
Abstract
More than 85% of the malaria burden in the Yunnan Province is caused by imported vivax malaria, and Yunnan is also where the majority of vivax malaria patients are diagnosed in China. Timely removal of the infection sources of Plasmodium vivax and its breeding environment remains the key to eliminating the secondary transmission of imported malaria. To that end, blood samples were collected from cases diagnosed and revalidated as single species infection with P. vivax in the Yunnan Province from 2013 to 2020. Specifically, samples from vivax malaria patients with suspected relapses episodes were subjected to PCR amplification, product sequencing, and analysis of the P. vivax circumsporozoite protein (pvcsp) gene. In total, 77 suspected relapse patients were identified out of 2484 cases infected with P. vivax, with a total of 81 recurrent episodes. A total of 156 CDS (coding DNA sequence) chains were obtained through PCR amplification and sequencing of the pvcsp gene from 159 blood samples, 121 of which can be matched to the paired sequences of 59 vivax malaria patients with both primary attack and recurrent experience. Of the 59 pairs of pvcsp gene sequences, every one of 31 pairs showed only one haplotype and no variant sites (VS), meaning every two paired sequence was completely homologous. Every one of the remaining 28 paired sequences had two haplotypes but no length polymorphism, indicating that the paired sequences was “weakly heterologous” with no fragment insertions (or deletions). All 59 vivax malaria patients with recurrences were caused by the activation of P. vivax hypnozoites originated from the same population as the primary infection. The paired analysis of the similarity between high variant genes allowed the identification of relapse episodes caused by P. vivax homologous hypnozoites and also demonstrated pvcsp gene as one of the candidate molecular markers for tracing infection origin.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In recent years, as efforts to control malaria have increased, the proportion of P. vivax infections in many traditionally highly endemic areas, such as Sri Lanka, Thailand, and Brazil, has shown a counter-intuitive increasing trend. The cause may be related to the fact that previous prevention and control measures ignored the characteristics of P. vivax and indirectly contributed to the accumulation of P. vivax infection sources (Battle et al. 2015). It has been observed that mature gametophytes (stage V gametophytes) of P. vivax appear almost simultaneously with its asexual bodies in the early stages and continue to produce gametophytes throughout the infection period (Bockarie et al. 2006; Ghanchi et al. 2019). The high efficiency of transmission therefore also means that the most effective measures to remove the infection sources of Plasmodium falciparum, such as early diagnosis and timely treatment, may not be able to contain the danger of an early episode of P. vivax infection. In addition, P. vivax develops more rapidly inside Anopheles mosquitoes and can easily circumvent these interventions such as insecticide netting and indoor residual spraying. Thus, the maintenance of P. vivax populations is easier than those applicable to P. falciparum using the same vector control procedure (Golding et al. 2015). Moreover, due to the existence of a parasitic mechanism of hypnozoites, P. vivax can break through the local and seasonal limits of mosquito vector transmission (White et al. 2012; Battle et al. 2014). The accumulation of infection sources plays a negative role in increasing the complexity associated with P. vivax transmission.
The last indigenous malaria cases infected with P. vivax in China ceased in 2016. However, Yunnan remains the province with the largest number of imported vivax malaria cases, with instances primarily originating in Southeast Asian countries. To wit, in 2018, such cases accounted for 80.8% (172/213) of malaria cases caused by P. vivax infection throughout the province (Li et al. 2021), of which there was no shortage of groups with multiple episodes and suspected relapse of P. vivax infection (Huang et al. 2021). During the period spanning 2013–2019, suspected relapse events constituted approximately 3.5% (83 in total) of the 2364 vivax malaria patients diagnosed in the Yunnan Province. This was based on a minimum interval of 45 days between the first recurrence and the original episode (Llanos-Cuentas et al. 2014). Comparing other malaria endemic areas, this interval has an average duration of 41 days across those Southeast Asian countries where P. vivax is widely prevalent (Battle et al. 2014) and 7% of vivax malaria relapse within 6 months in Thailand (Llanos-Cuentas et al. 2014), suggesting the burden of vivax malaria relapse in Yunnan Province is no less than that of neighboring countries. Of course, other regions of the world are also facing the challenge posed by vivax malaria relapse, with the relapse rate in Papua New Guinea being as high as 80% (Betuela et al. 2012). Twenty-three percent of pregnant women in Brazil had a relapse episode due to failure to receive primaquine radical treatment (Corder et al. 2020); overcoming this negative result has become an unavoidable source of difficulty in the process towards the global elimination of malaria (Baird 2016; White et al. 2016). One of the three major technical bottlenecks in the control of vivax malaria is accurate identification of relapse infection caused by the activation of P. vivax hypnozoites (Baird 2016).
