
Abstract
Background
Hepatocellular carcinoma (HCC) is an aggressive cancer with a high rate of death globally. The use of bioinformatics may help to identify immune cell-related genes both as targets for potential immunotherapies and for their value associated with predicting therapy responses.
Methods
In this study, mRNA expression profiles of HCC samples from The Cancer Genome Atlas (TCGA) database were subjected to gene enrichment, cell type abundance, immune cell infiltration, and pathway enrichment analyses to determine immune cell gene features, cell type abundance, and functional annotation characteristics. We also evaluated their prognostic values using Cox regression and Kaplan–Meier analyses and assessed potential responses to chemotherapy. Four subgroups (Groups 1–4) were identified. Group 4 was associated with advanced clinical characteristics, high immune cell enrichment scores, and the poorest outcomes.
Results
Differentially expressed genes (DEGs) in the HCC samples were enriched in the following pathways: antigen binding, cell surface receptor signal transduction of the immune response, and cell surface activated receptor signal transduction of the immune response. Highly expressed genes in Group 4 were enriched in elements of the WNT signalling pathway. We identified five immune-related genes (SEMA3A, TNFRSF11B, GUCA2A, SAA1, and CALCR) that were significantly related to HCC prognosis. A prognostic model based on these five genes exhibited good predictive value, with 1-year and 5-year area under the curve (AUC) values of > 0.66. Group 4 was also potentially more sensitive to EHT 1864, FH535, and lapatinib chemotherapies than the other groups.
Conclusions
We identified and validated four HCC subgroups based on immune system-related genes and identified five genes that may be used for an immune-based prognostic model for HCC treatment.
Similar content being viewed by others

Background
Liver cancer ranks fifth in neoplasm frequency and has the second highest rate of cancer-associated mortality worldwide [1, 2]. Hepatocellular carcinoma (HCC) accounts for 80% of primary liver cancers and 90% of non-metastatic liver tumours [3,4,5], with approximately 8,54,000 new cases and 8,10,000 deaths per year globally [6, 7]. The pathology of HCC is multifactorial and involves many steps [8]. It has been reported that viral hepatitis infection, aflatoxins, alcohol, altered transcriptional regulation, and genetic susceptibility/polymorphisms are all considered significant factors (individually or synergistically) that contribute to the aetiology of HCC. The poor prognosis of HCC is mainly due to a lack of sensitive detection methods at its early stages and a high frequency of both recurrence and metastasis [9]. Currently, resection surgery, transplantation, targeted chemotherapy, radiation therapy, interventional therapy, and gene therapy are all effective alternative options for HCC [10, 11]. Although other advanced treatments have been explored, local and regional therapies are still recommended for early-stage disease because few effective options exist for advanced-stage HCC. Recently, however, immune system-associated therapies have been successfully tested in the clinic.
Tumour immunotherapy has revolutionized cancer treatments and has consistently been the focus of attention because of its promising outcomes for advanced HCC [12, 13]. Active and passive immunotherapies, immune checkpoint inhibitors (ICIs), and therapies targeting the tumour microenvironment constitute major breakthroughs for cancer treatments [13, 14]. Accumulating evidence has demonstrated that ICIs [e.g., those targeting programmed cell death ligand 1 (PD-1), T cell immunoglobulin mucin domain-containing-3 (TIM3), and cytotoxic T lymphocyte antigen 4 (CTLA4)] in combination with conventional therapies exhibited enhanced anti-tumour effects and broad applicability for cancer patients [15,16,17]. Despite these considerable achievements, questions remain as to how to improve the efficacy of immunotherapies, how to broaden their application range, and how to better predict immune responses [17, 18]. Immune system-based models can provide detailed mechanistic insights and be used to categorize patients into low, medium, and high immune response subgroups.
Bioinformatic analyses allow the identification of potential immune-sensitive therapeutic biomarkers, prognostic models can be constructed based on immune system-related genes [19, 20]. In this study, we performed a comprehensive analysis of HCC-related immune infiltration and constructed a gene-based immune response model for predicting immunotherapy responses and for identifying potential biomarkers for HCC-targeting therapies.
