
Abstract
Background
Drug resistance in Plasmodium falciparum is a major threat to malaria control efforts. Pathogen genomic surveillance could be invaluable for monitoring current and emerging parasite drug resistance.
Methods
Data from two decades (2000–2020) of continuous molecular surveillance of P. falciparum parasites from Senegal were retrospectively examined to assess historical changes in malaria drug resistance mutations. Several known drug resistance markers and their surrounding haplotypes were profiled using a combination of single nucleotide polymorphism (SNP) molecular surveillance and whole genome sequence based population genomics.
Results
This dataset was used to track temporal changes in drug resistance markers whose timing correspond to historically significant events such as the withdrawal of chloroquine (CQ) and the introduction of sulfadoxine-pyrimethamine (SP) in 2003. Changes in the mutation frequency at Pfcrt K76T and Pfdhps A437G coinciding with the 2014 introduction of seasonal malaria chemoprevention (SMC) in Senegal were observed. In 2014, the frequency of Pfcrt K76T increased while the frequency of Pfdhps A437G declined. Haplotype-based analyses of Pfcrt K76T showed that this rapid increase was due to a recent selective sweep that started after 2014.
Discussion (Conclusion)
The rapid increase in Pfcrt K76T is troubling and could be a sign of emerging amodiaquine (AQ) resistance in Senegal. Emerging AQ resistance may threaten the future clinical efficacy of artesunate-amodiaquine (ASAQ) and AQ-dependent SMC chemoprevention. These results highlight the potential of molecular surveillance for detecting rapid changes in parasite populations and stress the need to monitor the effectiveness of AQ as a partner drug for artemisinin-based combination therapy (ACT) and for chemoprevention.
Similar content being viewed by others


Background
The World Health Organization (WHO) estimated 247 million malaria cases and 619,000 malaria deaths in 2021 [1]. Children under five years of age are the most vulnerable to malaria, accounting for 80% of deaths worldwide, with 96% of all malaria cases and deaths in 2021 occurring in the African region [1]. Increased funding for malaria control has led to an estimated 30% reduction of malaria mortality since 2000, using a combination of vector control and drug-based interventions [1]. However, the development of parasite resistance to anti-malarials threatens to undermine control and elimination efforts and poses a significant threat to public health. Anti-malarials are used for both therapeutic treatment and chemoprevention. Chemoprevention includes intermittent preventive treatment for pregnant women (IPTp) and infants (IPTi), seasonal malaria chemoprevention (SMC), and mass drug administration (MDA) [2].
Pathogen molecular surveillance tracks emerging trends in parasite drug resistance, which the WHO defines as the ability of a parasite to survive or multiply despite the administration and absorption of a drug given in doses equal to or higher than the recommended therapeutic dose [3]. Therapeutic efficacy studies (TES) are the gold standard for evaluating clinical and therapeutic drug efficacy [3, 4] but are resource intensive and challenging to implement. Pathogen molecular surveillance of known drug resistance markers can be used to assess the risk of emerging drug resistance or changes in parasite fitness [5,6,7,8,9,10] before clinical drug efficacy is irreparably compromised. Of particular importance is the continuous evaluation of drug resistance mutations for artemisinin (ART) and partner drugs used in artemisinin-based combination therapy (ACT). ART (partial) resistance is associated with delayed clearance times in Southeast Asia and is defined as having more than 10% of patients with asexual parasites on the third day following treatment [3]. Molecular surveys of drug resistance include the monitoring of single nucleotide polymorphisms (SNPs), copy number variations associated with drug resistance [1, 10], and haplotype-based analyses of hard or soft selective sweeps [10, 11]. Drug resistance markers are often associated with selective sweeps [12] as they provide a strong survival advantage to parasites that harbor the drug resistance alleles under drug pressure.
Previous studies have identified multiple molecular markers associated with antimalarial drug resistance. Chloroquine (CQ) resistance has been associated with changes in the chloroquine resistance transporter gene Pfcrt, particularly at codon Pfcrt K76T [13, 14]. Sulfadoxine-pyrimethamine (SP) resistance is associated with mutations at codons 51, 59 and 108 of dihydrofolate reductase (Pfdhfr) and codons 437 and 540 of dihydropteroate synthase (Pfdhps) [15, 16]. Mutations in the multi-drug resistance Pfmdr1 gene that have been associated with resistance against many drugs, including amodiaquine (AQ), mefloquine, quinine, lumefantrine and other quinolines [17,18,19]. Mutations in the kelch propeller domain gene Pfkelch13 have been associated with ART resistance and delayed parasite clearance [3, 20].
This study examined a two-decades long collection of P. falciparum samples obtained from febrile individuals in Senegal to identify historical changes in drug resistance markers. Based on the historical anti-malarial policy in Senegal, this study focused surveying WHO-validated and verified [4] SNPs in Pfcrt, Pfdhfr, Pfdhps, and Pfmdr1 associated with resistance to CQ, AQ, and SP [3]. Molecular surveillance of Pfkelch13 was started in 2015 in response to the emergence of Pfkelch13 C580Y as a marker associated with partial ART resistance markers in Southeast Asia [20,21,22] and to monitor the frequency of Pfkelch13 A578S, which is no longer considered to be associated with resistance but at the time was the most frequent mutation reported in Africa [23]. The goal of this study was to determine whether there were changes in parasite population genetics that could serve as an early warning sign for emerging drug resistance or changes in parasite fitness.
Methods
Sampling
Parasite samples were collected from treatment seeking patients aged 3 months or older presenting with fever or history of fever within the prior 48 h at clinics in Pikine, Thiès, Kédougou, Diourbel, Kolda, and Kaolack between 2000 and 2020. Informed consent was obtained from all patients, or from their parents or guardians if the patient was a minor. All patients with positive tests for malaria received free malaria treatment with AL or ASAQ, in accordance with the National Health Development Policy in Senegal as recommended by the WHO.
Microscopy slides, rapid diagnostic tests, and finger stick blood samples, (dried blood sample, DBS) spotted on (Whatman Protein Saver FTA (Whatman® 3MM CHR CAT N° 3030–662) filter paper, were obtained from all consenting individuals. Samples were stored in plastic bags at room temperature and protected with silica gel desiccant before DNA isolation and molecular testing. All samples were coded and anonymous, as per the consent process.
Laboratory procedures
Sample collection and DNA extraction
For the SNP-based molecular surveillance data, a total of 3284 DNA samples were extracted from DBS using QIAamp DNA Mini Kit (QIAGEN, Valencia, CA, USA), according to manufacturer’s directions. These samples were genotyped using high-resolution melting (HRM) or targeted amplicon genome (TAG) sequencing (see below).
High-resolution melting
Drug resistance markers in Pfcrt, Pfmdr1, Pfdhfr, Pfdhps were assessed using a high resolution melting (HRM) assay with the Roche LightCycler 96 instrument (Roche Molecular systems) as previously described [24]. The HRM assay was set up in a total volume of 5 μl containing 2.5 μl of DNA and 2.5 × LightScanner mastermix LCGreen (Plus double-stranded DNA dye (Idaho Technology, Inc.)). The codons 76 in Pfcrt gene; 86, 184, 1042, 1246 in Pfmdr1 gene; 51, 59, 108 in Pfdhfr gene; 437, 540, 581, 613 in Pfdhps gene were used. The following reference DNA strains were used: 3D7, Dd2, HB3, and IPC_3445 (MRA-1236).
Amplification and sequencing of the Pfkelch13 gene
The Pfkelch13 propeller domain was amplified using a P. falciparum specific protocol described previously [25]. PCR products were visualized on a 2% agarose gel after electrophoresis. Sequencing of PCR products was performed using an ABI 3730 sequencer by Sanger using a protocol established at CDC/Atlanta Malaria Genomic laboratory (Applied Biosystems, Foster City, CA).