The WHO recommends analyzing the homology of single-copy antigenic genes of P. vivax as a method for the molecular identification of “new infection” and “recrudescence” events (Koepfli et al. 2009). Lin et al. (2015) identified 37.9% (11/29) of re-emergence vivax malaria episodes caused by the activation of P. vivax homologous hypnozoites based on analysis of the genetic similarity between paired strains from initial infection and the re-emergent strains. Craig et al. (1996), Imwong et al. (2012), and Chen et al. (2007) also assessed the differentiation characteristics of pvama1 (P. vivax apical membrane antigen 1), pvcsp, pvmsp1 (P. vivax merozoite surface protein 1), and other genes, which revealed the first vivax malaria relapse was mostly caused by the activation of P. vivax homologous hypnozoites. The feasibility of molecular identification of relapse events in P. vivax infection has been demonstrated from different perspectives. To establish a practical and reasonable method for evaluating the relapse episode of P. vivax infection, this study used nest polymerase chain reaction (PCR) for amplification of hypervariable genes (White et al. 2016), followed by DNA sequencing of PCR amplification product to identify vivax malaria recurrent events in the Yunnan Province. The results of the molecular characterization of the pvcsp gene and its marker role in identification of the relapse strains of P. vivax are reported below.
Materials and methods
Study subjects and blood samples
The study was conducted on cases from throughout Yunnan Province from January 2013 to December 2020 that were confirmed at the Yunnan Province Malaria Diagnosis Referent Laboratory (YPMDRL) using both examination by light microscopy of blood smears and genetic testing (SI 1) (Dong et al. 2020). All cases were officially registered and counted in the “China Disease Surveillance Information Reporting System,” from which suspected relapse individuals infected with P. vivax and with a history of re-emergence were further screened. The infection source of P. vivax was confirmed as indigenous or imported by epidemiological surveys at each outbreak of vivax malaria cases. The criteria are as follows: those with no history of movement outside Yunnan Province within 30 days prior to the onset of P. vivax infection were classified as indigenous cases in Yunnan Province, while those with a history of exposure to vivax malaria endemic areas such as Myanmar and Africa were classified as patients who imported Myanmar and African infections (SI 2). The following inclusion criteria were applied for the vivax malaria relapse: (1) patients who were clinically cured after an 8-day course of chloroquine/primaquine (in total 1550-mg chloroquine therapy within 3 days, followed by the subsequent 8-day course of 22.5-mg/day primaquine therapy) and had a second episode of P. vivax infection in the peripheral blood 28 days later; (2) no further history of exposure to malaria endemic areas was verified by epidemiological survey after the original episode. The data of recurrence from every vivax malaria patient are shown in SI 2.
Venous blood was collected from all vivax malaria patients during the primary infection and before treatment for recurrent episodes (day 0). The samples were prepared as filter paper dry blood drops for Plasmodium genetic analysis.
Extraction of Plasmodium genomic DNA and PCR amplification of the pvcsp gene
Three filter paper dry blood drops with a diameter of 5 mm were used to extract Plasmodium genomic DNA. This was carried out in accordance with the instructions of the QIAgen Mini Kit (QIAamp, Germany), and the samples were subsequently stored at − 20 °C.
In the current study, PCR amplification products were sequenced to obtain the DNA sequence of the whole pvcsp gene, and the nested-PCR method, having relatively good sensitivity and specificity, was adopted in order to stably amplify the target gene from the extracted Plasmodium genome samples for each storage period. The reference sequence (ID: NC_009913.1) was used as the template to design the primers and reaction conditions for the nested PCR amplification of the pvcsp gene. The first round of PCR amplified the region of 1,537,513–1,539,033 that contains approximately 1521 base pairs (bp), with the upstream and downstream primers as 5′-CCGTTCGAACAAGTTCTGTTC-3′ and 5′-GCGCATAATGTGTAAGAGGTGT-3′, respectively. The second round amplified a region of 1341 or so bp from 1,537,625–1,538,965, for which the upstream and downstream primers were 5′-GCTTAAG TTAAGCAAGCAAAACAGC-3′ and 5′-GCAGGGAACATTCATAAGAAAGGG-3′, respectively. The system for both PCR reactions was the following: 2.6 μl of DNA template; 14 μl of PCR mixed with 2 × Taq; and 0.7 μl of each of the upstream and downstream primers (10 umol/L), then ddH2O was added to reach a volume of 25 μl. It was conducted under the following sets of PCR reaction conditions: 94 °C for 3 m; 94 °C for 30 s, 49 °C for 90 s, 72 °C for 2 m, 35 cycles; or 72 °C for 7 m. The amplified products after second round PCR were carried out using 1.5% agarose gel electrophoresis for result observation, then the amplified products were sent to Shanghai Meiji Biomedical Technology Co. Ltd. for Sanger sequencing.
Gene polymorphism and homology analysis of pvcsp genes in paired blood samples
The sequencing results were collated in DNASTAR 11.0 and BioEdit 7.2.5. The obtained DNA sequences were compared with the reference sequences (XM_001616843.1) of the pvcsp gene. When the “query cover” and “identifies” were greater than 98%, it indicated that the sequences collated were the intended targets. DnaSP 6.11.01 software was used to confirm the locus mutations and haplotypes of the pvcsp gene, from which the expected heterozygosity (He) and nucleic acid diversity index (π) of the DNA sequences were calculated. The pvcsp gene sequences of paired primary and relapse isolates from the same individual patients with several episodes of P. vivax infection were identified (Craig et al. 1996; Chen et al. 2007) separately using MEGA 5.04 software for similarity matching and central repetitive region (CRR) analysis (Imwong et al. 2005; Coppi et al. 2011; Dias et al. 2013). These amino acid residue regions including N-terminal, KLKQP five amino acids, and the C-terminal deduced from the pvcsp gene sequence were named α-N, region I (RI), and thrombospondin repeat (TSR), respectively (Coppi et al. 2011). When the main peptide repeat motifs (PRMs) of the CRR were “GDRA[D/A]GQPA” and “ANGAGNQPG,” they were identified as VK210 and VK247 type sequences, respectively (Rosenberg et al. 1989; Imwong et al. 2005; Dias et al. 2013), while “APGANQ[E/G]GAA” was confirmed as the PRM of the P. simiovale-type sequences (Qari et al. 1993).
