S A. 2013;110:6997–7002.
Geng C, Rajapakshe K, Shah SS, Shou J, Eedunuri VK, Foley C, Fiskus W, Rajendran M, Chew SA, Zimmermann M, et al. Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer Res. 2014;74:5631–43.
Groner AC, Cato L, de Tribolet-Hardy J, Bernasocchi T, Janouskova H, Melchers D, Houtman R, Cato ACB, Tschopp P, Gu L, et al. TRIM24 is an oncogenic transcriptional activator in prostate Cancer. Cancer Cell. 2016;29:846–58.
Geng C, Kaochar S, Li M, Rajapakshe K, Fiskus W, Dong J, Foley C, Dong B, Zhang L, Kwon OJ, et al. SPOP regulates prostate epithelial cell proliferation and promotes ubiquitination and turnover of c-MYC oncoprotein. Oncogene. 2017;36:4767–77.
Theurillat JP, Udeshi ND, Errington WJ, Svinkina T, Baca SC, Pop M, Wild PJ, Blattner M, Groner AC, Rubin MA, et al. Prostate cancer. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science. 2014;346:85–9.
Zhu H, Ren S, Bitler BG, Aird KM, Tu Z, Skordalakes E, Zhu Y, Yan J, Sun Y, Zhang R. SPOP E3 ubiquitin ligase adaptor promotes cellular senescence by degrading the SENP7 deSUMOylase. Cell Rep. 2015;13:1183–93.
Zhang L, Peng S, Dai X, Gan W, Nie X, Wei W, Hu G, Guo J. Tumor suppressor SPOP ubiquitinates and degrades EglN2 to compromise growth of prostate cancer cells. Cancer Lett. 2017;390:11–20.
Ma J, Chang K, Peng J, Shi Q, Gan H, Gao K, Feng K, Xu F, Zhang H, Dai B, et al. SPOP promotes ATF2 ubiquitination and degradation to suppress prostate cancer progression. J Exp Clin Cancer Res. 2018;37:145.
Wu F, Dai X, Gan W, Wan L, Li M, Mitsiades N, Wei W, Ding Q, Zhang J. Prostate cancer-associated mutation in SPOP impairs its ability to target Cdc20 for poly-ubiquitination and degradation. Cancer Lett. 2017;385:207–14.
Gan W, Dai X, Lunardi A, Li Z, Inuzuka H, Liu P, Varmeh S, Zhang J, Cheng L, Sun Y, et al. SPOP promotes Ubiquitination and degradation of the ERG Oncoprotein to suppress prostate Cancer progression. Mol Cell. 2015;59:917–30.
Dai X, Gan W, Li X, Wang S, Zhang W, Huang L, Liu S, Zhong Q, Guo J, Zhang J, et al. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat Med. 2017;23:1063–71.
Janouskova H, El Tekle G, Bellini E, Udeshi ND, Rinaldi A, Ulbricht A, Bernasocchi T, Civenni G, Losa M, Svinkina T, et al. Opposing effects of cancer-type-specific SPOP mutants on BET protein degradation and sensitivity to BET inhibitors. Nat Med. 2017;23:1046–54.
Zhang P, Wang D, Zhao Y, Ren S, Gao K, Ye Z, Wang S, Pan CW, Zhu Y, Yan Y, et al. Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nat Med. 2017;23:1055–62.
Zhang J, Bu X, Wang H, Zhu Y, Geng Y, Nihira NT, Tan Y, Ci Y, Wu F, Dai X, et al. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature. 2018;553:91–5.
Ju LG, Zhu Y, Long QY, Li XJ, Lin X, Tang SB, Yin L, Xiao Y, Wang XH, Li L, et al. SPOP suppresses prostate cancer through regulation of CYCLIN E1 stability. Cell Death Differ. 2019;26:1156–68.
Yoo SK, Lee S, Kim SJ, Jee HG, Kim BA, Cho H, Song YS, Cho SW, Won JK, Shin JY, et al. Comprehensive analysis of the transcriptional and mutational landscape of follicular and papillary thyroid cancers. PLoS Genet. 2016;12:e1006239.
Jung SH, Kim MS, Jung CK, Park HC, Kim SY, Liu J, Bae JS, Lee SH, Kim TM, Lee SH, Chung YJ. Mutational burdens and evolutionary ages of thyroid follicular adenoma are comparable to those of follicular carcinoma. Oncotarget. 2016;7:69638–48.
