
Abstract
Background
A three-day course of chloroquine remains a standard treatment of Plasmodium vivax infection in Thailand with satisfactory clinical efficacy and tolerability although a continuous decline in in vitro parasite sensitivity has been reported. Information on the pharmacokinetics of chloroquine and its active metabolite desethylchloroquine are required for optimization of treatment to attain therapeutic exposure and thus prevent drug resistance development.
Methods
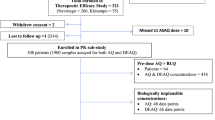
The study was conducted at Mae Tao Clinic for migrant worker, Tak province, Thailand. Blood samples were collected from a total of 75 (8 Thais and 67 Burmeses; 36 males and 39 females; aged 17–52 years) patients with mono-infection with P. vivax malaria [median (95 % CI) admission parasitaemia 4898 (1206–29,480)/µL] following treatment with a three-day course of chloroquine (25 mg/kg body weight chloroquine phosphate over 3 days). Whole blood concentrations of chloroquine and desethylchloroquine were measured using high performance liquid chromatography with UV detection. Concentration–time profiles of both compounds were analysed using a population-based pharmacokinetic approach.
Results
All patients showed satisfactory response to standard treatment with a three-day course of chloroquine with 100 % cure rate within the follow-up period of 42 days. Neither recurrence of P. vivax parasitaemia nor appearance of P. falciparum occurred. A total of 1045 observations from 75 participants were included in the pharmacokinetic analysis. Chloroquine disposition was most adequately described by the two-compartment model with one transit compartment absorption model into the central compartment and a first-order transformation of chloroquine into desethylchloroquine with an additional peripheral compartment added to desethylchloroquine. First-order elimination from the central compartment of chloroquine and desethylchloroquine was assumed. The model exhibited a strong predictive ability and the pharmacokinetic parameters were estimated with adequate precision.
Conclusion
The developed population-based pharmacokinetic model could be applied for future prediction of optimal dosage regimen of chloroquine in patients with P. vivax infection.
Similar content being viewed by others

Background
Malaria remains one of the major global public health problems in the tropics and subtropics including Southeast Asia. The most recent World Malaria Report revealed an estimated 3.3 billion people at risk, 198 million estimated cases, and 584,000 deaths, of which 90 % occurred in Africa [1]. Apart from drug resistance in Plasmodium falciparum, the “sleeping giant” in the Greater Mekong subregion is P. vivax malaria, which has now become resistant to the blood schizontocide chloroquine in some of the Southeast Asian countries, notably Indonesia [2, 3]. Chloroquine resistance is linked to increasing rates of anaemia and may be an important factor in severe P. vivax malaria [3]. The burden of P. vivax varies widely with the World Health Organization (WHO) estimating it being responsible for approximately 12–22 million cases worldwide annually [1]. The disease is rarely life-threatening, but morbidity from a prolonged illness and the possibility of relapses from a persistent hepatic form (hypnozoite) which occurs more frequently with the tropical form of P. vivax found in Southeast Asian countries, is of major concern and cause considerable economic loss.
In Thailand, chloroquine and the tissue schizontocide primaquine have remained the mainstay treatment of P. vivax infection for more than 60 years with a conserved clinical efficacy of virtually 100 % [4–7]. To date, there has been no clinico-parasitological evidence of chloroquine resistant P. vivax in Thailand, although a trend in gradual decline of in vitro sensitivity to the drug has been documented in some areas of the country, particularly along the Thai-Myanmar border [8, 9]. It is possible that resistant levels may remain obviously below the threshold of detectability by the in vivo assessment. The accumulating reports of chloroquine resistant P. vivax in other parts of the world during the past three decades particularly in Southeast Asian region such as Indonesia [10], Papua New Guinea [11–14], Irian Jaya [15–18], Myanmar [19–21] and Vietnam [22], emphasize the need for closely and continuously monitoring clinical efficacy in conjunction with in vitro sensitivity with confirmed adequacy of anti-malarial systemic drug exposure [23]. The information obtained would facilitate the early recognition of treatment failures and adjustment of treatment policy. Optimization of chloroquine treatment is essential to attain therapeutic exposure and thus prevent resistance development to the drug. Inadequate drug exposure may lead to subtherapeutic concentrations of chloroquine and an increased risk of severe vivax malaria as well as the development of resistant strains of P. vivax. The aim of the study was to investigate the pharmacokinetics of chloroquine and its active metabolite desethylchloroquine following treatment with a three-day standard course of chloroquine in patients with P. vivax infection on the Thai-Myanmar border.