Confirmation of the paired P. vivax strains and the nature of the recurrent episode
When the CRR repeat motifs deduced from two DNA sequence in each paired samples collected from the same vivax malaria patient showed the following characteristics, the P. vivax strains from both the original infection and the recurrent episodes were considered to be inoculated by the same mosquito, and the subsequent vivax malaria attacks in this patient were considered to be caused by the activation of P. vivax hypnozoites in this population. Then this kind of recurrent episodes were termed as relapse episodes (Imwong et al. 2007): (1) only one haplotype was shown, and both the expected heterozygosity (He) and quantity of VS were equal to 0. This indicated the P. vivax strains from paired samples were completely homologous single-clone strains (Khusmith et al. 1998; Chen et al. 2007; Koepfli et al. 2009; Imwong et al. 2012); (2) the number of haplotypes was greater than one, and both He and VS greater than 0. VS was confirmed by checking peak plots (SI 3), from which it was seen that the paired DNA sequences were of the same length and, with no fragment deletions (or insertions), suggesting the paired P. vivax strains were sibling strains and that pvcsp genes were weakly heterologous but did not undergo intra-helical recombination events (Brito et al. 2011; Dias et al. 2013).
Results
Sample information and PCR amplification product sequencing
In total, 77 reports (3.1%) with recurrent episodes were retrieved from 2484 cases infected with P. vivax, drawn from the period between 2013–2019, all of which patients were living in Yunnan Province (97°31′ E to 106°11′ E; 21°8′ N to 29°15′ N). The majority of patients could be traced to origins in Myanmar (98.7%, 76/77), and the male-to-female ratio was 5 males per 1 female for all patients in the study. The majority of patients had a single relapse (97.4%, 75/77), while one patient had episodes twice, and another patient had three episodes. General demographic information and original place of infection for the 77 patients are shown in Table 1 (SI 2).
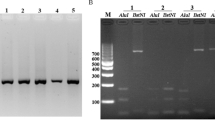
A total of 81 recurrent episodes occurred among these 77 patients infected with P. vivax, allowing a total of 159 blood samples to be obtained from all reported original infection and recurrent episodes. From these, 156 PCR amplification products of the pvcsp gene were successfully obtained, which showed strong signal target bands longer than 1000 bp and shorter than 1500 bp in the electrophoregram (SI 4), with a product acquisition rate of 98.1% (156/159). Of them, paired CDS full strands (807–1179 bp in length) of the pvcsp gene were obtained from blood samples in 75 patients (97.4%, 75/77), but only those from 59 patients could be used for homology analysis of the gene sequences (Fig. 1).

Paired pvcsp gene sequencing results obtained from relapse patients infected with P. vivax, in Yunnan Province from 2013 to 2020
The structural regions of the amino acid chains derived from the CDS strand conversions were completed, including the conserved region near the 5′-end (coding 1st–90th aa of PvCSP) (α-N region), the R I (KLKQP) encoding region, near the 3′-end (coding 276th–393th aa of PvCSP) (TSR) and the highly variable region in pvcsp middle for encoding CRR of PvCSP (96th–275th aa), etc. (SI 5).
Diversity of the pvcsp gene and the CRR array of PvCSP
The 121 CDS strands (GenBank ID: OP354287–OP354399) of pvcsp gene obtained from paired blood samples of 59 patients showed 32 double alleles at positions 112, 113, 233, 234, 240, 261, 264, 270, 274, 282, 295, 309, 327, 347, 354, 426, 491, 507, 511, 534, 545, 552, 572, 579, 615, 684, 742, 761, 769, 805, 892, and 999 (bimodal chart), with a nucleotide diversity index (π) equal to 0.1384 (± 0.0056). The sequences from the original infection and recurrent strains both call only one of the biallelic bases, usually the type with a strong sequencing signal (SI 3). The 32 double alleles were distributed in all seats of the pvcsp gene but were predominantly concentrated in the CRR region (62.5%, 20/32) (Table 2).
Furthermore, 56.3% (18/32) of the polymorphic sites belonged to the third base of the amino acid codon, and only 27.8% (5/18) of these resulted in amino acid variants. The proportion of the second base and first base were 17.6% (6/34) and 26.5% (9/34), respectively (Table 2), while the highest frequency of the double allele was 0.165 for c.234, the minor allele frequency (MAF) was 0.1488 for c.240, and 75.0% (24/32) of the double alleles were present in only one set of paired sequences (Table 2).
In addition to the aforementioned, 121 CDS strands were defined as 84 haplotypes (H01 to H84) with a He of 0.9940 (± 0.0040). Of these, haplotypes H05, H50, H51, H63, and H64 had CRR repeat units (PRMs) of the VK247 genotype (Fig. 2B), while the remaining 79 had the PRMs characteristic of the VK210 genotype (Fig. 2A).

