
Abstract
Background
Glutamate dehydrogenase of malaria parasites (pGDH) is widely used in rapid diagnostic tests for malaria. Variation in the pGDH gene among Korean isolates of Plasmodium vivax was analysed, and a recombinant pGDH protein was evaluated for use as antigens for the serodiagnosis of vivax malaria.
Methods
Genomic DNA was purified from blood samples of 20 patients and the pGDH gene of P. vivax was sequenced. Recombinant protein was prepared to determine the antigenicity of pGDH by enzyme-linked immunosorbent assay (ELISA).
Results
Partial sequence analysis of the P. vivax pGDH gene from the 20 Korean isolates showed that an open reading frame (ORF) of 1410 nucleotides encoded a deduced protein of 470 amino acids. The amino acid and nucleotide sequences were conserved among all the Korean isolates. This ORF showed 100% homology with P. vivax strain Sal-I (GenBank accession No. XP_001616617.1). The full ORF (amino acids 39–503), excluding the region before the intron, was cloned from isolate P. vivax Bucheon 3 (KJ726751) and subcloned into the expression vector pET28b for transformation into Escherichia coli BL21(DE3)pLysS. The expressed recombinant protein had a molecular mass of approximately 55 kDa and showed 84.8% sensitivity (39/46 cases) and 97.2% specificity (35/36 cases) in an ELISA. The efficacy of recombinant pGDH protein in seroepidemiological studies was also evaluated by ELISA using serum samples collected from 876 inhabitants of Gyodong-myeon, Ganghwa County, Incheon Metropolitan City. Of these samples, 91 (10.39%) showed a positive reaction with recombinant pGDH protein. Among the antibody-positive individuals, 13 (14.29%) had experienced malaria infection during the last 10 years.
Conclusion
The pGDH genes of P. vivax isolates from representative epidemic-prone areas of South Korea are highly conserved. Therefore, pGDH is expected to be a useful antigen in seroepidemiological studies. It was difficult to identify the foci of malaria transmission in Gyodong-myeon based on the patient distribution because of the very low parasitaemia of Korean vivax malaria. However, seroepidemiology with recombinant pGDH protein easily identified regions with the highest incidence of malaria within the study area. Therefore, recombinant pGDH protein may have a useful role in serodiagnosis.
Similar content being viewed by others

Background
Microscopic examination is the gold standard method for diagnosis of malaria. Despite the simplicity and low cost, however, it is not always possible to use this method [1]. During the last 20 years, the development of alternative diagnostic methods for malaria, such as rapid diagnostic tests (RDTs), has made it possible to extend biological diagnosis to remote areas that lack advanced medical services. RDTs are lateral-flow immunochromatographic tests that detect specific malaria antigens. They are rapid and simple enough to carry out without electricity, specific equipment, or intensive training of personnel [2,3,4]. Glutamate dehydrogenase (GDH), an enzyme involved in urea synthesis, catalyzes the reversible oxidative deamination of l-glutamate to form α-ketoglutarate and ammonia, using nicotinamide adenine dinucleotide phosphate (NADP(H)) or nicotinamide adenine dinucleotide (NAD(H)) as cofactor [5]. There are three types of GDH, depending on the cofactor. The enzymes specific for NAD(H) (EC 1.4.1.2) generally catalyze the oxidative deamination of l-glutamate (to generate α-ketoglutarate) and have an alkaline pH optimum, whereas the enzymes specific for NADP(H) (EC 1.4.1.4) usually carry out the reductive amination of α-ketoglutarate (to generate l-glutamate) and have a neutral pH optimum. The third type (EC 1.4.1.3), represented by the vertebrate GDH enzymes, can use both cofactors for the deamination of l-glutamate [6]. Plasmodium falciparum contains three genes encoding potential parasite glutamate dehydrogenase (pGDH) proteins; two are found on chromosome 14 (PF14_0164 and PF14_0286, encoding pGDHa and pGDHb, respectively) and one is present on chromosome 8 (PF08_0132, encoding pGDHc) [7, 8]. pGDHa and pGDHb are NADP(H) dependent, and the primary sequence of pGDHb suggests that the protein is located in the apicoplast, whereas the localization and cofactor specificity of pGDHc cannot be predicted. The presence of multiple pGDH proteins is reminiscent of the situation in plants and fungi [9,10,11,12]. pGDH is considered integral to the parasite’s antioxidant machinery and is thought to be a potential drug target [8, 13,14,15,16]. In recent years, pGDH has been used as an antigen for malaria detection.