Methods
Patients and study design
The study was conducted at Mae Tao clinic for migrant workers, Tak Province, Thailand. Prior to study, approval of the study protocol was obtained from the Ethics Committee of the Ministry of Public Health of Thailand. The study was part of the clinical study conducted during 2010–2011 to monitor the clinical efficacy and in vitro sensitivity of P. vivax isolates to chloroquine in an area along the Thai-Myanmar border [7]. Written informed consents were obtained from all patients before study participation. A total of 75 (8 Thai and 67 Burmese; 36 males and 39 females; aged 17–52 years) patients with P. vivax mono-infection [median (95 % CI) admission parasitaemia 4898 (1206–29,480)/µL] were included in the study [7]. In brief, patients were treated with the standard three-day chloroquine (Government Pharmaceutical Organization of Thailand, 250 mg chloroquine phosphate per tablet) regimen given at a total dose of 25 mg base/kg body weight over 3 days (10 and 5 mg/kg at 0 and 6–12 h on day 0, and 5 mg/kg each on day 1 and day 2) and primaquine (Government Pharmaceutical Organization of Thailand, 15 mg base per tablet) given at daily doses of 15 mg base/kg body weight daily for 14 days starting from the second day (day 1) of chloroquine treatment. Chloroquine and primaquine dose administration during the first 3 days (days 0, 1 and 2) were administered with a glass of 250 mL drinking water under the supervision of a medical staff. Patients were closely observed for at least 30 min after drug ingestion.
All patients were admitted to the clinic during the course of treatment or until signs and symptoms of malaria disappeared. Prior to treatment, a blood sample (5 mL) was collected from each patient for in vitro sensitivity testing of P. vivax isolates to chloroquine and determination of baseline anti-malarial drug concentrations (chloroquine and its active plasma metabolite desethylchloroquine). Patients were requested to return for follow-up on days 7, 14, 21, 28, 35 and 42, or at any time if fever or symptoms suggestive of malaria developed. At each visit, a parasite count was performed (Giemsa stain), and a detailed questionnaire for general symptoms was recorded. Malaria blood smears were obtained on enrollment and thereafter, twice daily until two consecutive slides were confirmed to be negative, as well as at every follow-up visit. Thick films were screened for 200 oil-immersion fields before declaring a slide negative. Asexual parasites and gametocytes were separately counted against 200 white blood cells (WBCs); if the parasite density was too numerous to count on the thick film, the number of parasites per 2000 red blood cells (RBCs) on the thin film were counted. Clinical efficacy of the three-day course of chloroquine was evaluated in the group of patients who completed the 42-day follow-up period. The classification of the therapeutic outcome was according to the WHO protocol [23].
Blood sampling and drug analysis
Blood samples were collected at specified time points, i.e., pre-dose and at 1, 6, 12, 24, 25, 36, 48 and 49 h after the first dose for measurement of chloroquine and desethylchloroquine concentrations. Blood samples were also collected during the follow-up period at day 7, 14, 21, 28, 35 and 42 after the initiation of the treatment. Concentrations of chloroquine and desethylchloroquine in plasma samples were measured using high performance liquid chromatography according to the method of Cheomung and colleagues [24]. The lower limit of quantification (LOQ) of the assay was 2 ng/mL for both chloroquine and desethylchloroquine. The assay accuracy (expressed as % relative error: % RE) of the quality control samples used during the sample analysis for both chloroquine and desethylchloroquine ranged from 0.25 to 5.7 %. The assay precision (expressed as coefficient of variation: %CV) were less than 5 % for both chloroquine and desethylchloroquine.
