
Abstract
Background
Malaria diagnosis in many malaria-endemic countries relies mainly on the use of rapid diagnostic tests (RDTs). The majority of commercial RDTs used in Africa detect the Plasmodium falciparum histidine-rich protein 2 (PfHRP2). pfhrp2/3 gene deletions can therefore lead to false-negative RDT results. This study aimed to evaluate the frequency of PCR-confirmed, false-negative P. falciparum RDT results in Monrovia, Liberia.
Methods
PfHRP2-based RDT (Paracheck Pf®) and microscopy results from 1038 individuals with fever or history of fever (n = 951) and pregnant women at first antenatal care (ANC) visit (n = 87) enrolled in the Saint Joseph’s Catholic Hospital (Monrovia) from March to July 2019 were used to assess the frequency of false-negative RDT results. True–false negatives were confirmed by detecting the presence of P. falciparum DNA by quantitative PCR in samples from individuals with discrepant RDT and microscopy results. Samples that were positive by 18S rRNA qPCR but negative by PfHRP2-RDT were subjected to multiplex qPCR assay for detection of pfhrp2 and pfhrp3.
Results
One-hundred and eighty-six (19.6%) and 200 (21.0%) of the 951 febrile participants had a P. falciparum-positive result by RDT and microscopy, respectively. Positivity rate increased with age and the reporting of joint pain, chills and shivers, vomiting and weakness, and decreased with the presence of coughs and nausea. The positivity rate at first ANC visit was 5.7% (n = 5) and 8% (n = 7) by RDT and microscopy, respectively. Out of 207 Plasmodium infections detected by microscopy, 22 (11%) were negative by RDT. qPCR confirmed absence of P. falciparum DNA in the 16 RDT-negative but microscopy-positive samples which were available for molecular testing. Among the 14 samples that were positive by qPCR but negative by RDT and microscopy, 3 only amplified pfldh, and among these 3 all were positive for pfhrp2 and pfhrp3.
Conclusion
There is no qPCR-confirmed evidence of false-negative RDT results due to pfhrp2/pfhrp3 deletions in this study conducted in Monrovia (Liberia). This indicates that these deletions are not expected to affect the performance of PfHRP2-based RDTs for the diagnosis of malaria in Liberia. Nevertheless, active surveillance for the emergence of PfHRP2 deletions is required.
Similar content being viewed by others

Background
Delay in diagnosis and treatment is a leading cause of death in malaria patients [1]. The recommendation issued in 2010 by the World Health Organization (WHO) to restrict malaria treatment to parasitological confirmed malaria infections has boosted the use of rapid diagnostic tests (RDTs), which have now become a critical component of management and surveillance of malaria. Indeed, it has been estimated that, in 2019, over 348 million RDTs were sold by manufacturers and 267 million distributed by national malaria programmes, at a cost of hundreds of millions of euros [2].
Most RDTs manufactured, purchased and used around the world are based on the detection of Plasmodium falciparum histidine-rich protein 2 (PfHRP2), alone or in combination with other antigens (Plasmodium lactate dehydrogenase (pLDH) and Plasmodium aldolase (pAldo)). The PfHRP2 is a parasite-specific protein produced only by P. falciparum (and not the other human malaria parasite species) throughout its asexual life cycle, and released during schizogony into the peripheral circulation [3], where it can persist for weeks after the elimination of parasites [4]. In 2010, it was shown that some isolates of P. falciparum in Peru lacked the pfhrp2 gene [5]. The pfhrp3 gene is highly homologous to pfhrp2 [6], and parasites lacking both pfhrp2 and pfhrp3 genes, or substantial parts of these genes, do not express functional proteins and are therefore not detected by PfHRP2-based RDTs [7]. Such false negative results pose a serious threat to case management, as patients truly infected with P. falciparum may be falsely identified as malaria-free, and thus not managed adequately. Recently, numerous studies have reported P. falciparum parasites lacking pfhrp2 and pfhrp3 genes in several countries in Africa [8,9,10,11,12,13,14,15,16], with pfhrp2 deletion having been identified by WHO as one of the biological challenges currently threatening malaria control and elimination efforts. A mathematical model identified that a low intensity of transmission and a high frequency of treatment based on RDT detection of infection are the two main drivers of selection of pfhrp2-deleted parasites [17]. Current WHO recommendations suggest switching to non-PfHRP2 RDTs when the prevalence of pfhrp2-deleted parasites reaches the lower 90% confidence interval for 5% prevalence, or a plan for change if deletions surpass a frequency of 5% [18]. Good quality data are required to guide this switch, particularly taking into account that non-PfHRP2-based RDTs can have lower sensitivity than those based on the detection of PfHRP2 [19]. Systematic monitoring of parasites with pfhrp2/3 deletions is therefore required to monitor the risk of false-negative RDT results.