In this study, variation of the pGDH genes of P. vivax isolates from 20 patients living in five malaria epidemic-prone areas of South Korea was investigated, and a recombinant protein was evaluated as a serodiagnostic tool.
Methods
Blood sample collection
Patients with clinically suspected malaria who had attended Public Health Centers in Bucheon-si, Gimpo-si, and Paju-si of Gyeonggi Province, Ganghwa County of Incheon Metropolitan City, and Cheorwon County of Gangwon Province, South Korea, were examined for malaria parasites. Blood was collected from symptomatic patients and thin and thick blood smears were prepared for microscopic examination. The usefulness of recombinant pGDH protein in seroepidemiological studies was evaluated using sera collected at Gyondong-myeon, Ganghwa County, Incheon Metropolitan City, in December 2012. The serum samples were stored at −80 °C until use.
Microscopic examination
Thin blood films were prepared to determine the infectivity of blood samples. The blood films were fixed with methanol and stained with Giemsa stain diluted with buffered water (pH 7.2) to emphasize the parasite inclusions in the red blood cells. The densities of blood-stage parasites were estimated by counting the number of asexual parasites relative to 200 white blood cells (WBCs) and then multiplying the parasite:WBC ratio by 8000 (the assumed number of WBCs per microlitre of blood) [3, 17].
Amplification of the pGDH gene
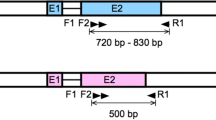
For amplification of the pGDH gene of P. vivax, genomic DNA was extracted from each patient’s whole blood using a QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). The polymerase chain reaction (PCR) was performed using TaKaRa Ex Taq DNA polymerase (Takara, Otsu, Japan), 50 ng of purified genomic DNA, and 40 pmol each of forward (PvGDH-F1: 5′-ATT TTA CCC CTC TCG GCC GTG GCC CTT TTC-3′) and reverse primers (PvGDH-R1: 5′-GGC GCC GTC GCA CTG CTC GTA GAT GCT CCT-3′) for DNA sequence analysis (Figs. 1, 2). The PvGDH-Fex1 primer (5′-AGC CAT ATG CGC GCC AAG GTG CGC GGC GC-3′) with an NdeI restriction enzyme site (underlined) and the PvGDH-Rex1 primer (5′-GTG CTC GAG CAG GCC GCC CTG CTC CAG GA-3) with an XhoI restriction enzyme site (underlined) were used to establish the recombinant protein expression vector. The total PCR volume was adjusted to 50 μL with distilled water. The thermocycling conditions were as follows: denaturation at 94 °C for 5 min; 35 cycles of 30 s at 94 °C, 60 s at 55 °C, and 90 s at 72 °C; and final extension at 72 °C for 5 min. All PCR products were analysed on a 1% agarose gel, confirmed under a UV transilluminator, and purified with a QIAquick Gel Extraction Kit (Qiagen).

Structure of the plasmid (pVpGDH) used for DNA sequence analysis and expression of the Plasmodium vivax parasite glutamate dehydrogenase gene

Multiple amino acid sequence alignment of parasite glutamate dehydrogenases among Plasmodium species. The deduced amino acid sequence of the type strain of Korean P. vivax isolates (Pv Kor, Bucheon strain, GenBank accession No. KJ726751) was aligned with sequences from other Plasmodium species. Computer analysis was performed using the multiple sequences alignment program MegAlign. All amino acid sequences were obtained from GenBank using BLAST (http://www.ncbi.nlm.nih.gov). Py, P. yoelii yoelii strain 17XNL (XM_719434.1); Pk, P. knowlesi strain H (XM_002260680.1); Pf, P. falciparum strain FCC1/HN (AY040586.1); Pch, P. chabaudi chabaudi (XM_738585.1); Pc, P. cynomolgi strain B (XM_004224427.1); Pb, P. berghei strain ANKA (XM_673901.1); Pv Sal-1, P. vivax strain Sal-1 (XP_001616667)
DNA sequencing and analysis
For genotyping of the pGDH gene of P. vivax, the PCR product of the gene was ligated into a pCR2.1 cloning vector (Invitrogen, Carlsbad, CA, USA) and transformed into E. coli TOP10 cells. Cells containing recombinant plasmid were selected on ampicillin-containing medium [18]. The transformants were confirmed by gel electrophoresis of EcoRI-digested plasmid DNA prepared with a plasmid isolation kit (Qiagen) according to the manufacturer’s protocol. The pGDH gene sequence was determined using the ABI PRISM BigDye Terminator Ready Reaction Cycle Sequencing Kit FS (Perkin Elmer, Cambridge, MA, USA) according to the manual supplied by the manufacturer. The nucleotide and deduced amino acid sequences were analysed using EditSeq and Clustal in the MegAlign program, a multiple sequence alignment program in the DNASTAR package (DNASTAR, Madison, WI, USA). The internet-based BLAST program of the National Center for Biotechnology Information was used to search protein databases.