Population pharmacokinetics
Modelling and data handling
Concentration–time data of chloroquine and desethylchloroquine, transformed into their natural logarithms, were analysed using the mixed-effects modelling in NONMEM® (version 7.12; ICOM Development Solutions, Ellicot City, MD, USA) and the output results and graphical plots were handled using the statistical analysis programs R (version 2.15.1; Free Software Foundation, Boston, MA, USA) and R-package Xpose (version 4.3.5; Uppsala University, Uppsala, Sweden). The observations that were below the limit of quantification were excluded from the pharmacokinetic analysis. The first-order conditional estimation (FOCE) method was used throughout the modeling. Model evaluation was based on visual inspection of diagnostic plots, precision of parameters and the objective function value (OFV; proportional to −2 Log likelihood) [25]. For nested models, the difference in OFV is approximately Chi squared distributed and it can therefore be used in model discrimination.
For a one parameter difference between models, 3.84 correspond to a p value of 0.05. Population pharmacokinetic models were constructed to evaluate the concentration–time data for chloroquine and desethylchloroquine and to identify any covariates that could describe between subject variability (BSV). A metabolite model was implemented to describe the pharmacokinetics of chloroquine and desethylchloroquine. One, two-and three compartment models were initially investigated both for the parent drug and metabolite. Different models for the elimination of chloroquine and its metabolite were evaluated. A first-order absorption model and a transit compartment absorption model with 1–10 transit compartments were investigated to describe the absorption of chloroquine. Relative bioavailability was added with a typical value of 100 % with an estimate of between-subject variability. Between-subject variability was added exponentially, resulting in log-normal distributed parameters:
where Pi is the true value of the parameter for the individual and Pp is the typical or population value of the parameter. Pp is the fixed effect parameter estimated from the structural model and \(\upeta_{\text{i}}\) represents the difference between Pi and Pp.
An additive residual variability (RUV) model was applied according to:
where Cobs is the observed drug or metabolite concentration and Cp is the concentration predicted by the model and εad represents the difference between these values. An additive model on log-transformed data is equivalent to an exponential model.
The most adequate structural model with random effects (base model) was further developed to include covariates using a stepwise forward addition (p = 0.05) of covariates, followed by a stepwise backward elimination procedure (p = 0.001). Relationships between all parameters estimated in the base model and covariates, i.e., body weight (BW), age, sex, parasite clearance time (PCT) and fever clearance time (FCT) were evaluated. The covariate was retained in the final model if its removal resulted in an increase in the objective function of ≥10.83 points (p < 0.001) from the full model. BW was applied as a covariate on all CL and V values as a power model according to equation:
where Pt is the typical population value of the parameter for the population; θ1 represents the estimate of P in an individual with median BW; and θ2 is the fractional change in Pt with each kilogram change in BW from median BW. BW was allometrically scaled and θ2 was defined as 0.75 and 1 when applied on CL and V, respectively.
The covariate model for continuous covariates such as FCT was exemplified by the following equation:
where Pt is the typical value of parameter P; θ1 represents the estimate of P in an individual with median FCT; and θ2 the fractional change in P with each change in unit of FCT from median FCT.
All clearance and distribution parameters are reported as the ratio of the parameter and bioavailability since oral dosing was not accompanied by an intravenous dose. The pharmacokinetic population parameters estimated from the final covariate model were used to calculate terminal half-life for chloroquine and desethylchloroquine.
Bootstrap diagnostics were performed using 1000 re-sampled datasets. The precision was described as a relative standard error. A visual predictive check (VPC) is a tool for the evaluation of the predictive ability and the appropriateness of a model and was done by performing simulations of 1000 observations at each time point for the real observations in the data set with the final covariate model. The median and 95 % prediction intervals of the simulated data and the true observations were plotted against time. The predictive ability of the model was assumed to be adequate if less than 10 % of the observed concentrations fell outside the prediction interval.