RDT-negative but microscopy-positive results can occur due to operator error, inappropriate storage, limited performance of specific RDT brands and lots, low-parasite density infections, and pfhrp2/pfhrp3 deletions. Mutant parasites carrying the deletion are usually identified by a discrepancy between positive microscopy results and negative results of the PfHRP2-based RDT in patients undergoing both tests [5, 20]. The detection of parasite DNA by polymerase chain reaction (PCR) offers the possibility of detecting low density infections that are not readily detected by RDT and the genomic confirmation of complete or partial deletions of the pfhrp2/3 gene [7]. This study aimed to assess the frequency of true (PCR-confirmed) false-negative P. falciparum PfHRP2 RDT results among symptomatic patients and pregnant women at first antenatal care (ANC) visit attending a public hospital in Monrovia, Liberia.
Methods
Study site and population
The study was conducted at the Outpatients Department, Emergency and Antenatal Consultation of the not-for-profit Saint Joseph’s Catholic Hospital (SJCH) in Congo Town neighbourhood, Monrovia. The SJCH provides general services to the population in Monrovia. Although the SJCH applies a cost recovery system for the general public, the institution has a charity arm to subsidize healthcare-related costs for the most deprived.
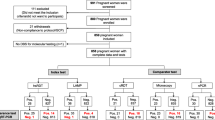
In the time-period 21 March to 21 July 2019, all patients who presented at the facilities with fever (temperature ≥ 37.5 °C) or history of fever during the preceding week, as well as pregnant women attending ANC for the first time during their pregnancy (irrespective of their fever status), were eligible for inclusion in the study. No individuals meeting inclusion criteria were excluded based on their race, social or economic status, religion, ethnic affiliation, nationality, political affiliation, or sexual orientation.
Recruitment
Eligible patients were invited to participate and informed of the study objectives and specimen collection procedures. After providing written informed consent, they were queried on basic socio-demographic and malaria prevention-related data. Their forehead temperature was measured with an infrared thermometer. For the participants attending their first ANC, the gestational age was assessed by date of last menstrual period and by measurement of fundal height. Data were manually captured by the recruiting research team using individual standardized paper-based case report forms.
Parasitological assessments
Participants were finger pricked for malaria testing. Five μl of blood were used to perform malaria testing using Paracheck Pf® (Orchid Biomedical Systems, Goa, India), a PfHRP2-based malaria RDT. Another 5 μl of blood was used to prepare a thick blood film for malaria parasite microscopy examination. If present, malaria parasites were detected and semi-quantified through the microscopic examination of Field’s stained thick blood film. A total of 9 technicians were in charge of the microscopic diagnosis. The diagnosis was made by a single technician without double confirmation. In case of any doubt, the expert opinion of the laboratory manager was used for a final decision. A 50-μl blood drop was spotted on Whatman 903 filter papers, dried for 24 h and stored in plastic bags with silica gel at -20ºC. One of the prepared filter papers was shipped to the Barcelona Institute of Global Health (Barcelona, Spain) for molecular detection of P. falciparum. All the samples with microscopy-, and RDT-discrepant results, plus a 25% random selection of the rest of samples, were selected for molecular assessment. DNA was extracted from filter papers following the Chelex method [21] and used for quantitative real-time PCR targeting P. falciparum 18S ribosomal RNA (rRNA) gene [21, 22]. In brief, purified DNA templates were amplified in an ABI PRISM 7500 Real-Time System (Applied Biosystems) following a previously described method [13]. Briefly, a 20-μl PCR mixture was performed using 5 μl of template, 10 μl of 2 × TaqMan® Universal PCR Master Mix (Applied Biosystems), a 300-nM concentration of each primers specific for 18S rRNA gene of P. falciparum, and a 150-nM concentration of probe labelled with 6-carboxy-fluorescein (FAM) as a reporter and 6-carboxytetramethylrhodamine (TAMRA) as a quencher. Amplification and detection were performed under the following conditions: 2 min at 50 °C, 10 min at 95 °C, and 40 cycles of 15 s at 95 °C and 1 min at 60 °C. The results were automatically analysed by the ABI Prism SDS2.0.6 software. Each specimen was run in duplicate. Parasitaemia was calculated by extrapolation against a standard curve of 5 serially diluted points prepared with known numbers of 3D7 ring-infected erythrocytes [23]. Samples without amplification (no Ct detected) were considered negative. A negative control (uninfected erythrocytes) and a blank control (no template) were run in all reactions.