Construction of the pGDH expression vector
For expression of recombinant pGDH in E. coli BL21(DE3)pLysS, the pGDH gene was amplified from a blood sample that was confirmed to be infected with the dormant type of P. vivax. Amplification was performed as described above with PvGDH-Fex1 and PvGDH-Rex1, which contain NdeI and XhoI sites at their 5′ ends, respectively. The amplified PCR product was digested with NdeI and XhoI, purified with a QIAquick Gel Extraction Kit, and integrated into the NdeI and XhoI sites of the pET28b expression vector (Novagen). The resulting plasmid (named pVKtype3) was subsequently used for the expression of a pGDH-(His)6 fusion protein in E. coli. The transformants were confirmed both by gel electrophoresis of the plasmid DNA after digestion with NdeI and XhoI and by DNA sequencing.
Expression and purification of recombinant pGDH
Escherichia coli BL21(DE3)pLysS carrying pVKtype3 was grown to the logarithmic phase in Luria broth containing 50 μg/mL kanamycin, and then expression of the recombinant protein was induced by adding 1 mM isopropyl-1-thio-β-d-galactopyranoside (IPTG) to the culture. The pGDH-(His)6 fusion protein was purified by affinity chromatography [19]. The purification was performed under native conditions according to the supplier’s protocol (Novagen). After each purification step, the protein was analysed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) with Coomassie brilliant blue staining.
Western blot analysis
The recombinant pGDH-(His)6 fusion protein was separated on a 12% SDS-PAGE gel and then transferred to a nitrocellulose membrane. After the transfer, the membrane was cut into appropriately sized strips and blocked with 5% skim milk in phosphate-buffered saline (PBS) for 12 h at 4 °C. The membrane was then washed three times for 10 min each with 0.15% Tween 20-PBS. The strips were allowed to react with serum from a patient with malaria or an uninfected patient (diluted 1:100, v/v) for 4 h and then washed as described above. The membrane was subsequently incubated with diluted horseradish peroxidase-conjugated goat anti-human IgG secondary antibody (1:1000, v/v; Sigma) for 3 h at room temperature. For color development, a solution containing 0.2% diaminobenzidine and 0.02% H2O2-PBS was applied to each well [20, 21].
Enzyme-linked immunosorbent assay
Enzyme-linked immunosorbent assays (ELISAs) were used to determine whether the blood samples contained antibodies against pGDH antigens of P. vivax. Briefly, capture antigen solution (50 μL, 0.5 μg/mL) with recombinant pGDH was placed in a 96-well plate (Corning, Lowell, MA, USA) and incubated for 12 h at room temperature (RT). After aspiration of the antigen solution, the wells were filled with blocking buffer (1% bovine serum albumin, 0.05% Tween 20-PBS) and the plates were incubated for 1 h at RT. The wells were washed three times with 0.05% Tween 20-PBS, and then human serum samples diluted in blocking buffer (1:100, v/v) were added to the wells. Four positive and four negative control serum samples were also added to each plate. After 2 h of incubation at RT, the plates were washed three times with 0.05% Tween 20-PBS, and then horseradish peroxidase-conjugated anti-human IgG (1:2000, v/v; Sigma) diluted in blocking buffer was added. The plates were incubated again for 1 h at RT. The reaction was stopped by washing the plates as described above. The color was developed by adding 100 µL of the peroxidase substrate 2,2′-azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (Kirkegaard & Perry Laboratories, Gaithersburg, MD, USA) and incubating the plates for 30 min. Optical density was measured at 450 nm, and the cutoff value for positivity was defined as the mean + 2 standard deviations of the negative control samples.