Data analysis
The HRM result was analyzed using the LightCycler 96 application software version 1.1.0.1320. The Pfkelch13 sequence data was analyzed using the Geneious software (version 9.0.5, www.geneious.com). A cutoff of quality score HQ (High Quality) > 30% was applied to all sequences. Polymorphisms were considered if both the forward and reverse strands carried a mutation and matched the quality score cut off.
Haplotype analyses
A subset of 231 monogenomic (single-strain) samples collected from febrile, clinic-reporting patients between 2006 and 2019 from Pikine, Thiès, and Kédougou were previously whole genome sequenced using next-generation Illumina short reads [26,27,28]. These sequences excluded polygenomic (multiple strain) infections and avoided repeated sequencing of clones using a 24-SNP molecular barcode [29]. Short-reads were aligned to the P. falciparum 3D7 reference genome (PlasmoDB v. 28) using BWA-mem and Picard Tools. An initial set of 577487 genome-wide SNPs were called using HaplotypeCaller in GATK v3.5 using the quality filters and standards for best practices as described by the Pf3k project [30, 31]. Bifurcation plots and extended haplotypes were calculated using rehh 2.0 [32].
The genomic regions surrounding Pfcrt, Pfdhfr, and Pfdhps were defined as the genomic region 100 kb upstream and downstream of the starting and ending boundaries (obtained from PlasmoDB, Plasmodb.org) of each gene. For each genomic region, samples and variant sites were filtered by sequentially applying the following filters: (1) first removing samples with > 50% unusable data (missing, heterozygous, or triallelic sites), (2) then retaining sites with < 10% unusable data, and (3) finally removing samples with > 5% unusable data. These filters were applied separately across all sample years, for samples collected before or during 2014 (pre-SMC), and for samples collected after 2014 (post-SMC).
Generalized additive model
Binomial generalized additive models were fit using the R package mcgv (v1.8–40). The structure of the GAM was defined as:
where \(proportion_{mutant}\) is the proportion of samples with the mutant allele, s(year,k) is a smoothing spline function for the sample year. The k is the degrees of freedom used in the smoothing spline function. The other covariates are binary categorical variables specifying sample origin. Kolda and Kaolack were excluded because they were sampled for less than three years.
H12 and H2/H1 analyses
A total of 43386 SNPs were used for H12 and H2/H1 analyses. These SNPs were optimized to accurately estimate genetic relatedness based on identity-by-descent (IBD) estimation and are described in detail in a previous publication [28]. Briefly, SNPs were identified after dividing the genome into 2 kb windows and choosing SNPs such that each window contained a maximum of 12 SNPs, requiring SNPs have a minimum allele frequency of 0.5%, requiring that markers be at least 20 bp apart [28]. These SNPs were used to identify haplotypes based on the shared chromosome segments that were IBD using the program hmmIBD (version 2.0.4) [33] after setting the genome-wide IBD fraction parameter to 20% to better assess local IBD status in parasites that otherwise have high genome-wide IBD sharing.
At each SNP locus, haplotypes were defined by clustering samples that were related to one another. Clustering was performed via a greedy algorithm as follows. For a pair of samples IBD at a SNP site:
-
If neither is already in a cluster, form a new cluster.
-
If both are already in the same cluster, do nothing.
-
If they are already in different clusters, merge the clusters if 40% of pairwise comparisons are IBD.
-
If one is in a cluster, add the other if it is IBD with at least 40% of existing samples in the cluster. Otherwise, start a new cluster.
The resulting clusters are defined as the set of haplotypes at that locus. H1, H2, and H12 [34, 35] were calculated as:
where pi is the frequency of the i-th most frequent haplotype. H12 treats the two most common haplotypes as a single haplotype. Singleton haplotypes were omitted since their population frequency is likely to be much lower than their sample frequency.
Results
Study design and SNP-based molecular surveillance sampling
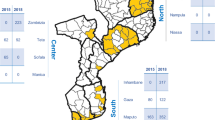
Plasmodium falciparum samples from febrile patients collected between 2000 and 2020 were genotyped or sequenced. A total of 3,284 samples were collected from six sampling locations within Senegal: Pikine, Thiès, Kédougou, Diourbel, Kaolack, and Kolda (Fig. 1A). Kédougou and Kolda are high transmission regions in southeast Senegal. In 2021, reported annual incidence was 536.5 cases per 1000 in Kédougou and 214.5 cases per 1000 in Kolda [36]. Kaolack (10.9 cases per 1000) and Diourbel (19.4 cases per 1000) are intermediate transmission regions in central Senegal. Thiès (2.8 cases per 1000) and Pikine (4.9 cases per 1000) are low transmission sites in western Senegal.

Study design and Senegal drug history A Map of Senegal highlighting the different study locations and their corresponding transmission levels in 2019. Each region is colored by transmission intensity, with red indicating high transmission and green indicating low. Squares denote high transmission sites that started SMC in 2014, triangles are moderate transmission sites that started SMC in 2019, and circles indicate low transmission sites that have not implemented SMC. B Therapeutic and chemoprevention drug use in Senegal. Therapeutic drug use in blue/green, chemoprevention drug use in red/purple. SP Sulfadoxine-Pyrimethamine, AQ amodiaquine, ASAQ Artesunate/Amodiaquine, Coartem/AL Coformulated Artemether Lumefantrine, IPTp-SP Intermittent preventative therapy in pregnant women using SP, SMC Seasonal malaria chemoprevention using SP + AQ.
The sample collection varied throughout time and space (Fig. S1, Supplemental Table 1). The 444 samples collected between 2000 and 2005 came exclusively from Pikine, the 800 samples collected between 2006 and 2014 came from Thiès, and the 340 samples collected between 2015 and 2017 came from Kédougou. Between 2017 and 2020, 1700 samples were collected from Thiès, Kaolack (starting in 2020), Diourbel, Kolda (starting in 2019), and Kédougou (continuously since 2015).
The sample collection spanned several important changes in official drug use policy (Fig. 1B). Changes in therapeutic anti-malarial treatment include the withdrawal of CQ, and successively, the introductions of SP, AQ, and ACT. Changes in chemoprevention strategies include the introduction of IPTp and SMC (Supplemental Tables 2 and 3). IPTp coverage at each of the examined study sites ranged between 33.3 and 73.2% from 2013 to 2022. SMC was introduced to Kolda and Kédougou in 2014, with SMC coverages ranging from 56.9 to 100%. Diourbel and Kaolack started implementing SMC in 2019. The reported SMC coverage for the health facility in Diourbel ranged from 44.9 to 53.9% and the health facility in Kaolack ranged from 57.3 to 82.2%.
Based on this drug policy history, several WHO-verified and validated SNP-based drug resistance markers in Pfcrt, Pfdhfr, Pfdhps, and Pfmdr1 were examined (Table 1). For Pfkelch13, only Pfkelch13 C580Y and A578S were examined. Because resistance to SP involves multiple mutations in Pfdhfr and Pfdhps, the frequency of Pfdhfr triple mutant IRN (N51I, C59R, and S108N) [37] and “quadruple” mutants (Pfdhfr triple mutant IRN + Pfdhps A437G) was examined. Likewise, the frequency of the Pfmdr1 NFD (N86Y, Y184F, D1246Y) haplotype was also examined because this haplotype is associated with resistance to multiple drugs [19, 38]. Due in part to the long timescale of the molecular surveillance study, two different molecular genotyping approaches were used. High resolution melting assays were used for the mutations in Pfcrt, Pfdhfr, Pfdhps (Methods) while the mutations in Pfkelch13 were examined using a more sensitive assay based on Sanger sequencing [25].