CRR repeats of PvCSP deduced from each haplotype of the pvcsp gene in P. vivax strains (A VK210; B VK247) made in Excel. (1) *: including H09, H10, H25, H42, H43, H13, H35, and H69. (2) #: Including H31, H32, H33, H34, and H15. (3) &: Including H01, H12, H14, H27, H58, and H59. (4) @: Including H03, H18, H61, H62, and H24. %: Including H24, H60, H38, and H39
Among the haplotypes of the VK210 type, there were 39 CRR forms consisting of peptide repeat motifs (PRMs) (Fig. 2A). Of these, there were 15 PRM unit types, with GDRAAGQPA and GDRADGQPA occurring most often, with frequencies of 0.470 (987/2100) and 0.3833 (805/2100), respectively. The remaining 13 PRMs, included the five newly detected PRMs GNRANGQPA (0.0033, 7/2100), GNRANGQAA (0.0001, 1/2100), GDRADGQTA (0.0001, 1/2100), GDRADGHPA (0.0001, 1/2100), and GNGAAGQPA (0.0001, 1/2100) (Fig. 2A). Generally, the CRRs of VK210 type consisted of 14–20 PRMs with 18 being the most common, and 96.8% (38/39) ended with GNGAGGQAA units (Fig. 2A). The CRRs of the paired sample from imported patient 24 infected in Africa (SI 1) were defined as Hap-23 of VK210 type, which did not show any divergent whit Myanmar strains (Fig. 2A).
Of the five haplotypes of type VK247, three types of CRR consisted of 17–21 PRMs (Fig. 2B) in which there were eight unit types of PRMs. Those with the highest frequency of occurrence were ANGAGNQPG (0.7414, 86/116), ANGAGGQAA (0.0517, 6/116), and ANGDDQPGA (0.0172, 2/116), and the remaining two were newly detected PRMs (Fig. 2B).
Comparison of paired blood samples of the pvcsp gene and confirmation of relapse episodes
The results of alignment of the paired pvcsp gene CDS chains of the 59 paired blood samples showed that every paired CDS chains of 31 groups (52.5%, 31/59) had only one haplotype and no VS, and the He and VS values were both 0. This indicated that each of the 31 pairs was homologous and the source of the paired P. vivax strains was a single clone with complete genetic homology, belonging to the same mosquito bite–inoculated population (Table 3). Subsequent episodes of P. vivax infection were caused by the activation of hypnozoites from the same population as primary infection strains. Every paired CDS chains from the other 28 paired blood samples (47.5%) had varying numbers of polymorphic sites (1–6 loci) between each pair. However, there were two exceptions, at c.39 (0.0082, 1/121) and c.1027 (0.0082, 1/121), which were true base substitutions (Table 3, SI 3), while the remaining sites were all double allelic bases (Table 2). These 28 paired sequences did not show the trace of DNA fragment insertion (or deletion), suggesting they were heterologous with few base substitutions, but did not experience intra-helical recombination events. Also, their heterogeneity resulted from sibling strains that were not completely homologous genetically and belonged to the latter generations bred from the common ancestor inoculated at one time by mosquito bite (Table 3). Subsequent recurrent episodes of P. vivax infection were still caused by the activation of hypnozoites in the same population as primary infection strains.
However, similarity matching of two randomly sequences from the 121 CDS strands showed there were significantly more base substitutions between the two sequences. Furthermore, length polymorphism presented between the two unpaired CDS strands due to the presence of different length oligonucleotide strand insertions (or deletions) (Table 3). This indicated that the two sequences of the unpaired samples were more heterologous and their corresponding genomic donors, P. vivax, were not more likely to belong to the same population inoculated by the same mosquito (Table 3).
Discussion
The single-copy pvcsp gene commences at the tip of P. vivax chromosome 8 (Brito et al. 2011; Dias et al. 2013; NCBI 2022), extends for 807–1179 bp, and does not possess introns but has a CDS that encodes polypeptides between 269 and 393 aa long. The structural diversity of the pvcsp gene is concentrated in the mid-segment encoding CRR repeat region from the 90th to 275th aa and its flanks (Fig. 1) (Imwong et al. 2005; Dias et al. 2013; Võ et al. 2020). It is generally characterized by insertions and deletions of repetitive units, mostly due to sexual recombination during meiosis or intra-helical strand-slippage events during mitotic DNA replication (McConkey et al. 1990; Kim et al. 2006; Bousema et al. 2011), as well as frequent base substitutions within the locus. Using the repeat units of the CRR as markers, P. vivax strains can be defined as three genotypes: VK210; VK247; and the P. simiovale, which is P. vivax-like (Qari et al. 1993; Tripathi et al. 2011). Therefore, pvcsp gene is commonly used as a molecular marker for the evolution of population genetic differentiation of P. vivax (di Giovanni et al. 1990; Brito et al. 2011; Kosaisavee et al. 2011; Etemadi et al. 2020).