Calculation of annual parasite incidence
The annual parasite incidence (API) was calculated as the number of malaria-positive patients per 1000 inhabitants for each study area, that is, API = (number of microscopically proven malaria cases/population of administrative area) × 1000.
Data analysis
Data analyses were performed using GraphPad software (GraphPad Software Inc., La Jolla, CA, USA). A two-tailed t test was performed to examine the significance of differences between the malaria patient group and the normal group in the ELISA test. One-way ANOVA followed by a Kruskal–Wallis test was performed to examine the significance of differences between parasitaemia and optical density of ELISA. Pearson’s correlation analysis was performed to examine the relationship between positive rate and API in 2012 and 2013. A P value of <0.05 was considered statistically significant.
Results
Sequence variation of Plasmodium vivax pGDH genes from Korean isolates
The geographical locations of blood sample collection were Ganghwa County (37.31N, 125.33E) of Incheon Metropolitan City; Gimpo-si (37.33N, 126.48E), Bucheon-si (37.29N, 126.46E), and Paju-si (37.88N, 126.76E) of Gyeonggi Province; and Cheorwon County (38.10N, 127.30E) of Gangwon Province. Four blood samples from patients infected with indigenous P. vivax were collected from each location during 2010–2011. Amplification of the pGDH gene from blood genomic DNA yielded a product of approximately 1400 bp. After purification, the amplified gene fragment was ligated into the pCR2.1 cloning vector (3.9 kb). The plasmid containing the PCR product was named pVpGDH (5.3 kb) and was used for DNA sequence analysis (Fig. 1). Based on the DNA sequencing results, the cloned pGDH gene was 1410 bp (excluding the start codon, ATG, and the intron) and encoded 470 amino acids, which were deduced using DNASIS.
The amino acid and nucleotide sequences were identical to all the Korean isolates (KJ726749–KJ726768). Among the isolates, Pv Kor (Pvk), isolated from Bucheon-si, was designated as the type strain on the basis of pGDH gene sequence analysis. The open reading frame showed 100.0% homology with P. vivax strain Sal-I (GenBank accession No. XM_001616617.1), 83.0% homology with Plasmodium cynomolgi strain B (PCYB_132800, XM_004224305.1), 78.6% homology with Plasmodium knowlesi strain H (PKH_131950, XM_002260679.1), 66.6% homology with Plasmodium chabaudi chabaudi (XM_738585.1), 64.5% homology with Plasmodium berghei strain ANKA (XM_673901.1), 58.5% homology with Plasmodium falciparum (AF269241.1), and 56.2% homology with Plasmodium yoelii yoelii strain 17XNL (XM_719434.1) (Figs. 2, 3).

a Phylogenetic relationships among parasite glutamate dehydrogenases of several species of Plasmodium and b percent identity. The deduced amino acid sequence of the type strain of Korean P. vivax isolates (Pvk, Bucheon strain, GenBank accession No. KJ726751) was aligned with sequences from other Plasmodium species. Computer analysis was performed using the multiple sequences alignment tool of MegAlign. All amino acid sequences were obtained from GenBank using BLAST (http://www.ncbi.nlm.nih.gov). Pb, P. berghei strain ANKA (XM_673901.1); Pc1, P. cynomologi strain B (PCYB_132800, XM_004224305.1); Pc2, P. cynomolgi strain B (PCYB_134020, XM_004224427.1); Pch, P. chabaudi chabaudi (XM_738585.1); Pf1, P. falciparum (AF269241.1); Pf2, P. falciparum strain FCC1/HN (AY040586.1); Pk1, P. knowlesi strain H (PKH_131960, XM_002260680.1); Pk2, P. knowlesi strain H (PKH_131950, XM_002260679.1); Pv Sal-1, P. vivax strain Sal-1 (XM_001616617.1); Py, P. yoelii yoelii strain 17XNL (XM_719434.1)
Expression of pGDH in Escherichia coli
For generation of the expression plasmid, the P. vivax pGDH gene sequence from after the intron to before the stop codon was amplified from genomic DNA isolated from blood of an infected patient. The amplified DNA was digested with NdeI and XhoI and subcloned into the same restriction sites of the pET28b expression vector. The resulting plasmid, pVKtype3, was used to express pGDH fused to a (His)6-tag (Fig. 4a). The recombinant plasmid was transformed into E. coli BL21(DE3)pLysS and protein expression was induced with IPTG. As evident on the SDS-PAGE gel, the recombinant pGDH protein had a molecular mass of about 55 kDa under native purification conditions (Fig. 4b).