Samples positive by microscopy and negative by the 18S P. falciparum qPCR were analysed by qPCR for Plasmodium spp using one set of generic primers targeting a highly conserved region of the 18S rRNA gene of the genus Plasmodium [24]. Amplification and detection of the amplified product were performed on an ABI Prism 7500 (Applied Biosystems). A negative control (uninfected erythrocytes) and a blank control (no template) as well as 4 positive controls (one for P. falciparum, P. vivax, P. ovale, and P. malariae) were included. The sample was considered positive by identifying the threshold cycle number (Ct) at which normalized reporter dye emission raised above background noise. If the fluorescent signal did not increase within 40 cycles (Ct = 40), the sample was considered negative.
Samples that were positive by 18S rRNA qPCR but negative by PfHRP2-RDT were subjected to multiplex qPCR assay for detection of pfhrp2 and pfhrp3 following previously described method [25] with minor modifications. In brief, an ABI 7500 real-time machine was used, with the following QSY probes (Applied Biosystems): Pfhrp2_probe 6FAM 5′-ATGCAAAAGGACTTAATTTAAATAAGAGATT-3′; Pfhrp3_probe VIC 5′-ACAATTCCCATACTTTACATCATGCA-3′; Pfldh_probe ABY 5′-GTAATAGTAACAGCTGGATTTACCAAGGCCCCA-3′; HumanTuBB_P JUN 5′-TTAACGTGCAGAACAAGAACAGCAGCT-3′.
To optimize the final reaction, pfhrp2 primers and probe were increased to 900 nM and 250 nM, respectively. TaqMan® Multiplex Master Mix was used. The thermocycling conditions were 20 s at 95 °C, followed by 45 cycles of 3 s at 95 °C and 33 s at 60 °C. Samples with HumTuBB positive and pfldh positive but negative for pfhrp2 or pfhrp3 were considered as pfhrp2-deleted and pfhrp3-deleted, respectively. Samples with HumTuBB positive but pfldh negative were defined as having a parasitaemia too low to be detected by this method. Finally, samples with a HumTuBB negative result were considered to be invalid and indicated the need to repeat the DNA extraction and/or PCR experiment.
Data management and statistical analysis
The study participants were assigned with a sequential unique identification number (UIN) that linked the signed consent forms to the case report forms. The case report forms did not include personal identifiers and were used to collect socio-demographic and malaria care-related data. The laboratory technologists were oriented to document all laboratory test results and report both the blood film and RDT results in standard reporting forms. All information contained in the case report forms was double-captured (by a trained laboratory technician and by a member of the research team) into a research database built in Microsoft Excel. The point-of-access to the Excel spreadsheets were designed to protect the confidentiality and integrity of the data and included authorization, authentication, auditing, and availability features to safeguard the access and usage of the data. The Excel spreadsheets were in a password-protected computer sited at the SJCH laboratory.
Data entered into Excel was converted to STATA (version 8.0, STATA Corporation, College Station, TX, USA) for further analyses. The sensitivity, specificity, false negative, and false positive values of Paracheck Pf® were calculated using microscopy as the gold standard. Briefly, sensitivity was calculated as the proportion of positive test results against true positives; specificity was calculated as a proportion of negative test results against true negatives. Negative RDT results were considered false-negatives if microscopy result was positive. Positive RDTs were considered false-positives if microscopy was negative. True false-negatives and false-positives were considered if the qPCR was positive and negative, respectively. Proportions were compared using Chi2 test and differences with a probability of less than 0.05 (P < 0.05) were accepted as significant.