This study provides preliminary pathogenic evidence on vivax malaria relapse episodes based on the analysis of pvcsp gene differentiation in the sample group which occurred in Yunnan Province. Although the P. vivax population included in this study was not truly a natural population, 97.4% of the strains in the sample were nonetheless harvested from patients infected in Myanmar. Consequently, the pvcsp genes of the entire sample group showed similar findings to those reported by Thanapongpichat et al. (2013) and Võ et al. (2020), which related to the Myanmarese population. These included the CRR region, the same significant polymorphism in length and composition of PRMs, the same predominance of VK210 types (95.0%, 115/121), and the absence of P. vivax-like variants. Furthermore, there was the same extremely low DNA sequence homology, with up to 84 haplotypes in 121 pvcsp gene sequences, though the expected heterozygosity (He, 0.9940 ± 0.0040) was greater than that observed by Võ et al. (He, 0.096 ± 0.034) (2020). However, at the same time, the RI of the pvcsp gene in this set of samples had only one type of arrangement of KLKOP, far fewer than the seven types described by Võ et al. (2020). In addition to this, there was a reduction in variety of PRMs units constituting the CRR region (VK210: 15, VK247: 8) and less complex arrangements and length polymorphisms than those reported in the aforementioned research. These may be justified by this study having involved a relatively homogeneous population of primarily Myanmarese strains of P. vivax, unlike the wider range and a more complex population composition of samples taken by Võ et al. However, the displayed high variability of the pvcsp gene was largely consistent with the results obtained in the current study. Furthermore, it is agreed that the CRR is suitable as a molecular marker for separating genetic differences in P. vivax populations due to its poor conservation, whereas the α-N and TSR regions on both flanks are suitable as candidate antigen genes for polyclonal antibody serum preparation because of their highly conserved sequences (Coppi et al. 2011; Dias et al. 2013).
In addition to confirming the high degree of polymorphism in the pvcsp gene structure at the population level, the similarity comparison performed with paired blood samples from the vivax malaria recurrent patients (n = 59) showed that the P. vivax strains with nearly identical gene sequences were always present rather than an accidence. In 52.5% (31/59) of the patients, the paired pvcsp gene sequences of P. vivax strains from the original infection and relapse were not only highly consistent in length but also completely similar in sequence structure, with both He and VS equal to 0, indicating complete homology of the paired sequences. Even among the 28 sets of paired sequences with double allelic expression (Tables 2 and 3), the pvcsp gene sequences were not only highly concordant in length. Conversely, there were notable oligonucleotide fragment insertions (or deletions) in any two of the pvcsp gene sequences from non-paired blood samples and there was also a significant increase in the number of base substitutions (Table 3). It is suggested that the length polymorphism in the pvcsp gene may be an important point of differentiation between populations of P. vivax, compared to the degree of single nucleotide polymorphism.
Besides the possible causes of reinfection and recrudescence (De Niz et al. 2018; Brito et al. 2022),
it is necessary to validate whether the activation of the intrahepatic P. vivax hypnozoites result in the vivax malaria frequent recurrence after antimalaria treatment (Looareesuwan et al. 1987; Imwong et al. 2007; Orjuela-Sánchez et al. 2009). In the 77 vivax malaria patients with recurrent episodes included in this study, history of exposure in malaria endemic areas outside the country occurred before the original infection episode only. Therefore, the possibility of recurrence caused by reinfection is basically excluded. The molecular identification of relapse in only 59 (76.6%, 59/77) patients was conducted by alignment of paired pvcsp gene sequences. In 31 of these patients, including those with two or three relapses, the paired pvcsp gene sequences of P. vivax strains from the original infection and the relapse episode were identical in length and had no VS within the sequences. This observation suggested that single clone strains of a genetically identical P. vivax existed in every paired blood samples, and the vivax malaria relapse could be attributed to the activation of intrahepatic hypnozoites with identical gene structure to the original infection strain (Lin et al. 2015; Craig et al. 1996; Chen et al. 2007; Imwong et al. 2007; Kirchgatter et al. 1998). In the other 28 patients, there was still a large degree of reproducibility in gene structure between the paired pvcsp gene sequences, which were identical in length, and the single nucleotide polymorphic sites were also mostly double alleles (Tables 2 and 3). Such an appearance of locus polymorphism may have been due to differential calling of double allelic bases in paired sequences by first generation sequencing (Orjuela-Sánchez et al. 2009), suggesting the sibling strains (Brito et al. 2011; Dias et al. 2013; Bright et al. 2014) with identical genetic structure existed in every group of paired blood samples, and the two batches of P. vivax populations from primary infection and relapse actually reflect parallel homology of polyclonal strains. Therefore, the authors suggested that the relapse of these 28 patients infected with P. vivax still belonged to the activation event of intrahepatic hypnozoites coming from common ancestor strains inoculated by a single mosquito bite occurrence (Conway et al. 1991; Sutton et al. 2009; Lin et al. 2015; Bright et al. 2014). It is worth examining whether the polymorphism form resulting from the differential mobilization of the double alleles is equivalent to the previously mentioned “weakly heterologous” genes (Chan et al. 2012; Nair et al. 2014; Friedrich et al. 2016; Popovici et al. 2018).