a Conformation of the cloned pET28b vector containing the Plasmodium vivax parasite glutamate dehydrogenase (pGDH) gene (pVKtype3). M, molecular size marker; lane 1 undigested plasmid; lane 2 NdeI-digested plasmid; lane 3 NdeI- and XhoI-digested plasmid. b Expression of the pGDH gene in Escherichia coli BL21(DE3)pLysS. Lane M molecular weight protein marker; lane 1, Non-induction clone, lanes 2–4 induction clones of recombinant pGDH protein. c Purification of the pGDH protein from Escherichia coli BL21(DE3)pLysS. Lane M molecular weight protein marker; lane 1 purified recombinant pGDH protein
Antigenicity of the recombinant pGDH protein
The antigenicity of the purified recombinant pGDH protein was assessed by western blot analysis and ELISA. The tested samples contained malaria parasites, as confirmed by microscopic examination, but the parasites had not been counted. Negative sera were collected from volunteer staff from the Korea National Research Institute of Health. All sera of P. vivax-infected patients (n = X) exhibited a positive reaction with pGDH by western blotting, whereas sera from the normal control group (n = 5), who had never been exposed to malaria, tested negative. In addition, pGDH did not react with the sera of P. falciparum-infected patients (n = 11) (Fig. 5). The antigenicity of the recombinant pGDH protein was then evaluated by ELISA using a larger number of samples. Thirty-nine of the 46 microscopically confirmed malaria-positive serum samples reacted with the recombinant pGDH protein (sensitivity of 84.8%). Only 1 of the 36 samples from the normal control group reacted with the recombinant protein (specificity of 97.2%, Fig. 6a). Positive and negative sera could be differentiated from 1:80 (v/v) serum dilution (Fig. 6b). Parasitaemia decreased the intensity of the reaction, but the difference was not significant (Fig. 7, P = 0.6309).

Western blot analysis of recombinant Plasmodium vivax parasite glutamate dehydrogenase. a Pv patients (Plasmodium vivax-infected patients). b Normal, healthy volunteers. c Pf patients (Plasmodium falciparum-infected patients). M molecular weight marker

Enzyme-linked immunosorbent assay of immune responses of the vivax malaria patient group and normal individuals to recombinant parasite glutamate dehydrogenase (a). Twofold serum dilution of malaria patients (P1–P5) and normal person (N1–N4) (b)

Relationship between parasitaemia and optical density of enzyme-linked immunosorbent assay
Usefulness of recombinant pGDH protein in seroepidemiology
The usefulness of the purified recombinant pGDH protein was evaluated by ELISA with the serum samples collected from Gyodong-myeon, Ganghwa County, Incheon Metropolitan City, in 2012. A total of 876 serum samples (28.49%) were collected from 3074 inhabitants of 13 villages in Gyodong-myeon (Fig. 8). Ninety-one samples (10.39%) were positive in the ELISA. Nanjeong-ri showed the highest positive rate (16.81%, 19/113), followed by Jiseok-ri (16.39%, 10/61). The lowest positive rate was found in Seohan-ri (2.60%, 2/77) (Fig. 8; Table 1). Among the antibody-positive individuals, 13 (14.29%) had experienced malaria infection during the last 10 years, while 78 (85.71%) had newly acquired antibodies against pGDH. Among the 876 inhabitants, 49 (5.59%) had experienced malaria infection. Nine malaria cases were reported in 2012 and four were reported in 2013; therefore, the API was reduced from 2.93 in 2012 to 1.30 in 2013. Jiseok-ri showed the highest API in 2012 (9.66), in accordance with its high positive rate (16.39%), but the API decreased to 0 in 2013. Nanjeong-ri showed the next highest API in 2012 (7.58), in agreement with its high positive rate (16.81%) in comparison with other villages. Only two villages, Najeong-ri and Samseon-ri, had the API in both years, while the APIs of the other villages were up and down between years. The relationship between the pGDH-positive rate and API was stronger in 2013 than in 2012, although the difference was not significant (Table 1).