Results
A total of 75 patients with P. vivax malaria were included in the analysis. All had completed a 42 days follow-up period. All patients showed good response following treatment with no reappearance of parasitaemia. The treatment was well-tolerated. All patients showed satisfactory response to treatment with 100 % cure rate within the follow-up period of 42 days. Median (95 % CI) parasite clearance time (PCT: the time taken for the parasite count to fall below the level of microscopic detection) and fever clearance time (FCT: the time taken for the temperature to return to normal, i.e., <37.3 °C) were 30 (18–36) and 24 (12–42) hours, respectively. Neither recurrence of P. vivax parasitaemia nor appearance of P. falciparum occurred.
Population pharmacokinetic models
The final data set included in the pharmacokinetic modeling consisted of 1405 observations of both chloroquine and desethylchloroquine from 75 individuals (less than 5 % of the samples below the lower limit of quantification). The final model for chloroquine and desethylchloroquine following a three-day chloroquine dose regimen was a two-compartment model for both chloroquine and its metabolite (p < 0.01) with a one transit compartment model for the absorption of chloroquine (Fig. 1). The parameter describing the transformation of chloroquine into desethylchloroquine (CLm) was fixed to 18 % of the transformation clearance from parent drug to metabolite [26]. One transit compartment described the absorption phase adequately and the relative bioavailability were retained in the final model (p < 0.05). In the final model, BSV was kept on the apparent volume of distribution of desethylchloroquine, relative bioavailability and the apparent volume of distribution in the peripheral compartment of chloroquine and desethylchloroquine. Adding between-subject variability on the mean transit time resulted in high relative standard errors on this parameter (149 %, based on 59 successful bootstraps runs out of 100) and was not kept in the model.

A two-compartment model (central and peripheral) with a one transit compartment model for the absorption of chloroquine into the central compartment and a first-order transformation of chloroquine into desethylchloroquine with an additional peripheral compartment added to desethylchloroquine. CQ is chloroquine compartments and DCQ represents desethylchloroquine compartments. k represents the rate constant between different compartments
No covariates were added in the final model. FCT was a significant covariate for VP CQ/F in the forward step but was not retained in the backward step. The basic goodness of fit plots exhibited adequate description of the data (Fig. 2). Parameter estimates and their precision for the final model and estimates from the non-parametric bootstrap are listed in Table 1. The calculated half-lives are presented in Table 1. The visual predictive check indicated a strong predictive ability of the model for the dataset where less than 10 % of the observations were outside the 95 % prediction interval (Figs. 3, 4).

Goodness of fit plots of chloroquine. Plots of the observed versus population predicted concentrations (a) and observed versus individual predicted concentrations (b). Weighed individual residuals versus individual predictions (c) and weighed residuals versus time (d). The black line is a non-parametric smoother describing the trend and the black line is the line of unity

Goodness of fit plots of desethylchloroquine. Plots of the observed versus population predicted concentrations (a) and observed versus individual predicted concentrations (b). Weighed individual residuals versus individual predictions (c) and weighed residuals versus time (d). The black line is a non-parametric smoother describing the trend and the black line is the line of unity

Plots from the visual predictive check for chloroquine (a) and desethylchloroquine (b) observations. The middle solid lines represent the median of simulated predictions by the final model. The dashed black lines represent the corresponding percentiles for the true observations. The black dots are the true observations and the grey shaded areas are the 95 % confidence intervals for the simulations. The decline in the upper percentiles of desethylchloroquine is due to base line values in the subjects
Discussion
Chloroquine remains the anti-malarial drug which is widely used in the tropics due to its safety, availability and low cost. The drug has now been rendered completely ineffective for treatment and prophylaxis of P. falciparum, but for P. vivax, P. ovale and P. malariae, it is still in use. For optimization of dosage regimen of chloroquine for the treatment or prophylaxis of these infections, plasma/blood drug concentrations and pharmacokinetic analysis are necessary. Nevertheless, the pharmacokinetics of chloroquine is not well understood. Previous studies involved small number of subjects and in some cases, with limitation of sensitivity of analytical methods and pharmacokinetic modeling techniques [27–32]. In addition, a wide range of inter-individual variability in the estimated pharmacokinetic parameters hurdles optimization of dose regimen of chloroquine for both clinical applications particularly in patients infected with P. vivax.