Results
Clinical and demographic characteristics of study participants
One-thousand and forty participants meeting inclusion criteria were invited to participate between 21 March and 21 July, 2019, all of whom consented. Of the 1040 enrolled participants, two discontinued their participation before blood specimen collection. The analysis presented is based on data from 1038 participants (951 febrile individuals and 87 pregnant women) with peripheral blood samples available and analysed for P. falciparum infection. From the 951 febrile participants (Table 1), 459 were males (48%), 330 (35%) under 5 years of age, 387 (41%) above 20 years, and 541 (57%) reported primary education or below. Use of insecticide-treated nets and household indoor residual spraying was reported by 441 (46%) and 532 (56%) of the study participants, respectively. Three-hundred and sixty-five (38%) presented with fever and 725 (76%) reported fever during the preceding week. Malaria signs and symptoms ranged from 13% in the case of diarrhoea (n = 119) to 44% in the case of headache (n = 420). Seven-hundred and six of the study participants (74%) reported history of a previous malaria episode.
Out of the 87 women at first ANC visit, 4 (5%) and 7 (8%) had fever or reported any sign/symptom of malaria at their first ANC visit, respectively (Table 2).
Plasmodium falciparum positivity rate and association with clinical data
One-hundred and eighty-six (19.6%) and 200 (21.0%) of the 951 febrile participants had a P. falciparum-positive result by RDT and microscopy, respectively (Table 1). Positivity rate increased with age and with the reporting of joint pain, chills and shivers, headache, vomiting and weakness. In contrast, it was lower in patients with cough and nausea compared to those with other signs of malaria. The positivity rate among pregnant women at first ANC visit was 5.7% (5 out of 87 women) and 8.0% (7 out of 87) by RDT and microscopy, respectively (Table 2). None of the clinical variables tested were associated with positivity by RDT, microscopy or qPCR.
RDT performance
Compared to microscopy (Table 3), RDT sensitivity was 89% (185/207), with a false negativity rate of 11% (22/207). RDT specificity was 99% (825/831) and the false positivity rate was < 1% (6/831). Among the 28 samples with discordant RDT and microscopy results, 21 (5 RDT-positive but microscopy-negative, and 16 RDT-negative but microscopy-positive) were available for molecular analysis to screen for P. falciparum DNA using qPCR. Three (60%) of the 5 samples found to be positive by RDT but negative by microscopy were negative by qPCR, while 2 (40%) of them were positive by qPCR. The 16 (100%) samples, which were positive by microscopy but negative by RDT, were also negative by qPCR. Among the 22 samples positive by microscopy and negative by the 18S P. falciparum qPCR, only one was positive for Plasmodium spp. (Ct = 39.8) [24]. Among the randomly selected 224 samples that were negative by RDT and microscopy, 14 (6.2%) were confirmed positive by qPCR. Plasmodium falciparum densities (as quantified by qPCR) were higher among RDT-positive infections (n = 72, geometric mean: 767.9 parasites/μL; SD: 2,938.2) than RDT-negative infections (n = 14, 2.1, SD: 2.9; p < 0.001). The 14 samples were tested for the presence of hrp2/3 deletions using a multiplex qPCR assay that targets pfhrp2, pfhrp3, pfldh_ HumanTuBB_ [25]. The 14 samples were positive for the human gene (HumTuBB), but only 3 amplified pfldh. Among these 3 samples with enough parasitaemias for the detection of single copy parasite genes (pfpldh), all were positive for hrp2 and hrp3.
Discussion
This study provides evidence of the absence of qPCR-positive, false-negative RDT results and therefore of pfhrp2/3 deletions in P. falciparum isolates circulating in Monrovia (Liberia). Among the 1,038 individuals included in the study, only 22 had a negative RDT and a positive microscopy. Sixteen of these samples tested by qPCR confirmed the absence of P. falciparum DNA, therefore indicating a false positive result by microscopy. Results of this study suggest that P. falciparum parasites circulating in Monrovia do not yet carry pfhrp2/hrp3 deletions and are, therefore, conveniently detectable using PfHRP2-based RDTs. However, continuous monitoring for the emergence of PfHRP2 deletions is needed to avoid RDT failures that could potentially compromise malaria control programmes in Liberia.