Seroepidemiological study areas of Gyodong-myeon, Ganghwa County, Incheon Metropolitan City. A Bongso-ri; B Gogu-ri; C Insa-ri; D Jiseok-ri; E Muhak-ri; F Nanjeong-ri; G Seohan-ri; H Dongsan-ri; I Yanggap-ri; J Samseon-ri; K Daeryong-ri; L Eumnae-ri; M Sangyong-ri. DMZ demilitarized zone; DPRK Democratic People’s Republic of Korea; ROK Republic of Korea
Discussion
pGDH is a target malaria antigen that is widely used to develop monoclonal antibodies for non-microscopic immunochromatographic assays (i.e., RDTs). However, little is known about the ability to detect antibodies to pGDH in serum of patients with malaria. Therefore, the sequence variation of pGDH and its antigenicity was investigated among Korean isolates.
Plasmodium vivax has been prevalent in Korea for a long time. However, as the result of a national malaria eradication programme and with help from the World Health Organization, the incidence of vivax malaria rapidly decreased during the 1960s and 1970s [22,23,24,25]. Following the report of two cases of malaria in 1985 [25], there were no additional reported cases in Korea until the emergence of one case in 1993 [26] and two cases in 1994 [27]. The incidence of malaria then increased rapidly until approximately 2000 [28]. After that, reported cases of malaria declined again for several years, owing to nationwide efforts to reduce the incidence of this disease. However, malaria has not been eradicated from the Korean peninsula because many travellers and workers come from areas where malaria is prevalent, including North Korea [29]. For these reasons, serological diagnostic tools are needed to support both traditional microscopic diagnosis and antibody testing on a population level to gain an estimate of exposure to malaria in Korea. Currently, the immunofluorescence antibody test (IFAT) is used as the standard serological diagnostic method owing to its high sensitivity. However, the sensitivity of this test can be affected by the training and ability of users [30,31,32]. Therefore, a new antigen is needed for serodiagnosis. Several recombinant proteins cloned from Korean isolates of P. vivax have been tested for use as antigens for serodiagnosis, including Circumsporozoite protein (CSP) subtypes Pv210 [18] and Pv247 [33], merozoite surface protein (MSP) [34], CSP and MSP chimeric proteins [35], aldolase [36], and parasite lactate dehydrogenase [37]. None of these antigens has enabled replacement of the IFAT method because of their comparatively low sensitivity. Therefore, there has been a focus on pGDH. Monoclonal antibodies against pGDH have been used in several RDTs and exhibit a relatively high sensitivity for detection of malaria parasites. However, in the present study the ELISA detected only 84.8% (39/46) of microscopy-positive samples, even though the pGDH gene was cloned from a Korean P. vivax strain (pVKtype3, Fig. 2). Therefore, antibody detection using the recombinant pGDH protein is not superior to antigen detection using monoclonal antibodies against pGDH. Nevertheless, it was worth investigating whether the recombinant pGDH protein could be used to detect antibodies in asymptomatic patients or symptomatic patients with low parasitaemia (under 50/µL), using antibody detection methods such as ELISA or western blotting. The recombinant pGDH protein was evaluated for its usefulness in seroepidemiology using sera collected from Gyodong-myeon, Ganghwa County, Incheon Metropolitan City, in 2012. This study area is an island consisting of several hills surrounded by rice fields and two large irrigation reservoirs. Serum samples from 28.74% of the inhabitants of this island (876 cases) were tested by ELISA with recombinant pGDH protein as antigen. The villages with a positive rate above 10% are adjacent to the main rice field located in the center of the island and the two irrigation reservoirs, which are potential habitats for anopheline mosquitoes. In contrast, villages with a low positive rate are located in coastal areas and are separated by a hill from the main rice field and the two irrigation reservoirs. In conclusion, the results of this study indicate that antibody detection using recombinant pGDH may provide useful information regarding the prevalence of malaria in certain areas and individuals.
Conclusion
The findings of the present study suggest that seroepidemiological studies with recombinant pGDH protein, which displayed no amino acid sequence variation among 20 investigated Korean isolates, may be useful for understanding the epidemiology of malaria in Korea.
References
Reyburn H, Mbatia R, Drakeley C, Carneiro I, Mwakasungula E, Mwerinde O, et al. Overdiagnosis of malaria in patients with severe febrile illness in Tanzania: a prospective study. BMJ. 2004;329:1212.
Wongsrichanalai C. Rapid diagnostic techniques for malaria control. Trends Parasitol. 2001;17:307–9.
Moody A. Rapid diagnostic tests for malaria parasites. Clin Microbiol Rev. 2002;15:66–78.