In summary, this paper analyzes the possibility that relapse episodes caused by homologous hypnozoites could be identified by finding the strong similarity between highly variable genes in P. vivax. Thus, methodological implications are provided for the identification of relapse episodes in malaria-free transmission areas, although there are some shortcomings. First, the study may still be incomplete in excluding recrudescence events caused by chloroquine-resistant strains or continued proliferation of myelo-plasmodium (Obaldia et al. 2018; Silva-Filho et al. 2020) due to the lack of feasible experimental verification means, although the chloroquine resistance associated molecular marker has been detected in some P. vivax strains from individual paired samples (SI 6). Secondly, the relapse event was confirmed by the similarity of P. vivax genes between the paired samples from the original infection and the reactivation. It included two results of 100% homology and “weakly heterologous” with only a few base substitutions between paired gene sequences, and it remains to be confirmed whether or not the latter was part of the activation of heterologous hypnozoites populations that scholars have previously studied. Thirdly, no definitive confirmation could be provided concerning the nature of the paired P. vivax strains in 23.3% (18/77) of patients. Although the authors are continuing to mitigate the limitations of single genetic markers to identify homologous strains, as well as in other investigations of these relapse causes, the length of this article does not allow for further elaboration, which may somewhat reduce the apparent completeness of this paper.
Conclusion
In conclusion, this study identified vivax malaria relapse episodes caused by homologous hypnozoites using deep sequencing and analyzing the similarities of pvcsp genes in paired P. vivax strains from primary attack and relapse. The pvcsp gene is suitable as a candidate molecular marker to identify P. vivax homologous strains due to being simple in structure and easy to find in the traces of recombined gene. This study also demonstrated that most of vivax malaria relapse patients reported in Yunnan Province resulted from the single clone strains or sibling strains homologous with the original P. vivax infection. It is suggested that alignment of the gene similarity between paired P. vivax strains is suitable for identifying homologous hypnozoites activation.
Data availability
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request. Nucleotide sequence data reported in this paper are available in GenBank under accession numbers OP354287 to OP354399.
Abbreviations
- pvcsp :
-
Plasmodium vivax Circumsporozoite protein gene
- P. vivax:
-
Plasmodium vivax
- P. falciparum:
-
Plasmodium falciparum
- CDS:
-
coding DNA sequence
- pvama1:
-
P. vivax apical membrane antigen 1
- pvmsp1:
-
P. vivax merozoite surface protein 1
- PCR:
-
polymerase chain reaction
- YPMDRL:
-
Yunnan Province Malaria Diagnosis Referent Laboratory
- bp:
-
base pairs
- He:
-
expected heterozygosity
- π:
-
nucleic acid diversity index
- PvCSP:
-
P. vivax circumsporozoite protein
- CRR:
-
central repetitive region
- PRMs:
-
peptide repeat motifs
- α-N:
-
N-terminal of PvCSP
- RI:
-
region I, codons region of KLKQP five amino acids
- TSR:
-
thrombospondin repeat
- VS:
-
variable sites
- MAF:
-
minor allele frequency
- Com-Homo:
-
completely Homologous
- non-re-Sibl:
-
non-recombinated sibling
- dif-Popu:
-
different population
- pvcrt-o:
-
Plasmodium vivax chloroquine resistance protein ortholog
References
Baird JK (2010) Eliminating malaria—all of them. Lancet 376(9756):1883–1885. https://doi.org/10.1016/s0140-6736(10)61494-8
Baird JK (2016) Attacking Plasmodium vivax. Am J Trop Med Hyg 95(6 Suppl):1–3. https://doi.org/10.4269/ajtmh.16-0517
Battle KE et al (2014) Geographical variation in Plasmodium vivax relapse. Malar J 13(1):144. https://doi.org/10.1186/1475-2875-13-144
Battle KE et al (2015) Global database of matched Plasmodium falciparum and P. vivax incidence and prevalence records from 1985–2013. Sci Data 2:150012. https://doi.org/10.1038/sdata.2015.12
Betuela I et al (2012) Relapses contribute significantly to the risk of Plasmodium vivax infection and disease in Papua New Guinean children 1–5 years of age. J Infect Dis 206(11):1771–1780. https://doi.org/10.1093/infdis/jis580
Bockarie MJ et al (2006) Are insecticide-treated bednets more protective against Plasmodium falciparum than Plasmodium vivax-infected mosquitoes? Malar J 5(1):15. https://doi.org/10.1186/1475-2875-5-15
Bousema T et al (2011) Adjusting for heterogeneity of malaria transmission in longitudinal studies. J Infect Dis 204(1):1–3. https://doi.org/10.1093/infdis/jir225
Bright AT et al (2014) A high resolution case study of a patient with recurrent Plasmodium vivax infections shows that relapses were caused by meiotic siblings. PLoS Negl Trop 8(6):e2882. https://doi.org/10.1371/journal.pntd.0002882
Brito CF et al (2011) Molecular markers and genetic diversity of Plasmodium vivax. Mem Inst Oswaldo Cruz 106(Suppl 1):12–26. https://doi.org/10.1590/s0074-02762011000900003