Molecular surveillance detects rapid changes in Pfcrt, Pfdhfr, Pfdhps, and Pfmdr1 mutations over time
Mutation frequencies in regions with more than three years of continuous sampling (Pikine, Thiès, and Kédougou, Fig. S2-S4) were first examined to identify any time-dependent trends. Pikine and Thiès are urban sites with low transmission that have not implemented SMC while Kédougou is a rural site with high transmission that has used SMC since 2014. The statistical significance of any temporal changes in mutation frequencies was assessed based on the p-values of the slope of a fitted, piecewise binomial generalized linear model. A binomial generalized additive model (GAM) was used to identify and highlight Senegal-wide trends in mutation frequency (Methods, Fig. 2). A GAM was used to summarize the data because it 1) provided a flexible framework to account for the stratified sampling of the molecular surveillance study, and 2) because the temporal changes in mutation frequency were not always monotonic and could either increase or decrease depending on the sampling period.

SNP-based Molecular Surveillance Results. Frequencies for A Pfcrt K76T, B Pfdhfr triple mutant (N51I, C59R, S108N), C Pfdhps A437G, D Pfmdr1 N86F184D1246 haplotype (N86Y, Y184F, D1246Y), E Pfkelch13 A578S, and F) Pfkelch13 C580Y. The scatterplots show the observed frequencies and their 95% binomial confidence interval. Model predictions from a calibrated generalized additive model and the 95% confidence intervals are shown in orange. The model was calibrated with data from Pikine, Thiès, Diourbel, and Kédougou (denoted with circles). The data from Kolda and Kaolack (denoted with X) were not used for model calibration. Model predictions were not generated for the Pfkelch13 mutations due to their complete or near-complete absence in the data.
Increase in Pfcrt K76T mutation frequency after 2014
As expected, due to a fitness cost associated with Pfcrt K76T [39], a statistically significant decline in Pfcrt K76T mutation frequency was observed between 2000 and 2014 (p-value < 0.001, Binomial generalized linear model) following the withdrawal of CQ in 2003 (Fig. 2A). Unexpectedly, this was followed by an increase in Pfcrt K76T frequency after 2014 (p-value < 0.001, Binomial generalized linear model). In 2000, the Senegal-wide GAM model estimated a Senegal-wide mutation frequency of 0.76 (95% CI 0.59—0.87), which fell to 0.26 (95% CI 0.21—0.31) in 2014. However, the frequency began rising after 2014, and the model estimated the frequency to be 0.49 (95% CI 0.41—0.57) in 2020. When evaluating the goodness-of-fit of the model, the adjusted R-squared for the model was 0.466 and the deviance explained was 54.2%. To account for differences in sample size, down-sampled estimates of mutation frequency were generated by re-estimating allele frequencies after down-sampling the data from each site-year to 29 samples, which was the smallest number of samples collected across all examined site-years involving Thies, Pikine, Kédougou, and Diourbel for the Pfcrt K76T mutation. Down-sampling had no effect on the trends in average mutation frequency over time (Fig. S5).
Closer examination of the changes in Pfcrt K76T in each of the individual populations revealed as a statistically significant increase in Pfcrt K76T in both Thiès and Kédougou (p-value < 0.001 for both, binomial generalized linear model) after 2014. When examining the sites not included in the Senegal-wide GAM (Kolda, and Kaolack), the frequency of Pfcrt K76T were consistent with those predicted by the GAM. However, the frequency of Pfcrt K76T in Diourbel declined (p-value < 0.001, Binomial generalized linear model) from 0.45 (95% CI 0.55—0.40) to 0.20 (95% CI 0.11—0.29) and 0.22 (95% CI 0.13—0.31) in 2018, 2019, and 2020, respectively.
Changes in Pfdhfr triple mutants following the withdrawal of CQ and introduction of SP
Based on the introduction of pyrimethamine in SP therapy in 2003 and IPTp chemoprevention in 2004, an increase in Pfdhfr IRN triple mutants over time was expected. The data revealed a sharp rise in the frequency of Pfdhfr triple mutants starting in 2003 (p-value < 0.001, Binomial generalized linear model), coinciding with the replacement of CQ with SP as the first-line antimalarial treatment in Senegal (Fig. 2B). The increase in the Pfdhfr triple mutant corresponded to a decrease in Pfdhfr triple sensitive alleles (N51N, C59C, S108S) (Fig. S6A) and only a few parasites with mixed haplotypes (different combinations of wild-type or mutant at the three amino acid sites) were observed. Across the entire dataset, only 0.06 (95% CI 0.057—0.075) of the samples had mixed haplotypes. In 2003, the GAM predicted that the frequency of triple mutants in Senegal was 0.42 (95% CI 0.27—0.59). By 2020, the predicted frequency was 0.95 (95% CI 0.90—0.97). The goodness of fit for the GAM model was evaluated using the adjusted R-squared and deviance, which were 0.881 and 89.4%, respectively.
Multiple increases and decreases in Pfdhps A437G frequency over time
Mutations in Pfdhps were expected to increase due to sulfadoxine exposure from either SP therapy or SP use in chemoprevention (IPTp or SMC). Pfdhps K540E, A581G, A613T/S were rare (< 5%) or undetected and only Pfdhps A437G was detected at a high frequency (Fig. 2C, Fig. S2-S4). Unlike with the Pfdhfr triple mutants, the Pfdhps A437G allele trajectory and “quadruple” mutant haplotype trajectory changed directions multiple times (Fig. S6B). Unusually, several inflection points were identified (at 2003, 2008, and 2014) where the trajectory of Pfdhps A437G prevalence changed direction.
Between 2000 and 2003, an increase in Pfdhps A437G was observed (p-value = 0.008, Binomial generalized linear model). The overall, Senegal-wide frequency of Pfdhps A437G as predicted by the GAM rose from 0.26 (95% CI 0.14—0.44) in 2000 to 0.47 (95% CI 0.19—0.77) in 2003. After 2003, Pfdhps A437G decreased until 2008, when its predicted frequency was 0.17 (95% CI 0.11—0.27). Between 2008 and 2014 (p-value = 0.005, Binomial generalized linear model), the frequency of Pfdhps A437G rose until 2014, where its GAM-predicted mutation frequency was 0.72 (95% CI 0.58—0.83). After 2014, the frequency of Pfdhps A437G declined (p-value = 0.022, Binomial generalized linear model), and the GAM-predicted mutation frequency in 2020 was 0.33 (95% CI 0.25—0.40). Overall, the adjusted R-squared and deviance explained by the model were 0.554 and 69.1%, respectively.
Molecular surveillance detects changes in Pfmdr1 NFD haplotype over time
Expectations for the Pfmdr1 NFD haplotype were less certain as mutations in Pfmdr1 have been associated with resistance against multiple drugs. Between 2008 and 2012, a rapid increase in the Pfmdr1 NFD (N86Y, Y184F, D1246Y) haplotype (Fig. 2D) was observed. The corresponding model-predicted frequencies were 0.27 (95% CI 0.20—0.37) in 2004 and 0.74 (95% CI 0.57—0.79) in 2012. After 2012, the predicted frequency of the Pfmdr1 NFD haplotype declined to 0.56 (95% CI 0.48—0.63) in 2016, where it remained relatively stable until 2020 [0.56 (95% CI 0.49—0.63)]. The adjusted R-squared and deviance explained by the model were 0.817 and 87.2%, respectively.
Infrequent detection of Pfkelch13 A578S in Senegal
The Pfkelch13 A578S mutation was observed in Kédougou samples and identified in one of the 89 genotyped samples collected in 2015 and in two of the 123 genotyped samples collected in 2017, but in no other year or other site (Fig. 2E–F).
Genomic haplotype analyses reveal differences in selection acting on Pfcrt, Pfdhps, and Pfdhfr
To determine whether the allele frequency changes observed at Pfcrt K76T, Pfdhps A437G, and the Pfdhfr triple mutant IRN could be the result of selection, the genomic haplotypes surrounding these genes in a set of 231 samples collected from Thiès and Kédougou between 2006 and 2019 were examined from whole genome sequencing data (Fig. S7, Fig. S8). Strong, directional selection could result in selective sweeps that reduce the genomic haplotype diversity surrounding the mutation under selection. This reduction in genomic haplotype diversity can be quantified using a statistic known as the extended haplotype homozygosity (EHH) [11]; elevated EHH indicates a reduction in diversity (elevation of homozygosity) that could be the result of a selective sweep.