The prevalence of P. falciparum infections among individuals with fever or history of fever during the preceding week was 19.6% by RDT and 21.0% by microscopy. Among those individuals who were negative by both diagnostic tests, the prevalence of P. falciparum infection by qPCR was 5.3%, indicating a moderate level of low-density malaria infections, which are undetected among febrile individuals. The carriage of sub-patent infections might be higher among afebrile individuals, as observed in pregnant women at first ANC visit (9.2%), who tend to carry asymptomatic low-density infections [26]. Overall, the low malaria positivity rates in Monrovia compared to estimates from other African countries [27, 28] might be due to the relatively lower risk of malaria infection among the population residing in Monrovia compared to the rural areas in Liberia. Positivity rate is higher among individuals reporting joint pain, vomiting, chills and shivers and weakness. In contrast, cough and nausea were associated with lower malaria positivity rates, suggesting these clinical signs may appear to be resulted by other diseases such as respiratory infections. Positivity rate was also higher among older individuals, suggesting that occupational or motility factors may contribute to increased risk of exposure to malaria parasites in areas outside Monrovia with higher transmission.
The specificity of RDT, compared to microscopy, was high (99%), with most of the false-positive results being negative by qPCR, suggesting HRP2 persistence after a recently cleared P. falciparum infection [4]. False-negative results were higher, with 11% of the microscopic-positive subjects negative for RDT. This is below the overall estimate of 19.9% obtained from community-based malaria surveys in 19 sub-Saharan African countries [13]. Importantly, qPCR confirmed the absence of P. falciparum DNA in the 16 RDT-negative and microscopy-positive samples that were available for molecular testing. All these samples were also negative for other Plasmodium human species [24]. Only one of the 6 samples which were positive by both RDT and microscopy was positive for Plasmodium spp, therefore indicating a non-falciparum Plasmodium spp. that was missed by the first qPCR targeting P. falciparum 18S rRNA. Finally, among the 14 samples that were positive by qPCR but negative by RDT and microscopy, 3 only amplified pfldh, and among these 3 all were positive for hrp2 and hrp3 [25]. The lack of amplification of pfldh in the 11 samples is probably due to very low density infections which are still amplified by the 18S rRNA (multigene) qPCR but not by the single-gene qPCR (pfldh). Independent of the reason above, this study rules out the possibility of true (qPCR-confirmed) parasitaemic cases undetected by the RDT. This thus provides evidence that none of the parasite isolates collected in this study was potential carriers of pfhrp2/hrp3 deletions.
This study has several limitations. First, a sub-set of dried blood spots (including 6 of the 22 which were collected from individuals with RDT-negative but microscopy-positive results) were not available for molecular testing. And second, the fees of consultation and malaria diagnostic tests in the institution of recruitment may have led to an under-representation of populations with low social-economic backgrounds who may be more prone to malaria infection.
Conclusions
PfHRP2-based RDTs are efficacious in detecting the majority of the malaria parasites in the Monrovia area, with no evidence of pfhrp2/3 deletions in this parasite population.
Availability of data and materials
The datasets generated during and/or analysed during the current study are not publicly available due to the agreements reached with the regulatory authorities of the country but are available from the corresponding author on reasonable request.
Abbreviations
- 18 S rRNA:
-
18 Small ribonuclease ribonucleic acid
- ANC:
-
Antenatal Carre
- pAldo:
-
Plasmodium aldolase
- PfHRP2/3 :
-
Plasmodium falciparum Histidine-rich protein 2 and 3
- Pldh:
-
Plasmodium lactate dehydrogenase
- qPCR:
-
Quantitative polymerase chain reaction
- RDTs:
-
Rapid diagnostic tests
- SJCH:
-
Saint Joseph’s Catholic Hospital
- WHO:
-
World Health Organization
References
Mousa A, Al-Taiar A, Anstey NM, Badaut C, Barber BE, Bassat Q, et al. The impact of delayed treatment of uncomplicated P. falciparum malaria on progression to severe malaria: a systematic review and a pooled multicentre individual-patient meta-analysis. PLoS Med. 2020;17:e1003359.
WHO. World malaria report 2020. Geneva, World Health Oganization, 2020. www.who.int/malaria.
Desakorn V, Silamut K, Angus B, Sahassananda D, Chotivanich K, Suntharasamai P, et al. Semi-quantitative measurement of Plasmodium falciparum antigen PfHRP2 in blood and plasma. Trans R Soc Trop Med Hyg. 1997;91:479–83.