Brito MAM et al (2022) Morphological and transcriptional changes in human bone marrow during natural Plasmodium vivax malaria infections. J Infect Dis 225(7):1274–1283. https://doi.org/10.1093/infdis/jiaa177
Chan ER et al (2012) Whole genome sequencing of field isolates provides robust characterization of genetic diversity in Plasmodium vivax. PLoS Negl Trop Dis 6(9):e1811. https://doi.org/10.1371/journal.pntd.0001811
Chen N et al (2007) Relapses of Plasmodium vivax infection result from clonal hypnozoites activated at predetermined intervals. J Infect Dis 195(7):934–941. https://doi.org/10.1086/512242
Conway DJ et al (1991) The epidemiology of multiple-clone Plasmodium falciparum infections in Gambian patients. Parasitology 103(Pt 1):1–6. https://doi.org/10.1017/s0031182000059217
Coppi A et al (2011) The malaria circumsporozoite protein has two functional domains, each with distinct roles as sporozoites journey from mosquito to mammalian host. J Exp Med 208(2):341–356. https://doi.org/10.1084/jem.20101488
Corder RM et al (2020) Quantifying and preventing Plasmodium vivax recurrences in primaquine-untreated pregnant women: an observational and modeling study in Brazil. PLoS Negl Trop Dis 14(7):e0008526. https://doi.org/10.1371/journal.pntd.0008526
Craig AA et al (1996) Molecular analysis of strains of Plasmodium vivax from paired primary and relapse infections. J Infect Dis 174(2):373–379. https://doi.org/10.1093/infdis/174.2.373
De Niz M et al (2018) Plasmodium gametocytes display homing and vascular transmigration in the host bone marrow. Sci Adv 4(5):eaat3775. https://doi.org/10.1126/sciadv.aat3775
di Giovanni L et al (1990) On the evolutionary history of the circumsporozoite protein in plasmodia. Exp Parasitol 70(4):373–381. https://doi.org/10.1016/0014-4894(90)90120-2
Dias S et al (2013) Population genetic structure of the Plasmodium vivax circumsporozoite protein (Pvcsp) in Sri Lanka. Gene 518(2):381–387. https://doi.org/10.1016/j.gene.2013.01.003
Dong Y et al (2020) Analysis of initial laboratory diagnosis of malaria and its accuracy compared with re-testing from 2013 to 2018 in Yunnan Province. China Malar J 19(1):409. https://doi.org/10.1186/s12936-020-03477-1
Etemadi S et al (2020) Genotyping and phylogenetic analysis of Plasmodium vivax circumsporozoite protein (PvCSP) gene of clinical isolates in South-Eastern Iran. Iran J Public Health 49(5):981–988
Friedrich LR et al (2016) Complexity of infection and genetic diversity in Cambodian Plasmodium vivax. PLoS Negl Trop Dis 10(3):e0004526. https://doi.org/10.1371/journal.pntd.0004526
Ghanchi NK et al (2019) Hematological profile and gametocyte carriage in malaria patients from Southern Pakistan. Cureus 11(3):e4256. https://doi.org/10.7759/cureus.4256
Golding N et al (2015) Integrating vector control across diseases. BMC Med 13:249. https://doi.org/10.1186/s12916-015-0491-4
Huang H et al (2021) The association of CYP2D6 gene polymorphisms in the full-length coding region with higher recurrence rate of vivax malaria in Yunnan Province. China Malar J 20(1):160. https://doi.org/10.1186/s12936-021-03685-3
Imwong M et al (2005) Practical PCR genotyping protocols for Plasmodium vivax using Pvcs and Pvmsp1. Malar J 4(1):20. https://doi.org/10.1186/1475-2875-4-20
Imwong M et al (2007) Relapses of Plasmodium vivax infection usually result from activation of heterologous hypnozoites. J Infect Dis 195(7):927–933. https://doi.org/10.1086/512241
Imwong M et al (2012) The first Plasmodium vivax relapses of life are usually genetically homologous. J Infect Dis 205(4):680–683. https://doi.org/10.1093/infdis/jir806
Khusmith S et al (1998) Antigenic disparity of Plasmodium vivax causing initial symptoms and causing relapse. Southeast Asian J Trop Med Public Health 29(3):519–524
Kim JR et al (2006) Genetic diversity of Plasmodium vivax in Kolkata. India Malar J 5(1):71. https://doi.org/10.1186/1475-2875-5-71
Kirchgatter K et al (1998) Molecular analysis of Plasmodium vivax relapses using the MSP1 molecule as a genetic marker. J Infect Dis 177(2):511–515. https://doi.org/10.1086/517389
Koepfli C et al (2009) Evaluation of Plasmodium vivax genotyping markers for molecular monitoring in clinical trials. J Infect Dis 199(7):1074–1080. https://doi.org/10.1086/597303
Kosaisavee V et al (2011) The genetic polymorphism of Plasmodium vivax genes in endemic regions of Thailand. Asian Pac J Trop Med 4(12):931–936. https://doi.org/10.1016/S1995-7645(11)60221-6
Li XH et al (2021) Seven decades towards malaria elimination in Yunnan. China Malar J 20(1):147. https://doi.org/10.1186/s12936-021-03672-8
Lin JT et al (2015) Using amplicon deep sequencing to detect genetic signatures of Plasmodium vivax relapse. J Infect Dis 212(6):999–1008. https://doi.org/10.1093/infdis/jiv142
Llanos-Cuentas A et al (2014) Tafenoquine plus chloroquine for the treatment and relapse prevention of Plasmodium vivax malaria (DETECTIVE): a multicentre, double-blind, randomised, phase 2b dose-selection study. Lancet 383(9922):1049–1058. https://doi.org/10.1016/s0140-6736(13)62568-4