Strong evidence of a selective sweep was identified around Pfcrt K76T. The haplotype structure surrounding Pfcrt K76T was far less diverse than that surrounding the ancestral Pfcrt K76 (Figs. 3A-3B). Pfcrt K76 haplotypes rapidly diversified within the first 20 kb. In contrast, Pfcrt K76T was surrounded by a dominant extended haplotype to its left (upstream, 5′) and two major extended haplotypes to its right (downstream, 3′). These dominant haplotypes extended out nearly 50 kb away from the Pfcrt K76T locus and included parasites collected before and after 2014. The EHH for Pfcrt K76T was elevated relative to Pfcrt K76 (Fig. 3C).

Evidence of selection at Pfcrt, Pfdhps, and Pfdhfr. Extended haplotype plots for the A–C Pfcrt K76T locus, D–F Pfdhps A437G locus, and G-I the Pfdhfr triple mutant. The first two columns are bifurcation plots that show the decay in haplotype structure around the focal locus. The first column (A, D, and G, blue) shows the haplotype structure surrounding the sensitive allele (Pfcrt K76, Pfdhps A437, Pfdhfr triple sensitive) while the second column (B, E, and H, red) shows the haplotype structure surrounding the resistance allele (Pfcrt K76T, Pfdhps A437G, and Pfdhfr triple resistant mutant. Starting from the center and extending outwards, each branch represents a bifurcation point where a new haplotype emerges. The thickness of the branch is proportional to the number of samples that contain that haplotype. The third column (C, F, and I) shows the EHH surrounding the sensitive (blue) and resistant (red) alleles. The solid red and blue lines are the EHH estimates obtained from using all available samples in the category. The lighter pink and blue traces are bootstrapped EHH estimates obtained by randomly downsampling fifty samples. For G–I, samples with mixed Pfdhfr genotypes were excluded.
For Pfdhps, there was little evidence of a hard selective sweep (selection of a single or small number of haplotypes). Multiple long-range haplotypes extended outwards from either Pfdhps A437G or Pfdhps A437A for 30–50 kb and there was no significant elevation in EHH (Fig. 3D–F). The lack of EHH surrounding Pfdhps A437G, coupled with the strong changes in Pfdhps A437G mutation frequency over time, could indicate variable selective pressures that act as "soft sweeps" on multiple genomic haplotype backgrounds [12, 40]. To test this, two statistics were examined: H12, the expected haplotype homozygosity that assess the frequency of the two most common haplotypes, and H2/H1, the ratio of the second and the first most frequent haplotype (Fig. S9) [35]. Elevated H12 and a low H2/H1 ratio indicates a hard selective sweep while elevated H12 and a high H2/H1 ratio suggests a soft selective sweep (Methods). H12 around Pfcrt was high but H2/H1 was low, indicating a hard selective sweep (Fig. S9A). Both H12 and the H2/H1 ratio around Pfdhps were elevated (Fig. S9B), which is consistent with a soft selective sweep.
Strong extended haplotype structure was detected around the Pfdhfr triple mutant. Most Pfdhfr triple mutant parasites shared the same dominant haplotype (Fig. 3G–I). This haplotype extended out more than 50 kb to the left and 20 kb to the right of the Pfdhfr gene. The EHH of the Pfdhfr was likely due to a hard selective sweep, like that detected for Pfcrt K76T. However, this study lacked the sensitivity to accurately estimate the EHH surrounding the Pfdhfr triple sensitive genotype due to the rarity of Pfdhfr triple sensitive parasites. Most samples with usable Pfdhfr sequences were collected after 2010 (Fig. S6C), after the rapid increase in the Pfdhfr triple mutant identified by SNP-based molecular surveillance (Fig. 2C). Pfdhfr triple mutants comprised 0.83 (95% CI 0.77—0.89) of the whole genome sequenced samples; 0.07 (95% CI 0.03—0.11) were mixed mutants, and only 0.10 (95% CI 0.05—0.14) of the samples were Pfdhfr triple sensitive parasites.
Haplotype analyses reveal temporal changes in selection at Pfcrt K76T before and after 2014
The rapid increase in Pfcrt K76T observed in the SNP-based molecular surveillance data after 2014 (Fig. 2A) suggested that the selective sweep surrounding Pfcrt K76T (Figs. 3C and 4A) was recent. To time the emergence of this selective sweep, two additional EHH analyses were performed on the whole genome sequencing data using data collected before and after 2014 (Fig. 4B, C). Prior to 2014, the EHH directly proximal to the Pfcrt K76T resistance mutation was similar to that observed surrounding the sensitive Pfcrt K76. However, elevated EHH was detected 10 kb upstream and downstream of the Pfcrt K76T locus, which is likely a legacy of the historical selective sweep that occurred prior to CQ withdrawal in 2003 (Fig. 4B) [41]. After 2014, the EHH surrounding Pfcrt K76T was elevated (Fig. 4A–C) and consistent with the expectations for a new selective pressure acting on the mutation occurring after 2014. These results further indicate that the elevated EHH surrounding Pfcrt K76T observed across all the samples (Figs. 3C and 4A) was driven by the post-2014 changes in EHH driven by this new selective sweep.

Temporal shifts in selection for Pfcrt K76T. Extended haplotype plots for the Pfcrt K76T locus using A all collected samples, B samples collected before or during 2014, and C samples collected after 2014. The solid red and blue lines are the EHH estimates obtained from using all available samples in the category. The lighter pink and blue traces are bootstrapped EHH estimates obtained by randomly downsampling fifty samples. Note that Fig. 4A is the same as Fig. 3C.
Discussion
Accurate assessments of drug resistance in parasite populations are needed to ensure continued success of drug-based malaria control efforts. In this study, rapid changes in the mutation frequencies of drug resistance alleles were observed. These contrast with the stable allele trajectories seen at 24 neutral SNPs when surveying the genetic diversity of parasites in Thiès, Kédougou, and Richard Toll between 2012 and 2020 [42]. These changes are alarming and stress the need for follow-up experiments to confirm the drug resistance phenotypes of these parasites and new data collection strategies to attribute these changes in mutation frequencies to either difference in population structure (such as host age ranges) or therapeutic/prophylactic drug use.
In Senegal, Pfdhfr triple mutant approached near-fixation in 2013, which likely reflects pyrimethamine-mediated drug resistance from either anti-malarial SP therapy or from IPTp chemoprevention. A similar rise in Pfdhfr triple mutant frequency was previously reported by a study carried out in Thiès in 2003 and 2013 [43, 44]. The haplotype analyses show that the rapid fixation of Pfdhfr likely originated as a hard sweep acting on a single haplotype. The near-fixation of Pfdhfr triple mutant suggest that majority of parasites in Senegal have some degree of pyrimethamine resistance. Based on these results, SP chemoprevention efficacy should be closely monitored despite current WHO chemoprevention guidelines that recommend IPTp with SP in regions with high Pfdhps K540E frequencies [45].
These molecular surveillance results revealed two distinct trends in Pfcrt K76T allele frequency over time. Between 2003 and 2014, a decline in Pfcrt K76T was observed, which is consistent with the withdrawal of CQ the first-line anti-malarial therapy in Senegal due to widespread CQ resistance in Africa [41, 46, 47]. In the absence of CQ, Pfcrt K76T confers a fitness cost [48] and previous genomic surveillance studies have observed declines in Pfcrt K76T after the withdrawal CQ [39, 49, 50] and similar reductions in EHH (Fig. 4B) surrounding the Pfcrt K76T mutation as parasites begin outcrossing more frequently with CQ sensitive parasites [51, 52].