Bell D, Peeling RW. Evaluation of rapid diagnostic tests: malaria. Nat Rev Microbiol. 2006;4:S34–8.
Storm J, Perner J, Aparicio I, Patzewitz EM, Olszewski K, Llinas M, et al. Plasmodium falciparum glutamate dehydrogenase is dispensable and not a drug target during erythrocytic development. Malar J. 2011;10:193.
Hudson RC, Daniel RM. l-Glutamate dehydrogenases: distribution, properties and mechanism. Comp Biochem Physiol B. 1993;106:767–92.
Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511.
Aparicio IM, Marin-Menendez A, Bell A, Engel PC. Susceptibility of Plasmodium falciparum to glutamate dehydrogenase inhibitors-A possible new antimalarial target. Mol Biochem Parasitol. 2010;172:152–5.
Avendano A, Deluna A, Olivera H, Valenzuela L, Gonzalez A. GDH3 encodes a glutamate dehydrogenase isozyme, a previously unrecognized route for glutamate biosynthesis in Saccharomyces cerevisiae. J Bacteriol. 1997;179:5594–7.
DeLuna A, Avendano A, Riego L, Gonzalez A. NADP-glutamate dehydrogenase isoenzymes of Saccharomyces cerevisiae. Purification, kinetic properties, and physiological roles. J Biol Chem. 2001;276:43775–83.
Miyashita Y, Good AG. Glutamate deamination by glutamate dehydrogenase plays a central role in amino acid catabolism in plants. Plant Signal Behav. 2008;3:842–3.
Miyashita Y, Good AG. NAD(H)-dependent glutamate dehydrogenase is essential for the survival of Arabidopsis thaliana during dark-induced carbon starvation. J Exp Bot. 2008;59:667–80.
Wagner JT, Ludemann H, Farber PM, Lottspeich F, Krauth-Siegel RL. Glutamate dehydrogenase, the marker protein of Plasmodium falciparum—cloning, expression and characterization of the malarial enzyme. Eur J Biochem. 1998;258:813–9.
Werner C, Stubbs MT, Krauth-Siegel RL, Klebe G. The crystal structure of Plasmodium falciparum glutamate dehydrogenase, a putative target for novel antimalarial drugs. J Mol Biol. 2005;349:597–607.
Vander Jagt DL, Hunsaker LA, Kibirige M, Campos NM. NADPH production by the malarial parasite Plasmodium falciparum. Blood. 1989;74:471–4.
Walter RD, Nordmeyer JP, Konigk E. NADP-specific glutamate dehydrogenase from Plasmodium chabaudi. Hoppe Seylers Z Physiol Chem. 1974;355:495–500.
McKenzie FE, Prudhomme WA, Magill AJ, Forney JR, Permpanich B, Lucas C, et al. White blood cell counts and malaria. J Infect Dis. 2005;192:323–30.
Lee HW, Lee WJ, Lee JS, Lee HS. DNA sequencing and expression of the Circumsporozoite protein of Plasmodium vivax Korean isolate in Escherichia coli. Korean J Microbiol. 1999;37:234–42.
Lim KJ, Park JW, Sohn MJ, Lee S, Oh JH, Kim HC, et al. A direct sandwich ELISA to detect antibodies against the C-terminal region of merozoite surface protein 1 could be a useful diagnostic method to identify Plasmodium vivax exposed persons. Parasitol Res. 2002;88:855–60.
Tsang VCW, Peralta JM, Simons AR. Enzyme-linked immunoelectrotransfer blot techniques (EITB) for studying the specificities of antigens and antibodies separated by gel electrophoresis. Methods Enzymol. 1983;92:377–91.
Gao YH, Li HL, Lu Y, Gao FM, Lin YH, Zhou HC, et al. Identification of a vaccine candidate antigen, PfMAg-1, from Plasmodium falciparum with monoclonal antibody M26-32. Parasitol Res. 2009;105:1723–32.
Hasegawa. Malaria in Korea. Chosen Igakkai Zasshi. 1913;4:53–69.
National Malaria Eradication Service, Ministry of Health and Social Affairs, ROK. Malaria pre-eradication programme in Korea, 1961–1965. Progress report 1966, p. 44–70.
Paik YH, Rhee HI, Shim JC. Malaria in Korea. Jpn J Exp Med. 1988;58:55–66.