The large variability between individuals in the pharmacokinetic parameters of chloroquine makes population approaches a convenient method to assess the pharmacokinetic characteristics of the drug. Inclusion of concentration data of the active metabolite desethylchloroquine in the analysis is of further relevance as this metabolite has been shown to exhibit significant anti-malarial activity [33]. The final covariate model included a two-compartmental disposition for both chloroquine and desethylchloroquine with an adequate accuracy in the estimated parameters. The multi-exponential declines for both parent compound and metabolite are in consistency with previous reports [34–37]. For chloroquine, the estimates of absorption rate constant, the total apparent volume of distribution, and elimination half-life of chloroquine are in agreement with previously reported values. The apparent elimination clearance is lower compared to previous studies. This is probably due to the use of different sample matrixes [26–41]. Estimates of desethylchloroquine parameters such as apparent elimination clearance, apparent total volume of distribution and elimination half-life were relatively lower than reported values [26, 34–39]. The fact that reports of desethylchloroquine parameter estimations have been limited, might offer a skewed distribution of the values of the parameters. The underestimation of the parameters could also be due to the fixation of CLm to 18 %, an estimation based on the fraction desethylchloroquine of the total chloroquine dose recovered in urine. In the study reported by Karunajewa et al. [35], the same approximation of CLm was used and might also have underestimated their obtained pharmacokinetic parameters of desethylchloroquine. Besides desethylchloroquine, bidesethylchloroquine is produced by secondary metabolism of desethylchloroquine [42] and the formation of the third metabolite might also be a result of further metabolism of bidesethylchloroquine. This suggests that the amount of chloroquine transformed into desethylchloroquine should be assessed by estimating the amount of all secondary metabolites and including them in the estimation of the fraction desethylchloroquine formed. As chloroquine is metabolized up to 30–50 % by the liver and the mainly formed metabolites are (mono) desethylchloroquine and bidesethylchloroquine, the fraction of desethylchloroquine formed needs to be reassessed.
The quantification of a relationship between the drug concentration and response (pharmacokinetic-pharmacodynamic relationship) enables the identification of drug target levels. Biomarkers of response for the treatment of malaria have been collected and since the target of action of chloroquine is in infected red blood cells, it would most probably be uncomplicated and straightforward to characterize the relationship between blood concentrations and parasitaemia. The concentrations of chloroquine or desethylchloroquine associated with adequate treatment of P. vivax malaria have not been established rigorously. The minimum effective concentration (MEC) of chloroquine in plasma or serum of 15–30 ng/mL or in whole blood of 90 ng/mL have been suggested [45, 46]. Furthermore, patients with parasitaemia in the presence of chloroquine and desethylchloroquine concentration in whole blood of greater than 100 ng/mL is considered chloroquine resistant [16, 47, 48]. Unfortunately, this threshold level could not be determined in this group of patients as none had treatment failure following this standard regimen of chloroquine. In a recent study conducted in Bolivia, South America, chloroquine resistance P. vivax was reported in 6.5 % patients [49]. Chloroquine and desethylchloroquine in whole blood on day 7 and the day of parasite recrudescence in ten patients were 197–535 and 75–223 ng/mL, respectively. Six out of these ten patients has drug concentrations above the MEC.
Conclusion
A population pharmacokinetic model for chloroquine incorporating desethylchloroquine has been developed and validated with an adequate precision on the parameters. The determination of a more realistic fraction of desethylchloroquine formed would also be needed for future studies. This would create the possibility of more optimal exposure prediction for both compounds and optimization of malaria treatment in P.vivax monoinfection. Prompt and effective treatment would lead to efficacious killing of the malaria parasites and the prevention of resistance development to chloroquine.
References
WHO. World Malaria Report. Geneva: World Health Organization; 2014.
Ratcliff A, Siswantoro H, Kenangalem E, Wuwung M, Brockman A, Edstein MD, et al. Therapeutic response of multidrug-resistant Plasmodium falciparum and P. vivax to chloroquine and sulfadoxine-pyrimethamine in southern Papua, Indonesia. Trans R Soc Trop Med Hyg. 2007;101:351–9.