After 2014, this study showed an increase in Pfcrt K76T mutation frequency that was accompanied by evidence of a new selective sweep starting in 2014. Pfcrt K76T is most associated with CQ resistance, but it is unlikely that CQ use is responsible for the observed rise in Pfcrt K76T. While unofficial CQ use during the COVID-19 pandemic has been reported [53], the rise of Pfcrt K76T predates the COVID-19 pandemic. Instead, the increase in Pfcrt K76T likely reflects increasing AQ resistance [54,55,56]. These results are troubling, as AQ is used as a partner drug for ACT (as artesunate-amodiaquine [ASAQ]) and for chemoprevention in SMC. Mathematical models predict a decline in ASAQ treatment efficacy when Pfcrt K76T frequencies are high [57] or when partner drug resistance is high [58]. Fortunately, recent TES studies in Senegal show that ASAQ remains effective [59, 60], but these molecular surveillance results suggest that the efficacy of AQ as a partner drug for ACT or for chemoprevention may be at risk of decline in the future. Given these risks, therapeutic ASAQ treatment in regions targeted for SMC should be avoided.
A variety of mutations and copy number variations in Pfmdr1 gene have been shown to modulate resistance against multiple drugs as well as CQ and AQ [19]. The Pfmdr1 haplotype NFD was the most dominant genotype in this Senegal parasite population with an overall increase in frequency from 0.2 in 2004 to 0.56 in 2020. This haplotype has previously been reported to be associated with artemether-lumefantrine (AL) treatment failure, associated with in vitro reduced AL sensitivity [61,62,63], and used as a surrogate marker for AQ and lumefantrine resistance [17]. This result suggests that the use of AL in this region might be selecting for these specific Pfmdr1 alleles and the changes in haplotype frequency may reflect the combined impact of multiple drugs. The factors driving the changes in Pfmdr1 and their implications for future therapeutic and chemoprevention strategies is a subject for future study.
Likewise, the rapid frequency changes at Pfdhps A437G was unexpected and different from the accumulation of “quadruple” mutant parasites (parasites that are Pfdhfr triple mutant and Pfdhps A437G) observed in African countries where SP is administered [64]. These results are more difficult to interpret but could reflect fluctuating drug-mediated pressure over time. While Pfdhps A437G is associated with sulfadoxine resistance, it is possible that the various additional introductions of SP-AQ, ASAQ, and Coartem/AL [65] during this time period have altered the fitness benefits and costs associated with Pfdhps A437G over time. The data also suggested a possible rebound in Pfdhps A437G starting in 2018 that could not be captured by the current Senegal-wide GAM model because there is insufficient data after 2018 to properly fit the model. Refitting the Senegal-wide model to additional data from after 2020 will be needed to determine the significance of this rebound and determine whether it continues into the future. More detailed assessments of drug use and phenotypic studies are needed to determine whether these changes are related to an AQ-induced adaptation cost or other adaptive change.
Previous studies across the African continent have identified changes in Pfcrt, mutations in Pfdhps and Pfdhfr that are associated with the use of SMC [66,67,68]. High frequencies of Pfdhps A437G, as part of the Pfdhps VAGKS (431 V-436A-437G-540 K-581G-613S) haplotype, and Pfcrt K76T, as part Pfcrt CVIET (72C-73 V-74I-75G-76 T) haplotype have been reported in countries using SMC. This study showed that the near fixation of Pfdhfr triple mutant in Senegal predated SMC in Senegal and showed that the rise in Pfcrt K76T coincided with a new selective sweep in 2014, the same year that SMC was first implemented in Senegal. Detailed follow-up analysis of the malaria clinical epidemiology and drug usage patterns are needed to confirm to what extent these changes in drug resistance markers can be attributed to drug usage in Senegal.
A major limitation of this study was that sample collection was uneven across space and time. Despite this, common changes in mutation frequency across multiple sites in Senegal were observed. For example, changes in Pfdhps A437G and Pfcrt K76T were observed in both Kédougou where SP-AQ (SMC) is applied and AL is used therapeutically, and in the non-SMC region of Thiès where ASAQ is used as treatment. Reports from the 2014 Senegal census show significant levels of human movement from the higher transmission, southeastern corner of the country (Kédougou) to the lower transmission western sections of the country (Thiès) [69], which could explain why similar changes in drug resistance mutation frequency were observed in multiple collection sites. Previous parasite population genetic analyses of neutral or highly polymorphic markers and patterns of genetic relatedness in Senegal have consistently suggested a well-mixed parasite population [28, 42, 70], unlike the fragmented parasite populations observed in the Greater Mekong region of Southeast Asia [71].
The consistency of the changes in allele frequency across sampling sites motivated the use of a single, Senegal-wide GAM model to identify Senegal-wide trend across all the study sites. Assuming a Senegal-wide trend was based on the drug resistance marker data from Pikine, Thiès, and Kédougou, the geographical proximity of (~ 30 miles) Pikine and Thiès, and the consistency of post-2014 trends across all examined study sites, including Kolda and Kaolack. Overall, the model-estimated allele frequency trajectories mirrored those observed in the actual data and can be useful for summarizing molecular surveillance results in studies with significant stratification in sampling with respect to space and time. However, this approach should only be used when there is a high degree of connectivity between sampling locations because the model assumes that each location represents a sampling from a greater, country-wide trend. This framework could also be used to identify sites whose molecular surveillance results appear to be deviating from the general, country-wide trend. For example, Diourbel was the only study site where the frequency of Pfcrt K76T was observed to be declining since 2014 and suggests that there is something unique about Diourbel relative to the rest of Senegal. Previous population genomic analyses in Diourbel have revealed an unusually high clonal parasite population genetic structure relative to the rest of the Senegal population [28].
When examining mutations associated with ART resistance, this study was limited to only those mutations in the Pfkelch13 propeller domain. With the exception of Pfkelch13 A578S, no other mutations in the Pfkelch13 propeller domain were observed [20,21,22]. A previous study using targeted deep amplicon sequencing performed on the Pfkelch13 gene in Senegalese parasites did not identify any additional mutations associated with ART resistance [72]. Other Pfkelch13 mutations (C469Y and A675V) that have been associated with ART resistance in Uganda [23, 73, 74] were not observed. As of this study, TES studies in Senegal show that ACTs remain highly effective [59, 60], but the recent emergence of African ART resistance mutations highlight the need to continuously assess ART resistance risk in Senegal.
Molecular surveillance opens up a unique opportunity to assess changing drug risk before it causes a decline in clinical efficacy. Given that loss of ACT drug efficacy has historically been preceded by both ART resistance and partner drug resistance, molecular surveillance offers an opportunity to assess emerging drug resistance before efficacy has been compromised. In Senegal, the sudden increase in Pfcrt K76T after 2014 highlights the potential risk of emerging AQ resistance that could threaten the future efficacy of ASAQ-based therapy and SMC-based chemoprevention.
Availability of data and materials
All relevant data and code will be made available on publication. SNP data and relevant analyses code will be made available in a GitHub repository. Sequence data are being uploaded to the NCBI Sequence Read Archive under BioProject PRJNA972644. All genetic and genomic data from this study are provided by the Senegal Ministry of Health and Social Action and made publicly available, along with the population data used in this study. These population data include the date of sample collection, the name of the health facility where the sample was collected, and the latitude and longitude of the health facility. No other sample information (i.e., related to the individual who provided the sample) was utilized as per the ethical approval for the study.
Code availability
All relevant code for analysis will be made available on Github at (https://github.com/weswong/senegal_drugR_2000-2020) and archived in Zenodo upon publication.
Abbreviations
- ACT:
-
Artemisinin-based combination therapy
- AL:
-
Artemether-lumefantrine
- AQ:
-
Amodiaquine
- ART:
-
Artemisinin
- ASAQ:
-
Artesunate-amodiaquine
- CQ:
-
Chloroquine
- EHH:
-
Extended haplotype homozygosity
- GAM:
-
Generalized Additive Model
- IPTp:
-
Intermittent preventative treatment in pregnant women
- IPTi:
-
Intermittent preventative treatment in infants
- MDA:
-
Mass drug administration
- SMC:
-
Seasonal malaria chemoprevention
- SNP:
-
Single nucleotide polymorphism
- SP:
-
Sulfadoxine-pyrimethamine
- TES:
-
Therapeutic efficacy studies
- WHO:
-
World Health Organization
References
WHO. World Malaria Report 2021. Geneva: World Health Organization; 2021.