Ye L, Zhou X, Huang F, Wang W, Qi Y, Xu H, Yang S, Shen L, Fei X, Xie J, et al. The genetic landscape of benign thyroid nodules revealed by whole exome and transcriptome sequencing. Nat Commun. 2017;8:15533.
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
An J, Ren S, Murphy SJ, Dalangood S, Chang C, Pang X, Cui Y, Wang L, Pan Y, Zhang X, et al. Truncated ERG Oncoproteins from TMPRSS2-ERG fusions are resistant to SPOP-mediated proteasome degradation. Mol Cell. 2015;59:904–16.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29.
Li JJ, Zhang JF, Yao SM, Huang H, Zhang S, Zhao M, Huang JA. Decreased expression of speckle-type POZ protein for the prediction of poor prognosis in patients with non-small cell lung cancer. Oncol Lett. 2017;14:2743–8.
Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–95.
Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800.
Imai S, Guarente L. Ten years of NAD-dependent SIR2 family deacetylases: implications for metabolic diseases. Trends Pharmacol Sci. 2010;31:212–20.
Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schroter M, et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997;386:517–21.
Chinnaiyan AM, O'Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–12.
Gibcus JH, Menkema L, Mastik MF, Hermsen MA, de Bock GH, van Velthuysen ML, Takes RP, Kok K, Alvarez Marcos CA, van der Laan BF, et al. Amplicon mapping and expression profiling identify the Fas-associated death domain gene as a new driver in the 11q13.3 amplicon in laryngeal/pharyngeal cancer. Clin Cancer Res. 2007;13:6257–66.
Chen G, Bhojani MS, Heaford AC, Chang DC, Laxman B, Thomas DG, Griffin LB, Yu J, Coppola JM, Giordano TJ, et al. Phosphorylated FADD induces NF-kappaB, perturbs cell cycle, and is associated with poor outcome in lung adenocarcinomas. Proc Natl Acad Sci U S A. 2005;102:12507–12.
Dong Y, Zhang D, Cai M, Luo Z, Zhu Y, Gong L, Lei Y, Tan X, Zhu Q, Han S. SPOP regulates the DNA damage response and lung adenocarcinoma cell response to radiation. Am J Cancer Res. 2019;9:1469–83.
Jiang J, Hui CC. Hedgehog signaling in development and cancer. Dev Cell. 2008;15:801–12.
Hui CC, Angers S. Gli proteins in development and disease. Annu Rev Cell Dev Biol. 2011;27:513–37.
Briscoe J, Therond PP. The mechanisms of hedgehog signalling and its roles in development and disease. Nat Rev Mol Cell Biol. 2013;14:416–29.
Mill P, Mo R, Fu H, Grachtchouk M, Kim PC, Dlugosz AA, Hui CC. Sonic hedgehog-dependent activation of Gli2 is essential for embryonic hair follicle development. Genes Dev. 2003;17:282–94.
Grachtchouk M, Mo R, Yu S, Zhang X, Sasaki H, Hui CC, Dlugosz AA. Basal cell carcinomas in mice overexpressing Gli2 in skin. Nat Genet. 2000;24:216–7.
Hutchin ME, Kariapper MS, Grachtchouk M, Wang A, Wei L, Cummings D, Liu J, Michael LE, Glick A, Dlugosz AA. Sustained hedgehog signaling is required for basal cell carcinoma proliferation and survival: conditional skin tumorigenesis recapitulates the hair growth cycle. Genes Dev. 2005;19:214–23.
Lee SJ, Do IG, Lee J, Kim KM, Jang J, Sohn I, Kang WK. Gastric cancer (GC) patients with hedgehog pathway activation: PTCH1 and GLI2 as independent prognostic factors. Target Oncol. 2013;8:271–80.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34.
Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin. 2014;64:104–17.
Zhang S, Xiao J, Chai Y, Hong Z, Liu Z, Yuan R, Luo Z, Zhou X, Lucero-Prisno DE III, Huang K. Speckle-type POZ protein Down-regulates matrix metalloproteinase 2 expression via Sp1/PI3K/Akt signaling pathway in colorectal Cancer. Dig Dis Sci. 2018;63:395–402.
Seidel C, Schnekenburger M, Dicato M, Diederich M. Histone deacetylase 6 in health and disease. Epigenomics. 2015;7:103–18.
Aldana-Masangkay GI, Sakamoto KM. The role of HDAC6 in cancer. J Biomed Biotechnol. 2011;2011:875824.