Tjitra E, Anstey NM, Sugiarto P, Warikar N, Kenangalem E, Karyana M, et al. Multidrug-resistant Plasmodium vivax associated with severe and fatal malaria: a prospective study in Papua, Indonesia. PLoS Med. 2008;5:e128.
Vijaykadga S, Rojanawatsirivej C, Cholpol S, Phoungmanee D, Nakavej A, Wongsrichanalai C. In vivo sensitivity monitoring of mefloquine monotherapy and artesunate-mefloquine combinations for the treatment of uncomplicated falciparum malaria in Thailand in 2003. Trop Med Int Health. 2006;11:211–9.
Tasanor O, Ruengweerayut R, Sirichaisinthop J, Congpuong K, Wernsdorfer WH, Na-Bangchang K. Clinical-parasitological response and in vitro sensitivity of Plasmodium vivax to chloroquine and quinine on the western border of Thailand. Trans R Soc Trop Med Hyg. 2006;100:410–8.
Poravuth Y, Socheat D, Rueangweerayut R, Uthaisin C, Pyae Phyo A, Valecha N, et al. Pyronaridine-artesunate versus chloroquine in patients with acute Plasmodium vivax malaria: a randomized, double-blind, non-interiority trial. PLoS One. 2011;6:e14501.
Muhamad P, Ruengweerayut R, Chacharoenkul W, Rungsihirunrat K, Na-Bangchang K. Monitoring of clinical efficacy and in vitro sensitivity of Plasmodium vivax to chloroquine in area along Thai Myanmar border during 2009–2010. Malar J. 2011;10:44.
Congpuong K, Na-Bangchang K, Thimasarn K, Tasanor U, Wernsdorfer WH. Sensitivity of Plasmodium vivax to chloroquine in Sa Kaeo Province, Thailand. Acta Trop. 2002;83:117–21.
Bureau of Vector Borne Diseases DoDC, Ministry of Health. National Strategic Plan for Malaria Control and Elimination in Thailand; 2011.
Schwartz IK, Lackritz EM, Patchen LC. Chloroquine-resistant Plasmodium vivax from Indonesia. N Eng J Med. 1991;324:927.
Rieckmann KH, Davis DR, Hutton DC. Plasmodium vivax resistance to chloroquine? Lancet. 1989;2:1183–4.
Schuurkamp GJ, Spicer PE, Kereu RK, Bulungol PK. A mixed infection of vivax and falciparum malaria apparently resistant to 4-aminoquinoline: a case report. Trans R Soc Trop Med Hyg. 1989;83:607–8.
Whitby M, Wood G, Veenendaal JR, Rieckmann K. Chloroquine-resistant Plasmodium vivax. Lancet. 1989;2:1395.
Collignon P. Chloroquine resistance in Plasmodium vivax. J Infect Dis. 1991;164:222–3.
Baird JK, Basri H, Purnomo Bangs MJ, Subianto B, Patchen LC, et al. Resistance to chloroquine by Plasmodium vivax in Irian Jaya, Indonesia. Am J Trop Med Hyg. 1991;44:547–52.
Murphy GS, Basri H, Purnomo Andersen EM, Bangs MJ, Mount DL, et al. Vivax malaria resistant to treatment and prophylaxis with chloroquine. Lancet. 1993;341:96–100.
Fryauff DJ, Soekartono Tuti S, Leksana B, Suradi Tandayu S, et al. Survey of resistance in vivo to chloroquine of Plasmodium falciparum and P. vivax in North Sulawesi, Indonesia. Trans R Soc Trop Med Hyg. 1998;92:82–3.
Sumawinata IW, Bernadeta Leksana B, Sutamihardja A, Purnomo Subianto B, et al. Very high risk of therapeutic failure with chloroquine for uncomplicated Plasmodium falciparum and P. vivax malaria in Indonesian Papua. Am J Trop Med Hyg. 2003;68:416–20.
Marlar T. Myat Phone K, Aye Yu S, Khaing Khaing G, Ma S, Myint O. Development of resistance to chloroquine by Plasmodium vivax in Myanmar. Trans R Soc Trop Med Hyg. 1995;89:307–8.