Soh CT, Lee KT, Im KI, Min DY, Ahn MH, Kim JJ, et al. Current status of malaria in Korea. Yonsei Rep Trop Med. 1985;16:11–8.
Chai IH, Lim GI, Yoon SN, Oh WI, Kim SJ, Chai JY. Occurrence of tertian malaria in a male patient who has never been abroad. Korean J Parasitol. 1994;32:195–200.
Cho SY, Kong Y, Park SM, Lee JS, Lim YA, Chae SL, et al. Two vivax malaria cases detected in Korea. Korean J Parasitol. 1994;32:281–4.
Lee JS, Kho WG, Lee HW, Seo M, Lee WJ. Current status of vivax malaria among civilians in Korea. Korean J Parasitol. 1998;36:241–8.
Kim TS, Kim JS, Na BK, Lee WJ, Kim HC, Youn SK, et al. Decreasing incidence of Plasmodium vivax in the Republic of Korea during 2010–2012. Malar J. 2013;12:309.
Lee WJ, Kim HH, Hwang SM, Park MY, Kim NR, Cho SH, et al. Detection of an antibody against Plasmodium vivax in residents of Gimpo-si, South Korea, using an indirect fluorescent antibody test. Malar J. 2011;10:19.
Collins EC, Skinner JC. The indirect fluorescent antibody test for malaria. Am J Trop Med Hyg. 1972;121:690–5.
Wang DQ, Tang LH, Gu ZC, Zheng X, Yang MN. Application of the indirect fluorescent antibody assay in the study of malaria infection in the Yangtze River Three Gorges Reservoir, China. Malar J. 2009;8:199.
Kim TS, Kim HH, Lee SS, Oh CM, Choi KM, Lin K, et al. Molecular cloning and expression of the VK247 Circumsporozoite protein for serodiagnosis of variant form Plasmodium vivax. Parasitol Res. 2011;108:1275–82.
Kim TS, Sohn Y, Kim JY, Lee WJ, Na BK, Kang YJ, et al. Detection of antibodies against the CB9 to ICB10 region of merozoite surface protein-1 of Plasmodium vivax among the inhabitants in epidemic areas. Malar J. 2014;13:311.
Lee C, Chung KW, Kim TS, Choi KM, Choi YK, Chung NJ, et al. Trials for the co-expression of the merozoite surface protein-1 and Circumsporozoite protein genes of Plasmodium vivax. Exp Parasitol. 2011;129:227–33.
Kim JY, Kim HH, Shin HL, Sohn Y, Kim H, Lee SW, et al. Genetic variation of aldolase from Korean isolates of Plasmodium vivax and its usefulness in serodiagnosis. Malar J. 2012;11:159.
Shin HI, Kim JY, Lee WJ, Sohn Y, Lee SW, Kang YJ, et al. Polymorphism of the parasite lactate dehydrogenase gene from Plasmodium vivax Korean isolates. Malar J. 2013;12:166.
Authors’ contributions
HWL, YJK and TSK conceived and designed the study and contributed to the execution of the research; HWL wrote the manuscript; HIS, JHP, BYJ and JYK analysed the DNA sequence and expressed the recombinant pGDH protein; and BS performed ELISA and western blot analysis of samples from patients and inhabitants. All authors read and approved the final manuscript.
Acknowledgements
We are grateful to all blood donors and the staff of the Public Health Centers in Ganghwa County, Gimpo-si, Paju-si, Yeoncheon County, and Cheorwon County.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets supporting the conclusions of this paper are included within the paper. Raw data may be obtained from the corresponding author on request.
Consent for publication
This study reports no individual data. Data reported in this study are solely based on Plasmodium vivax genetics and bears no individual participant information.
Ethics approval and consent to participate
Informed consent was obtained from all participants according to the ethical standards established by the Human Ethics Committee of Inha University (Incheon, Korea). The study was conducted according to the principles expressed in the Declaration of Helsinki, and the study procedures, potential risks, and benefits were explained to all participants. Investigators were blinded during data analysis, and all patient names were excluded.
Funding
This work was supported by an Inha University research fund.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Bomin Seol and Hyun-Il Shin contributed equally as first author.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Seol, B., Shin, HI., Kim, JY. et al. Sequence conservation of Plasmodium vivax glutamate dehydrogenase among Korean isolates and its application in seroepidemiology. Malar J 16, 3 (2017). https://doi.org/10.1186/s12936-016-1653-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-016-1653-3