Li C, Cao L, Xu C, Liu F, Xiang G, Liu X, Jiao J, Niu Y. The immunohistochemical expression and potential prognostic value of HDAC6 and AR in invasive breast cancer. Hum Pathol. 2018;75:16–25.
Hou H, Zhao L, Chen W, Li J, Zuo Q, Zhang G, Zhang X, Li X. Expression and significance of cortactin and HDAC6 in human prostatic foamy gland carcinoma. Int J Exp Pathol. 2015;96:248–54.
Ai J, Wang Y, Dar JA, Liu J, Liu L, Nelson JB, Wang Z. HDAC6 regulates androgen receptor hypersensitivity and nuclear localization via modulating Hsp90 acetylation in castration-resistant prostate cancer. Mol Endocrinol. 2009;23:1963–72.
Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601–7.
Jiang X, Huang Y, Liang X, Jiang F, He Y, Li T, Xu G, Zhao H, Yang W, Jiang G, et al. Metastatic prostate cancer-associated P62 inhibits autophagy flux and promotes epithelial to mesenchymal transition by sustaining the level of HDAC6. Prostate. 2018;78:426–34.
De Craene B, Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat Rev Cancer. 2013;13:97–110.
Kapoor S. Inhibition of HDAC6-dependent carcinogenesis: emerging, new therapeutic options besides belinostat. Int J Cancer. 2009;124:509.
Jadhavar PS, Ramachandran SA, Riquelme E, Gupta A, Quinn KP, Shivakumar D, Ray S, Zende D, Nayak AK, Miglani SK, et al. Targeting prostate cancer with compounds possessing dual activity as androgen receptor antagonists and HDAC6 inhibitors. Bioorg Med Chem Lett. 2016;26:5222–8.
Seidel C, Schnekenburger M, Mazumder A, Teiten MH, Kirsch G, Dicato M, Diederich M. 4-Hydroxybenzoic acid derivatives as HDAC6-specific inhibitors modulating microtubular structure and HSP90alpha chaperone activity against prostate cancer. Biochem Pharmacol. 2016;99:31–52.
Gibbs A, Schwartzman J, Deng V, Alumkal J. Sulforaphane destabilizes the androgen receptor in prostate cancer cells by inactivating histone deacetylase 6. Proc Natl Acad Sci U S A. 2009;106:16663–8.
Hai L, Szwarc MM, He B, Lonard DM, Kommagani R, DeMayo FJ, Lydon JP. Uterine function in the mouse requires speckle-type poz protein. Biol Reprod. 2018;98:856–69.
Liu N, Liu X, Yu Q, Chen X, Ding Y, He J, Gao R, Wang Y, Liu X. SPOP regulates endometrial stromal cell Decidualization in mice. Reprod Sci. 2016;23:1565–74.
Liu X, Sun G, Sun X. RNA interference-mediated silencing of speckle-type POZ protein promotes apoptosis of renal cell cancer cells. Onco Targets Ther. 2016;9:2393–402.
DeSantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA Cancer J Clin. 2014;64:52–62.
Rhodes A, Jasani B, Balaton AJ, Barnes DM, Miller KD. Frequency of oestrogen and progesterone receptor positivity by immunohistochemical analysis in 7016 breast carcinomas: correlation with patient age, assay sensitivity, threshold value, and mammographic screening. J Clin Pathol. 2000;53:688–96.
Munoz J, Wheler J, Kurzrock R. Expression of estrogen and progesterone receptors across human malignancies: new therapeutic opportunities. Cancer Metastasis Rev. 2015;34:547–61.
Beato M, Herrlich P, Schutz G. Steroid hormone receptors: many actors in search of a plot. Cell. 1995;83:851–7.
Diep CH, Daniel AR, Mauro LJ, Knutson TP, Lange CA. Progesterone action in breast, uterine, and ovarian cancers. J Mol Endocrinol. 2015;54:R31–53.
Lanari C, Wargon V, Rojas P, Molinolo AA. Antiprogestins in breast cancer treatment: are we ready? Endocr Relat Cancer. 2012;19:R35–50.
Seraj MJ, Samant RS, Verderame MF, Welch DR. Functional evidence for a novel human breast carcinoma metastasis suppressor, BRMS1, encoded at chromosome 11q13. Cancer Res. 2000;60:2764–9.
Phadke PA, Vaidya KS, Nash KT, Hurst DR, Welch DR. BRMS1 suppresses breast cancer experimental metastasis to multiple organs by inhibiting several steps of the metastatic process. Am J Pathol. 2008;172:809–17.