Looareesuwan S et al (1987) High rate of Plasmodium vivax relapse following treatment of falciparum malaria in Thailand. Lancet 2(8567):1052–1055. https://doi.org/10.1016/s0140-6736(87)91479-6
McConkey GA et al (1990) The generation of genetic diversity in malaria parasites. Annu Rev Microbiol 44:479–498. https://doi.org/10.1146/annurev.mi.44.100190.002403
Nair S et al (2014) Single-cell genomics for dissection of complex malaria infections. Genome Res 24(6):1028–1038. https://doi.org/10.1101/gr.168286.113
NCBI (2022) https://www.ncbi.nlm.nih.gov/genome/gdv/browser/gene/?id=5472322
Obaldia N et al (2018) Bone marrow is a major parasite reservoir in Plasmodium vivax infection. mBio 9(3):e00625-18. https://doi.org/10.1128/mBio.00625-18
Orjuela-Sánchez P et al (2009) Recurrent parasitemias and population dynamics of Plasmodium vivax polymorphisms in rural Amazonia. Am J Trop Med Hyg 81(6):961–968. https://doi.org/10.4269/ajtmh.2009.09-0337
Popovici J et al (2018) Genomic analyses reveal the common occurrence and complexity of Plasmodium vivax relapses in Cambodia. mBio 9(1):e01888-17. https://doi.org/10.1128/mBio.01888-17
Qari SH et al (1993) Identification of Plasmodium vivax-like human malaria parasite. Lancet 341(8848):780–783. https://doi.org/10.1016/0140-6736(93)90559-y
Rosenberg R et al (1989) Circumsporozoite protein heterogeneity in the human malaria parasite Plasmodium vivax. Science 245(4921):973–976. https://doi.org/10.1126/science.2672336
Silva-Filho JL et al (2020) Plasmodium vivax in hematopoietic niches: hidden and dangerous. Trends Parasitol 36(5):447–458. https://doi.org/10.1016/j.pt.2020.03.002
Sutton PL et al (2009) Plasmodium falciparum and Plasmodium vivax infections in the Peruvian Amazon: propagation of complex, multiple allele-type infections without super-infection. Am J Trop Med Hyg 81(6):950–960. https://doi.org/10.4269/ajtmh.2009.09-0132
Thanapongpichat S et al (2013) Microsatellite genotyping of Plasmodium vivax infections and their relapses in pregnant and non-pregnant patients on the Thai-Myanmar border. Malar J 12(1):275. https://doi.org/10.1186/1475-2875-12-275
Tripathi V et al (2011) Evolutionary analysis of circumsporozoite surface protein and merozoite surface protein-1 (CSP and MSP-1) sequences of malaria parasites. Bioinformation 6(8):320–323. https://doi.org/10.6026/97320630006320
Võ TC et al (2020) Genetic polymorphism and natural selection of circumsporozoite protein in Myanmar Plasmodium vivax. Malar J 19(1):303. https://doi.org/10.1186/s12936-020-03366-7
White NJ et al (2012) Relapse. Adv Parasitol 80:113–150. https://doi.org/10.1016/B978-0-12-397900-1.00002-5
White MT et al (2016) Variation in relapse frequency and the transmission potential of Plasmodium vivax malaria. Proc Biol Sci 283(1827):20160048. https://doi.org/10.1098/rspb.2016.0048
Acknowledgements
Our sincere gratitude for the provided initial diagnosis information on malaria patients and their blood samples goes to the Centers for Disease Control and Prevention of each prefecture and county: Dehong, Baoshan, Kunming, Pu’er, Lincang, Dali, Nujiang, Lijiang, Xishuangbanna, Yuxi, Chuxiong, Honghe, Zhaotong, Diqing, Qujing, and Whenshan.
Funding
The current study was supported by the National Science Foundation, China (Nos. 81660559, 82160637).
Author information
Authors and Affiliations
Contributions
YX was responsible for the study design, carried out genetic testing, and wrote the contents of microscopy examination in the manuscript; YD was responsible for the study design and coordinated the project and statistical analysis, wrote the manuscript, and edited of the text including an English check; Yan Deng, HH, MC, YL, JW, CZ, and ZW performed the collection of blood samples and microscopy examination. All authors read and approved the final version.
Corresponding authors
Ethics declarations
Ethics approval
The design and protocol of the study was approved by the Yunnan Institute of Parasitic Diseases and Ethical Committee (Ethical Approval No.: 2019 Yunnan Ethics Auditing No. 5). The purpose of the study was explained to the subjects or their relatives, and genetic testing was performed on stored blood samples obtained as part of the routine diagnostic workup of patients with fever who were suspected to have malaria. The patients were fully informed on the aims of the study and signed an informed consent agreement after confirmation Plasmodium infection.
Consent for publication
All authors provided their consent for the publication of this report.
Competing interests
The authors declare no competing interests.
Additional information
Handling Editor: Una Ryan
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, Y., Dong, Y., Deng, Y. et al. Molecular identification of vivax malaria relapse patients in the Yunnan Province based on homology analysis of the Plasmodium vivax circumsporozoite protein gene. Parasitol Res 122, 85–96 (2023). https://doi.org/10.1007/s00436-022-07700-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00436-022-07700-7