Plucinski MM, Dimbu PR, Fortes F, Abdulla S, Ahmed S, Gutman J, et al. Posttreatment HRP2 clearance in patients with uncomplicated Plasmodium falciparum malaria. J Infect Dis. 2018;217:685–92.
Gamboa D, Ho MF, Bendezu J, Torres K, Chiodini PL, Barnwell JW, et al. A large proportion of P. falciparum isolates in the Amazon region of Peru lack pfhrp2 and pfhrp3: implications for malaria rapid diagnostic tests. PLoS ONE. 2010;5:e8091.
Wellems TE, Howard RJ. Homologous genes encode two distinct histidine-rich proteins in a cloned isolate of Plasmodium falciparum. Proc Natl Acad Sci USA. 1986;83:6065–9.
Cheng Q, Gatton ML, Barnwell J, Chiodini P, McCarthy J, Bell D, et al. Plasmodium falciparum parasites lacking histidine-rich protein 2 and 3: a review and recommendations for accurate reporting. Malar J. 2014;13:283.
Amoah LE, Abankwa J, Oppong A. Plasmodium falciparum histidine rich protein-2 diversity and the implications for PfHRP 2: based malaria rapid diagnostic tests in Ghana. Malar J. 2016;15:101.
Koita OA, Doumbo OK, Ouattara A, Tall LK, Konare A, Diakite M, et al. False-negative rapid diagnostic tests for malaria and deletion of the histidine-rich repeat region of the hrp2 gene. Am J Trop Med Hyg. 2012;86:194–8.
Parr JB, Verity R, Doctor SM, Janko M, Carey-Ewend K, Turman BJ, et al. Pfhrp2-deleted Plasmodium falciparum parasites in the Democratic Republic of the Congo: a national cross-sectional survey. J Infect Dis. 2017;216:36–44.
Wurtz N, Fall B, Bui K, Pascual A, Fall M, Camara C, et al. Pfhrp2 and pfhrp3 polymorphisms in Plasmodium falciparum isolates from Dakar, Senegal: impact on rapid malaria diagnostic tests. Malar J. 2013;12:34.
WHO. False-negative RDT results and implications of new reports of P. falciparum histidine-rich protein. Geneva, World Health Oganization, 2016. www.who.int/malaria.
Watson OJ, Sumner KM, Janko M, Goel V, Winskill P, Slater HC, et al. False-negative malaria rapid diagnostic test results and their impact on community-based malaria surveys in sub-Saharan Africa. BMJ Glob Health. 2019;4:e001582.
Agaba BB, Yeka A, Nsobya S, Arinaitwe E, Nankabirwa J, Opigo J, et al. Systematic review of the status of pfhrp2 and pfhrp3 gene deletion, approaches and methods used for its estimation and reporting in Plasmodium falciparum populations in Africa: review of published studies 2010–2019. Malar J. 2019;18:355.
Berzosa P, Gonzalez V, Taravillo L, Mayor A, Romay-Barja M, Garcia L, et al. First evidence of the deletion in the pfhrp2 and pfhrp3 genes in Plasmodium falciparum from Equatorial Guinea. Malar J. 2020;19:99.
Gupta H, Matambisso G, Galatas B, Cistero P, Nhamussua L, Simone W, et al. Molecular surveillance of pfhrp2 and pfhrp3 deletions in Plasmodium falciparum isolates from Mozambique. Malar J. 2017;16:416.
Watson OJ, Slater HC, Verity R, Parr JB, Mwandagalirwa MK, Tshefu A, et al. Modelling the drivers of the spread of Plasmodium falciparum hrp2 gene deletions in sub-Saharan Africa. Elife. 2017;6:e25008.
WHO. False-negative RDT results and implications of new reports of P. falciparum histidine-rich protein 2/3 gene deletions. Geneva, World Health Oganization. WHO/HTM/GMP/201718. 2017.
Mouatcho JC, Goldring JPD. Malaria rapid diagnostic tests: challenges and prospects. J Med Microbiol. 2013;62:1491–505.
WHO. Protocol for estimating the prevalence of pfhrp2/pfhrp3 gene deletions among symptomatic falciparum patients with false-negative RDT results. Geneva: World Health Oganization; 2018.