Kim B, Nam HJ, Pyo KE, Jang MJ, Kim IS, Kim D, Boo K, Lee SH, Yoon JB, Baek SH, Kim JH. Breast cancer metastasis suppressor 1 (BRMS1) is destabilized by the Cul3-SPOP E3 ubiquitin ligase complex. Biochem Biophys Res Commun. 2011;415:720–6.
Lajer H, Jensen MB, Kilsmark J, Albaek J, Svane D, Mirza MR, Geertsen PF, Reerman D, Hansen K, Milter MC, Mogensen O. The value of gynecologic cancer follow-up: evidence-based ignorance? Int J Gynecol Cancer. 2010;20:1307–20.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30.
Xing M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat Rev Cancer. 2013;13:184–99.
McHenry CR, Phitayakorn R. Follicular adenoma and carcinoma of the thyroid gland. Oncologist. 2011;16:585–93.
Kondo T, Ezzat S, Asa SL. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat Rev Cancer. 2006;6:292–306.
Gusyatiner O, Hegi ME. Glioma epigenetics: from subclassification to novel treatment options. Semin Cancer Biol. 2018;51:50–8.
Arndt CA, Rose PS, Folpe AL, Laack NN. Common musculoskeletal tumors of childhood and adolescence. Mayo Clin Proc. 2012;87:475–87.
Wei X, Fried J, Li Y, Hu L, Gao M, Zhang S, Xu B. Functional roles of speckle-type Poz (SPOP) protein in genomic stability. J Cancer. 2018;9:3257–62.
Richards EJ. Inherited epigenetic variation--revisiting soft inheritance. Nat Rev Genet. 2006;7:395–401.
Huang CJ, Chen HY, Lin WY, Choo KB. Differential expression of speckled POZ protein, SPOP: putative regulation by miR-145. J Biosci. 2014;39:401–13.
Xu J, Wang F, Wang X, He Z, Zhu X. miRNA-543 promotes cell migration and invasion by targeting SPOP in gastric cancer. Onco Targets Ther. 2018;11:5075–82.
Wang LQ, Yu P, Li B, Guo YH, Liang ZR, Zheng LL, Yang JH, Xu H, Liu S, Zheng LS, et al. miR-372 and miR-373 enhance the stemness of colorectal cancer cells by repressing differentiation signaling pathways. Mol Oncol. 2018;12:1949–64.
Zhang XJ, Jin Y, Song JL, Deng F. MiR-373 promotes proliferation and metastasis of oral squamous cell carcinoma by targeting SPOP. Eur Rev Med Pharmacol Sci. 2019;23:5270–6.
Chong PA, Forman-Kay JD. Liquid-liquid phase separation in cellular signaling systems. Curr Opin Struct Biol. 2016;41:180–6.
Bouchard JJ, Otero JH, Scott DC, Szulc E, Martin EW, Sabri N, Granata D, Marzahn MR, Lindorff-Larsen K, Salvatella X, et al. Cancer mutations of the tumor suppressor SPOP disrupt the formation of active, phase-separated compartments. Mol Cell. 2018;72:19–36 e18.
Zhu G, Liang L, Li L, Dang Q, Song W, Yeh S, He D, Chang C. The expression and evaluation of androgen receptor in human renal cell carcinoma. Urology. 2014;83:510 e519–524.
Guo ZQ, Zheng T, Chen B, Luo C, Ouyang S, Gong S, Li J, Mao LL, Lian F, Yang Y, et al. Small-molecule targeting of E3 ligase adaptor SPOP in kidney Cancer. Cancer Cell. 2016;30:474–84.
Acknowledgements
Not applicable.
Funding
This work was supported by grant from National Natural Science Foundation of China (NSFC 81572936 and 81773186) and the Science and Technology Planning Project of Wenzhou City (No. Y20180082 and ZS2017006) and the Research Fund for Lin He’s Academician Workstation of New Medicine and Clinical Translation.
Author information
Authors and Affiliations
Contributions
YS, YX, CP and LY searched literature regarding to SPOP and cancer. YS and YX made the figures and Tables. YS, ZW and XZ wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Song, Y., Xu, Y., Pan, C. et al. The emerging role of SPOP protein in tumorigenesis and cancer therapy. Mol Cancer 19, 2 (2020). https://doi.org/10.1186/s12943-019-1124-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-019-1124-x