Myat Phone K, Myint O, Myint L, Thaw Z, Kyin Hla A, Nwe Nwe Y. Emergence of chloroquine-resistant Plasmodium vivax in Myanmar (Burma). Trans R Soc Trop Med Hyg. 1993;87:687–90.
Guthmann JP, Pittet A, Lesage A, Imwong M, Lindegardh N, MinLwin M, et al. Plasmodium vivax resistance to chloroquine in Dawei, southern Myanmar. Trop Med Int Health. 2008;13:91–8.
Phan GT, de Vries PJ, Tran BQ, Le HQ, Nguyen NV, Nguyen TV, et al. Artemisinin or chloroquine for blood stage Plasmodium vivax malaria in Vietnam. Trop Med Int Health. 2002;7:858–64.
WHO. Assessment and monitoring of antimalarial drug efficacy for the treatment of uncomplicated falciparum malaria. Geneva: World Health Organization; 2002.
Cheomung A, Na-Bangchang K. HPLC with ultraviolet detection for determination of chloroquine and desethylchloroquine in whole blood and finger-prick capillary blood dried on filter paper. J Pharm Bioanal. 2011;55:1031–40.
Beal SL, Sheiner LB. Estimating population kinetics. Crit Rev Biomed Eng. 1982;8:195–222.
Karunajeewa H, Salman S, Mueller I, Baiwog F, Gomorrai S, Law I, et al. Pharmacokinetics of chloroquine and monodesethyl-chloroquine in pregnancy. Antimicrob Agents Chemother. 2010;54:1186–92.
Edwards G, Looareesuwan S, Davies AJ, Wattanagoon Y, Phillips RE, Warrell DA. Pharmacokinetics of chloroquine in Thais: plasma and red-cell concentrations following an intravenous infusion to healthy subjects and patients with Plasmodium vivax malaria. Br J Clin Pharmacol. 1988;25:477–85.
Gustafsson LL, Lindström B, Grahnén A, Alván G. Chloroquine excretion following malaria prophylaxis. Br J Clin Pharmacol. 1987;24:221–4.
Tett SE, Cutler DJ. Apparent dose-dependence of chloroquine pharmacokinetics due to limited assay sensitivity and short sampling times. Eur J Clin Pharmacol. 1987;31:729–31.
Titus EO. Recent developments in the understanding of the pharmacokinetics and mechanism of action of chloroquine. Br J Clin Pharmacol. 1988;25:477–85.
Lee SJ, McGready R, Fernandez C, Stepniewska K, Paw MK, Viladpai-Nguen SJ, et al. Chloroquine pharmacokinetics in pregnant and nonpregnant women with vivax malaria. Eur J Clin Pharmacol. 2008;64:987–92.
Zhao Q, Tensfeldt TG, Chandra R, Mould DR. Population pharmacokinetics of azithromycin and chloroquine in healthy adults and paediatric malaria subjects following oral administration of fixed-dose azithromycin and chloroquine combination tablets. Malar J. 2014;13:36.
Aderounmu AF. In vitro assessment of the antimalarial activity of chloroquine and its major metabolites. Ann Trop Med Parasitol. 1984;78:581–5.
Gustafsson LL, Walker O, Alván G, Beermann B, Estevez F, Gleisner L, et al. Disposition of chloroquine in man after single intravenous and oral doses. Br J Clin Pharmacol. 1983;15:471–9.
Karunajeewa HA, Ilett KF, Mueller I, Siba P, Law I, Page-Sharp M, et al. Pharmacokinetics and efficacy of piperaquine and chloroquine in Melanesian children with uncomplicated malaria. Antimicrob Agents Chemother. 2008;52:237–43.
Obua C, Hellgren U, Ntale M, Gustafsson LL, Ogwal-Okeng JW, Gordi T, et al. Population pharmacokinetics of chloroquine and sulfadoxine and treatment response in children with malaria: suggestions for an improved dose regimen. Br J Clin Pharmacol. 2008;65:493–501.