Plowe CV. Malaria chemoprevention and drug resistance: a review of the literature and policy implications. Malar J. 2022;21:104.
WHO. Report on antimalarial drug efficacy, resistance and response 10 years of surveillance (2010–2019). Geneva: World Health Organization; 2020.
Marwa K, Kapesa A, Baraka V, Konje E, Kidenya B, Mukonzo J, et al. Therapeutic efficacy of artemether-lumefantrine, artesunate-amodiaquine and dihydroartemisinin-piperaquine in the treatment of uncomplicated Plasmodium falciparum malaria in Sub-Saharan Africa: a systematic review and meta-analysis. PLoS ONE. 2022;17: e0264339.
Srimuang K, Miotto O, Lim P, Fairhurst RM, Kwiatkowski DP, Woodrow CJ, et al. Analysis of anti-malarial resistance markers in pfmdr1 and pfcrt across Southeast Asia in the tracking resistance to artemisinin collaboration. Malar J. 2016;15:541.
Cheng W, Song X, Tan H, Wu K, Li J. Molecular surveillance of anti-malarial resistance pfcrt, pfmdr1, and pfk13 polymorphisms in African Plasmodium falciparum imported parasites to Wuhan. China Malar J. 2021;20:209.
Nwakanma DC, Duffy CW, Amambua-Ngwa A, Oriero EC, Bojang KA, Pinder M, et al. Changes in malaria parasite drug resistance in an endemic population over a 25-year period with resulting genomic evidence of selection. J Infect Dis. 2014;209:1126–35.
Imwong M, Dhorda M, Myo Tun K, Thu AM, Phyo AP, Proux S, et al. Molecular epidemiology of resistance to antimalarial drugs in the Greater Mekong subregion: an observational study. Lancet Infect Dis. 2020;20:1470–80.
Conrad MD, Rosenthal PJ. Antimalarial drug resistance in Africa: the calm before the storm? Lancet Infect Dis. 2019;19:e338–51.
Ndiaye YD, Hartl DL, McGregor D, Badiane A, Fall FB, Daniels RF, et al. Genetic surveillance for monitoring the impact of drug use on Plasmodium falciparum populations. Int J Parasitol Drugs Drug Resist. 2021;17:12–22.
Stephan W. Selective sweeps. Genetics. 2019;211:5–13.
Anderson TJC, Nair S, McDew-White M, Cheeseman IH, Nkhoma S, Bilgic F, et al. Population parameters underlying an ongoing soft sweep in Southeast Asian malaria parasites. Mol Biol Evol. 2017;34:131–44.
Fidock DA, Nomura T, Talley AK, Cooper RA, Dzekunov SM, Ferdig MT, et al. Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell. 2000;6:861–71.
Djimdé A, Doumbo OK, Cortese JF, Kayentao K, Doumbo S, Diourté Y, et al. A molecular marker for chloroquine-resistant falciparum malaria. N Engl J Med. 2001;344:257–63.
Peterson DS, Walliker D, Wellems TE. Evidence that a point mutation in dihydrofolate reductase-thymidylate synthase confers resistance to pyrimethamine in falciparum malaria. Proc Natl Acad Sci USA. 1988;85:9114–8.
Brooks DR, Wang P, Read M, Watkins WM, Sims PFG, Hyde JE. Sequence variation of the hydroxymethyldihydropterin pyrophosphokinase: dihydropteroate synthase gene in lines of the human malaria parasite, Plasmodium falciparum, with differing resistance to sulfadoxine. Eur J Biochem. 1994;224:397–405.
Okell LC, Reiter LM, Ebbe LS, Baraka V, Bisanzio D, Watson OJ, et al. Emerging implications of policies on malaria treatment: genetic changes in the Pfmdr-1 gene affecting susceptibility to artemether–lumefantrine and artesunate–amodiaquine in Africa. BMJ Glob Health. 2018;3: e000999.
Mbaye A, Dieye B, Ndiaye YD, Bei AK, Muna A, Deme AB, et al. Selection of N86F184D1246 haplotype of Pfmrd1 gene by artemether-lumefantrine drug pressure on Plasmodium falciparum populations in Senegal. Malar J. 2016;15:433.
Shafik SH, Richards SN, Corry B, Martin RE. Mechanistic basis for multidrug resistance and collateral drug sensitivity conferred to the malaria parasite by polymorphisms in PfMDR1 and PfCRT. PLoS Biol. 2022;20: e3001616.
Rasmussen C, Ariey F, Fairhurst RM, Ringwald P, Ménard D. Role of K13 mutations in artemisinin-based combination therapy. Clin Infect Dis. 2016;63:1680–1.
Miotto O, Amato R, Ashley EA, MacInnis B, Almagro-Garcia J, Amaratunga C. Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat Genet. 2015;47:226–34.
Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois A-C, Khim N, et al. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature. 2014;505:50–5.
Schmedes S, Patel D, Dhal S, Kelley J, Svigel S, Dimbu PR, et al. Plasmodium falciparum kelch 13 mutations, 9 countries in Africa, 2014–2018. Emerg Infect Dis. 2021;27:1902.
Daniels R, Hamilton EJ, Durfee K, Ndiaye D, Wirth DF, Hartl DL, et al. Methods to increase the sensitivity of high resolution melting single nucleotide polymorphism genotyping in malaria. J Vis Exp. 2015;2015:52839.
Talundzic E, Chenet SM, Goldman IF, Patel DS, Nelson JA, Plucinski MM, et al. Genetic analysis and species specific amplification of the artemisinin resistance-associated Kelch propeller domain in P. falciparum and P. vivax. PLoS ONE. 2015. https://doi.org/10.1371/journal.pone.0136099.
Daniels RF, Schaffner SF, Wenger EA, Proctor JL, Chang H-H, Wong W, et al. Modeling malaria genomics reveals transmission decline and rebound in Senegal. Proc Natl Acad Sci USA. 2015;112:7067–72.
Chang H-H, Park DJ, Galinsky KJ, Schaffner SF, Ndiaye D, Ndir O, et al. Genomic sequencing of Plasmodium falciparum malaria parasites from senegal reveals the demographic history of the population. Mol Biol Evol. 2012;29:3427–39.
Schaffner SF, Badiane A, Khorgade A, Ndiop M, Gomis J, Wong W, et al. Malaria surveillance reveals parasite relatedness, signatures of selection, and correlates of transmission across Senegal. Nat Commun. 2023;14:7268.
Daniels R, Volkman S, Milner D, Mahesh N, Neafsey D, Park D, et al. A general SNP-based molecular barcode for Plasmodium falciparum identification and tracking. Malar J. 2008;7:223.
MalariaGen. Pf Community Project. 2016.
MalariaGEN AA, Ali M, Almagro-Garcia J, Amambua-Ngwa A, Amaratunga C, et al. An open dataset of Plasmodium falciparum genome variation in 7000 worldwide samples. Wellcome Open Res. 2021. https://doi.org/10.12688/wellcomeopenres.16168.1.
Gautier M, Klassmann A, Vitalis R. rehh 2.0: a reimplementation of the R package rehh to detect positive selection from haplotype structure. Mol Ecol Resour. 2017;17:78–90.
Schaffner SF, Taylor AR, Wong W, Wirth DF, Neafsey DE. hmmIBD: software to infer pairwise identity by descent between haploid genotypes. Malar J. 2018;17:196.
Garud NR, Messer PW, Buzbas EO, Petrov DA. Recent selective sweeps in North American Drosophila melanogaster show signatures of soft sweeps. PLoS Genet. 2015;11: e1005004.