Taylor SM, Mayor A, Mombo-Ngoma G, Kenguele HM, Ouedraogo S, Ndam NT, et al. A quality control program within a clinical trial Consortium for PCR protocols to detect Plasmodium species. J Clin Microbiol. 2014;52:2144–9.
Mayor A, Serra-Casas E, Bardaji A, Sanz S, Puyol L, Cistero P, et al. Sub-microscopic infections and long-term recrudescence of Plasmodium falciparum in Mozambican pregnant women. Malar J. 2009;8:9.
Gupta H, Macete E, Bulo H, Salvador C, Warsame M, Carvalho E, et al. Drug-resistant polymorphisms and copy numbers in Plasmodium falciparum, Mozambique, 2015. Emerg Infect Dis. 2018;24:40–8.
Rougemont M, Van Saanen M, Sahli R, Hinrikson HP, Bille J, Jaton K. Detection of four Plasmodium species in blood from humans by 18S rRNA gene subunit-based and species-specific real-time PCR assays. J Clin Microbiol. 2004;42:5636–43.
Grignard L, Nolder D, Sepulveda N, Berhane A, Mihreteab S, Kaaya R, et al. A novel multiplex qPCR assay for detection of Plasmodium falciparum with histidine-rich protein 2 and 3 (pfhrp2 and pfhrp3) deletions in polyclonal infections. EBioMedicine. 2020;55:102757.
Rogerson SJ, Desai M, Mayor A, Sicuri E, Taylor SM, van Eijk AM. Burden, pathology, and costs of malaria in pregnancy: new developments for an old problem. Lancet Infect Dis. 2018;18:e107–18.
Kamau A, Mtanje G, Mataza C, Malla L, Bejon P, Snow RW. The relationship between facility-based malaria test positivity rate and community-based parasite prevalence. PLoS ONE. 2020;15:e0240058.
Mpimbaza A, Sserwanga A, Rutazaana D, Kapisi J, Walemwa R, Suiyanka L, et al. Changing malaria fever test positivity among paediatric admissions to Tororo district hospital, Uganda 2012–2019. Malar J. 2020;19:416.
Acknowledgements
We are indebted to all the study participants the St Joseph’s Catholic Hospital’s staff participants who demonstrated much enthusiasm towards this study, as well as to the project manager Cristina Muñoz.
Funding
This study was conducted thanks to a grant from the European and Developing Countries Clinical Trials Partnership (EDCTP CSA2016ERC-1420). The EDCTP2 programme is supported under Horizon 2020, the European Union’s Framework Programme for Research and Innovation. ISGlobal receives support from the Spanish Ministry of Science and Innovation through the “Centro de Excelencia Severo Ochoa 2019–2023” Program (CEX2018-000806-S), and support from the Generalitat de Catalunya through the CERCA Program. This research is part of ISGlobal’s Program on the Molecular Mechanisms of Malaria, which is partially supported by the Fundación Ramón Areces. Alfredo Mayor is supported by the Department d’Universitats I Recerca de la Generalitat de Catalunya (AGAUR; 2017SGR 664). The funding body had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Contributions
AM and QB designed the study. AM wrote the draft of this manuscript. AG participated in fieldwork, supervising and monitoring the quality lab procedures in Liberia. DPL facilitated the SJCH research team and supported the collection of clinical and epidemiological data and laboratory analyses. MK, SO and CKT organized the recruitment of participants. PC, HC and BA carried out the molecular tests analysis and interpretation of molecular results. AS and QB revised and contributed intellectually the draft preparation for submission. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consents were obtained from all participants if 18 years of age or older. Parental consents in addition to minor assent were obtained from the participants aged younger than 18 years. Participants did not receive any retribution for their engagement as study subjects. Refusal to participate in this study did not affect service provision as per standard health care practice. This research protocol was approved by the local University of Liberia-Pacific Institute Research and Evaluation Institutional Review Board (UL-PIRE, Monrovia, Liberia) and by the Hospital Clínic Health Research Ethics Committee (CEIC, Barcelona, Spain). Study participants were treated following national guidelines.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
King, M., George, A.E., Cisteró, P. et al. No evidence of false-negative Plasmodium falciparum rapid diagnostic results in Monrovia, Liberia. Malar J 20, 238 (2021). https://doi.org/10.1186/s12936-021-03774-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-021-03774-3