Aderounmu F, Salako L, Lindström B, Walker O, Ekman L. Comparison of the pharmacokinetics of chloroquine after single intravenous and intramuscular administration in healthy Africans. Br J Clin Pharmacol. 1986;22:559–64.
Frisk-Holmberg M, Bergqvist Y, Termond E, Domeij-Nyberg B. The single dose kinetics of chloroquine and its major metabolite desethylchloroquine in healthy subjects. Eur J Clin Pharmacol. 1984;26:521–30.
Bustos D, Lazaro J, Gay F. Pharmacokinetics of sequential and simultaneous treatment with the combination chloroquine and sulfadoxine-pyrimethamine in acute uncomplicated Plasmodium falciparum malaria in the Philippines. Trop Med Int Health. 2002;7:584–91.
Vries PJ, Oosterhuis B, Boxtel C. Single-dose pharmacokinetics of chloroquine and its main metabolite in healthy volunteers. Drug Invest. 1994;8:143–9.
Wetsteyn JC, De Vries PJ, Oosterhuis B, Van Boxtel CJ. The pharmacokinetics of three multiple dose regimens of chloroquine: implications for malaria chemoprophylaxis. Br J Clin Pharmacol. 1995;39:696–9.
Projean D, Baune B, Farinotti R. In vitro metabolism of chloroquine: identification of CYP2C8, CYP3A4, and CYP2D6 as the main isoforms catalyzing N-desethylchloroquine formation. Drug Metab Disp. 2003;31:748–54.
Silamut K, Molunto P, Ho M, Davis TM, White NJ. Alpha 1-acid glycoprotein (orosomucoid) and plasma protein binding of quinine in falciparum malaria. Br J Clin Pharmacol. 1991;32:311–5.
Mansor SM, Molyneux ME, Taylor TE, Ward SA, Wirima JJ, Edwards G. Effect of Plasmodium falciparum malaria infection on the plasma concentration of alpha 1-acid glycoprotein and the binding of quinine in Malawian children. Br J Clin Pharmacol. 1991;32:317–21.
Most H, London IM, Kane CA, Lavietes PH, Schroeder EF, Hayman JM. Chloroquine for treatment of acute attacks of vivax malaria. J Am Med Assoc. 1964;131:963–7.
Gordon HH, Dieuaide FR, Marble A, Christianson HB, Dahl LK. Treatment of Plasmodium vivax malaria of foreign origin: a comparison of various drugs. Arch Intern Med. 1947;79:365–80.
Baird JK, Leksana B, Masbar S, Fryauff DJ, Sutanihardja MA, Suradi, et al. Diagnosis of resistance to chloroquine by Plasmodium vivax: timing of recurrence and whole blood chloroquine levels. Am J Trop Med Hyg. 1997;56:621–6.
Baird JK. Chloroquine resistance in Plasmodium vivax. Antimicrob Agents Chemother. 2004;11:4075–83.
Añez A, Moscoso M, Laguna A, Garnica C, Melgar V, Cuba M, et al. Resistance of infection by Plasmodium vivax to chloroquine in Bolivia. Malar J. 2015;14:261.
Authors’ contributions
KN and RH was involved in providing the conception, design of the study, data analysis and interpretation, and revised the manuscript critically for intellectual content and approved the final version of the manuscript. RH and AA were involved in providing the conception, design of the study, pharmacokinetic data analysis, and revised the manuscript. RR performed data collection. AC performed drug analysis. All authors read and approved the final manuscript.
Acknowledgements
The study was supported by The Commission on Higher Education, Ministry of Education of Thailand and The National Research University Project of Thailand (NRU), Office of Higher Education Commission and Thammasat University (Center of Excellence in Molecular Biology and Pharmacology of Malaria and Cholangiocarcinoma). We thank Ms. Kulaya Ruengweerayut and staff of Mae Tao malaria clinic, Tak Province for their kind support for sample collection.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Höglund, R., Moussavi, Y., Ruengweerayut, R. et al. Population pharmacokinetics of a three-day chloroquine treatment in patients with Plasmodium vivax infection on the Thai-Myanmar border. Malar J 15, 129 (2016). https://doi.org/10.1186/s12936-016-1181-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12936-016-1181-1