Harris AM, Garud NR, Degiorgio M. Detection and classification of hard and soft sweeps from unphased genotypes by multilocus genotype identity. Genetics. 2018;210:1429.
Programme National de lutte contre le Paludisme. Bulletin Epidemiologique Annuel 2021 du Paludisme au Senegal. 2021.
Lozovsky ER, Chookajorn T, Brown KM, Imwong M, Shaw PJ, Kamchonwongpaisan S, et al. Stepwise acquisition of pyrimethamine resistance in the malaria parasite. Proc Natl Acad Sci USA. 2009;106:12025–30.
Gil JP, Krishna S. pfmdr1 (Plasmodium falciparum multidrug drug resistance gene 1): a pivotal factor in malaria resistance to artemisinin combination therapies. Expert Rev Anti Infect Ther. 2017;15:527–43.
Laufer MK, Thesing PC, Eddington ND, Masonga R, Dzinjalamala FK, Takala SL, et al. Return of chloroquine antimalarial efficacy in Malawi. N Engl J Med. 2006;355:1959–66.
Hermisson J, Pennings PS. Soft sweeps: molecular population genetics of adaptation from standing genetic variation. Genetics. 2005;169:2335–52.
Wootton JC, Feng X, Ferdig MT, Cooper RA, Mu J, Baruch DI, et al. Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum. Nature. 2002;418:320–3.
Wong W, Volkman S, Daniels R, Schaffner S, Sy M, Ndiaye YD, et al. RH: a genetic metric for measuring intrahost Plasmodium falciparum relatedness and distinguishing cotransmission from superinfection. PNAS Nexus. 2022. https://doi.org/10.1093/pnasnexus/pgac187.
Ndiaye D, Dieye B, Ndiaye YD, van Tyne D, Daniels R, Bei AK, et al. Polymorphism in dhfr/dhps genes, parasite density and ex vivo response to pyrimethamine in Plasmodium falciparum malaria parasites in Thies, Senegal. Int J Parasitol Drugs Drug Resist. 2013;3:135–42.
Ndiaye D, Daily JP, Sarr O, Ndir O, Gaye O, Mboup S, et al. Mutations in Plasmodium falciparum dihydrofolate reductase and dihydropteroate synthase genes in Senegal. Trop Med Int Health. 2005;10:1176.
WHO. WHO. Geneva: Guidelines for malaria; 2023.
Ecker A, Lehane AM, Clain J, Fidock DA. PfCRT and its role in antimalarial drug resistance. Trends Parasit. 2012;28:504.
Trape JF, Pison G, Preziosi MP, Enel C, du Loû AD, Delaunay V, et al. Impact of chloroquine resistance on malaria mortality. C R Acad Sci. 1998;III(321):689–97.
Fröberg G, Ferreira PE, Martensson A, Ali A, Björkman A, Gilb JP. Assessing the cost-benefit effect of a Plasmodium falciparum drug resistance mutation on parasite growth in vitro. Antimicrob Agents Chemother. 2013;57:887–92.
Mulenga MC, Sitali L, Ciubotariu II, Hawela MB, Hamainza B, Chipeta J, et al. Decreased prevalence of the Plasmodium falciparum Pfcrt K76T and Pfmdr1 and N86Y mutations post-chloroquine treatment withdrawal in Katete District. Eastern Zambia Malar J. 2021;20:329.
Pelleau S, Moss EL, Dhingra SK, Volney B, Casteras J, Gabryszewski SJ, et al. Adaptive evolution of malaria parasites in French Guiana: reversal of chloroquine resistance by acquisition of a mutation in pfcrt. Proc Natl Acad Sci USA. 2015;112:11672–7.
Laufer MK, Takala-Harrison S, Dzinjalamala FK, Stine CO, Taylor TE, Plowe CV. Return of chloroquine-susceptible falciparum malaria in Malawi was a reexpansion of diverse susceptible parasites. J Infect Dis. 2010;202:801–8.
Verity R, Aydemir O, Brazeau NF, Watson OJ, Hathaway NJ, Mwandagalirwa MK, et al. The impact of antimalarial resistance on the genetic structure of Plasmodium falciparum in the DRC. Nat Commun. 2020;11:2107.
Taieb F, Mbaye KD, Tall B, Lakhe NA, Talla C, Thioub D, et al. Hydroxychloroquine and azithromycin treatment of hospitalized patients infected with SARS-CoV-2 in Senegal from March to October 2020. J Clin Med. 2021;10:2954.
Foguim FT, Bogreau H, Gendrot M, Mosnier J, Fonta I, Benoit N, et al. Prevalence of mutations in the Plasmodium falciparum chloroquine resistance transporter, PfCRT, and association with ex vivo susceptibility to common anti-malarial drugs against African Plasmodium falciparum isolates. Malar J. 2020;19:201.
Ochong EO, van den Broek IVF, Keus K, Nzila A. Association between chloroquine and amodiaquine resistance and allelic variation in the Plasmodium falciparum multiple drug resistance 1 gene and the chloroquine resistance transporter gene in isolates from the Upper Nile in Southern Sudan. Am J Trop Med Hyg. 2003;69:184–7.
Ehrlich HY, Bei AK, Weinberger DM, Warren JL, Parikh S. Mapping partner drug resistance to guide antimalarial combination therapy policies in sub-Saharan Africa. Proc Natl Acad Sci USA. 2021;118: e2100685118.
Bretscher MT, Dahal P, Griffin J, Stepniewska K, Bassat Q, Baudin E, et al. The duration of chemoprophylaxis against malaria after treatment with artesunate-amodiaquine and artemether-lumefantrine and the effects of pfmdr1 86Y and pfcrt 76T: a meta-analysis of individual patient data. BMC Med. 2020;18:47.
Slater HC, Griffin JT, Ghani AC, Okell LC. Assessing the potential impact of artemisinin and partner drug resistance in sub-Saharan Africa. Malar J. 2016;15:10.
Diallo MA, Yade MS, Ndiaye YD, Diallo I, Diongue K, Sy SA, et al. Efficacy and safety of artemisinin-based combination therapy and the implications of Pfkelch13 and Pfcoronin molecular markers in treatment failure in Senegal. Sci Rep. 2020;10:8907.
U.S. President’s Malaria Initiative. Senegal Malaria Operational Plan FY 2022. USAID, 2022.
Plucinski MM, Talundzic E, Morton L, Dimbu PR, Macaia AP, Fortes F, et al. Efficacy of artemether-lumefantrine and dihydroartemisinin-piperaquine for treatment of uncomplicated malaria in children in Zaire and Uíge provinces. Angola Antimicrob Agents Chemother. 2015;59:437–43.
Dokomajilar C, Nsobya SL, Greenhouse B, Rosenthal PJ, Dorsey G. Selection of Plasmodium falciparum pfmdr1 alleles following therapy with artemether-lumefantrine in an area of Uganda where malaria is highly endemic. Antimicrob Agents Chemother. 2006;50:1893–5.
Sisowath C, Strömberg J, Mårtensson A, Msellem M, Obondo C, Björkman A, et al. In vivo selection of Plasmodium falciparum pfmdr1 86N coding alleles by Artemether-Lumefantrine (Coartem). J Infect Dis. 2005;191:1014–7.
Quan H, Igbasi U, Oyibo W, Omilabu S, Chen SB, Shen HM, et al. High multiple mutations of Plasmodium falciparum-resistant genotypes to sulphadoxine-pyrimethamine in Lagos Nigeria. Infect Dis Poverty. 2020. https://doi.org/10.1186/s40249-020-00712-4.
U.S. President’s Malaria Initiative. Senegal Malaria Operational Plan FY 2020. USAID, 2020.
Baba E, Hamade P, Kivumbi H, Marasciulo M, Maxwell K, Moroso D, et al. Effectiveness of seasonal malaria chemoprevention at scale in west and central Africa: an observational study. Lancet. 2020;396:1829–40.
Mahamar A, Sumner KM, Levitt B, Freedman B, Traore A, Barry A, et al. Effect of three years’ seasonal malaria chemoprevention on molecular markers of resistance of Plasmodium falciparum to sulfadoxine-pyrimethamine and amodiaquine in Ouelessebougou. Mali Malar J. 2022;21:39.
Beshir KB, Muwanguzi J, Nader J, Mansukhani R, Traore A, Gamougam K, et al. Prevalence of Plasmodium falciparum haplotypes associated with resistance to sulfadoxine-pyrimethamine and amodiaquine before and after upscaling of seasonal malaria chemoprevention in seven African countries: a genomic surveillance study. Lancet Infect Dis. 2023;23:361–70.
Senegal Population and Housing Census 2013. 2013.
Ndiaye T, Sy M, Gaye A, Siddle KJ, Park DJ, Bei AK, et al. Molecular epidemiology of Plasmodium falciparum by multiplexed amplicon deep sequencing in Senegal. Malar J. 2020;19:403.
Jacob CG, Thuy-Nhien N, Mayxay M, Maude RJ, Quang HH, Hongvanthong B, et al. Genetic surveillance in the Greater Mekong Subregion and South Asia to support malaria control and elimination. Elife. 2021;10: e62997.
Gaye A, Sy M, Ndiaye T, Siddle KJ, Park DJ, Deme AB, et al. Amplicon deep sequencing of kelch13 in Plasmodium falciparum isolates from Senegal. Malar J. 2020;19:134.
Asua V, Conrad MD, Aydemir O, Duvalsaint M, Legac J, Duarte E, et al. Changing prevalence of potential mediators of aminoquinoline, antifolate, and artemisinin resistance Across Uganda. J Infect Dis. 2021;223:985.
Balikagala B, Fukuda N, Ikeda M, Katuro OT, Tachibana S-I, Yamauchi M, et al. Evidence of artemisinin-resistant malaria in Africa. N Engl J Med. 2021;385:1163–71.
Acknowledgements
We wish to thank the patients who have participated in these studies and the clinic nurses and staff who were involved with obtaining samples from the clinics.
Funding
The work was supported by the NIH/ICEMR, International Centre of Excellence for Malaria Research West Africa (U19AI089696), the Bill and Melinda Gates Foundation Grants (Investment ID 0PP1200177 and Investment ID INV-004036), by the Presidents’ Malaria Initiative, and the Islamic Development Bank.
Author information
Authors and Affiliations
Contributions
YDN and WW led the analyses and generated all figures with support from SFS. YDN, WW, JT, SKV wrote the main manuscript text. DN, DFW, SKV, JT designed the study, supervised data collection and analysis. All other authors were involved with data collection. All authors reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and content to participate
The study was reviewed and approved by the Ministry of Health and Social Action in Senegal (Protocol SEN14/49) and the Institutional Review Board at Harvard T.H. Chan School of Public Health (CR-16330-07). This study was registered at the Pan African Clinical Trials Registry on 09 March 2020 under the number PACTR 202003802011316.
Competing interests
The findings and conclusions in this paper are those of the authors and do not necessarily represent the official position of the U.S. Centers for Disease Control and Prevention. The authors declare no competing of interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
12936_2024_5024_MOESM1_ESM.docx
Supplementary Material 1: Fig. S1 Sample sizes for SNP-based molecular surveillance. Sample size per year per region for the SNP-based molecular surveillance. Fig. S2 SNP-based molecular surveillance in Pikine for A) Pfcrt, B) Pfdhfr, C) Pfdhps and D) Pfmdr1. The Pfdhfr I174L and Pfkelch13 SNPs were not examined in Pikine. Error bars indicate two binomial standard deviations from the mean. X’s denote years where samples were collected but the mutation was not observed. Gaps in the data were because samples were not collected for that year. Fig. S3 SNP-based molecular surveillance in Thiès for A) Pfcrt, B) Pfdhfr, C) Pfdhps, D) Pfmdr1, and E) Pfkelch13. Error bars indicate two binomial standard deviations from the mean. X’s denote years where samples were collected but the mutation was not observed. Gaps in the data were because samples were not collected for that year. Fig. S4 SNP-based molecular surveillance in Kédougou for A) Pfcrt, B) Pfdhfr, C) Pfdhps, D) Pfmdr1, and E) Pfkelch13. Error bars indicate two binomial standard deviations from the mean. X’s denote years where samples were collected but the mutation was not observed. Gaps in the data were because samples were either not collected or not genotyped for that year. Fig. S5 Down-sampled estimates of Pfcrt K76T for A) Thies, B) Pikine, C) Kédougou, and E) Diourbel. In blue are the estimated allele frequencies and 95% confidence intervals obtained from the raw data. In grey are average allele frequencies and 95% confidence intervals after down-sampling the data from each site-year to 29 samples, which was the smallest number of samples collected across all examined site-years involving Thies, Pikine, Kédougou, and Diourbel. Fig. S6 A) Frequency of Pfdhfr triple sensitive (N51, C59, S108) parasites. B) Frequency of “quadruple” (Pfdhfr triple mutant + Pfdhps A437G) parasites. The scatterplots show the observed frequencies and their 95% binomial confidence interval. Model predictions from a calibrated generalized additive model and the 95% confidence intervals are shown in orange. The model was calibrated with data from Pikine, Thiès, Diourbel, and Kédougou (denoted with circles). The data from Kolda and Kaolack (denoted with X) were not used for model calibration. Fig. S7 Sampling distribution for our whole genome sequence collection (A). Grey indicates that the sample came from Thiès. Green indicates the sample came from Kédougou. Sampling distributions for B) the Pfcrt genomic region, C) the Pfdhfr genomic region, and D) the Pfdhps genomic regions. For B and D, blue denotes samples with the sensitive allele and red indicates those with the resistance allele. For C, red denotes samples that are Pfdhfr triple mutant, blue indicates those that are Pfdhfr triple sensitive, and orange indicates those with a mix of resistant and sensitive alleles at the three examined Pfdhfr loci. Fig. S8 SNPs used to define genomic haplotypes. Genomic haplotypes surrounding the wild-type mutations: A) Pfcrt K76, B) Pfdhfr C59, C) Pfdhps A437 and the drug resistance mutations: D) Pfcrt K76T, E) Pfdhfr C59R, F) Pfdhps A437G. 171 samples and 173 SNPs were examined for Pfcrt, 170 samples and 85 SNPs for Pfdhfr, 182 samples and 83 SNPs for Pfdhps. Each row represents a sample. The left most column indicates whether the sample was collected before or during 2014 (green) or after 2014 (yellow). Alleles corresponding to the 3D7 reference are indicated by light blue and alleles corresponding to the alternative allele are indicated by dark blue. White corresponds to missing data. The orange boxes highlight the boundaries of the Pfcrt (A/D), Pfdhfr (B/E), and Pfdhps (C/F) genes. Fig. S9 Evidence of Hard and Soft Sweeps H12 (blue, left y-axis) and H2/H1 (orange, right y-axis) statistics for A) chromosome 7 and B) chromosome 8. The dotted green lines show the location of Pfcrt or Pfdhps. Table 1. Sample sizes for SNP-based molecular surveillance. Table 2. Regional and health facility IPTp coverage where samples were collected: Pikine (Deggo), Thiès (SLAP), Diourbel (Sessene), Kaolack (P. Assainies), Kolda (Bagadadji), and Kédougou (Bandafassi). Table 3. Regional SMC and health facility coverage where samples were collected: Pikine (Deggo), Thiès (SLAP), Diourbel (Sessene), Kaolack (P. Assainies), Kolda (Bagadadji), and Kédougou (Bandafassi).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Ndiaye, Y.D., Wong, W., Thwing, J. et al. Two decades of molecular surveillance in Senegal reveal rapid changes in known drug resistance mutations over time. Malar J 23, 205 (2024). https://doi.org/10.1186/s12936-024-05024-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-024-05024-8