Methods
Cell culture
The normal human liver cell line L02 and the HCC cell line Hep3B were purchased from the Chinese Academy of Sciences (Shanghai, China) and cultured in DMEM (Gibco, Carlsbad, CA, USA) supplemented with 10% foetal bovine serum and 1% penicillin. Cells were cultured at 37 °C in a 5% CO2 atmosphere.
Quantitative reverse-transcription PCR (qRT-PCR)
Total RNA was extracted from cells using an RNeasy Mini Kit (Qiagen, Valencia, USA) and then reverse transcribed into cDNA using the PrimeScript™ RT reagent Kit according to the manufacturer’s instructions. Relative mRNA expression levels were determined on an ABI 7500 Fast PCR instrument. GAPDH was used as the internal control. Relative expression levels of IL6, CCR3, SAA1, and GCG were quantified using the 2−ΔΔCt method. The primers are listed in Additional file 1: Table S1.
Public databases
We collected gene expression profile data (RNA-seq) from HCC patient samples in The Cancer Genome Atlas (TCGA) database (https://cancergenome.nih.gov/). In total, data from 371 patients with follow-up, status, and gene expression data were included in the liver hepatocellular carcinoma (LIHC) cohort. An additional 212 HCC datasets were downloaded from the International Cancer Genome Consortium (ICGC-LIRI-JP, https://dcc.icgc.org/projects/LIRI-JP) for further validation. The ICGC-LIRI-JP dataset was used to verify the prognosis-related immune biomarker genes in the HCC cohort.
Identification of immune cell-associated genes
We downloaded immune cell-related gene profiles from the ImmPort database [21] (http://www.immport.org). Immune cell-related genes were classified into different subgroups based on different immune cell characteristics, including cell type, functions, and associated pathways [22].
Single-sample gene set enrichment analysis (ssGSEA)
To evaluate immune cell infiltration characteristics in HCC patient samples, we performed ssGSEA to evaluate the degree of immune cell enrichment in the different samples. ssGSEA was performed using the gene set variation analysis (GSVA) package in R software [23].
Assessments of immune scores and stromal scores and identification of differentially expressed genes (DEGs)
We utilized the Estimation of STromal and Immune cells in MAlignant Tumour tissues using Expression data (ESTIMATE) method [24], as well as the CIBERSORT tool [25] and other algorithms to estimate cell type abundance from bulk tissue transcriptomes and to assess tumour immune scores and immune cell purities. The Microenvironment Cell Populations-counter (MCP-counter) [26], accessed through Connectivity Map at https://portals.broadinstitute.org/cmap/, was used to predict potential drug sensitivities. In addition, the DESeq package [27] in R software was applied to determine differential gene expression in HCC tumour tissue and adjacent normal tissue from the raw CGA datasets. We defined the false discovery rate (FDR) to be no more than 0.05 and the |log2 (fold change)| to be no less than one as the cut-off value.
Pathway enrichment analysis
To identify immune cell-related pathways, we utilized Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) analyses. Biological process (BP), molecular function (MF), and cellular component (CC) pathway analyses were included in the GO analysis. These analyses were performed using the clusterProfiler package in R software [28, 29].
Protein–protein interaction (PPI) analysis
To investigate the key regulatory genes and validate the connections between differentially expressed genes, a PPI network was constructed from gene expression datasets from the HCC cohort. We constructed the PPI network based on known and predicted PPIs. Furthermore, we defined the confidence score as ≥ 0.7, and the maximum number of interactors was identified as 0. These selected genes were input into Cytoscape (version 3.6.1) under the following parameters: degree cut-off = 2, haircut on, node score cut-off = 0.2, k-core = 2, and max. depth = 100.
Cox regression analysis
We used single-factor Cox regression analysis to identify prognosis-related genes and to assess the effects of the immune status on patient prognoses [30]. We set a p value < 0.01 as the significance level for immune cell-related genes. Multivariate Cox analysis was performed to calculate the risk scores. The median was defined as the dividing line between the high and low scores.
Evaluation of responses to clinical immunotherapies
Both a submap and the Tumour Immune Dysfunction and Exclusion (TIDE) algorithm were used to predict potential responses to ICIs [31].
Chemotherapy response predictions
A dataset from the Genomics of Drug Sensitivity in Cancer (GDSC, https://www.cancerrxgene.org/) [32] database was used to screen for potential chemotherapeutics for the treatment of HCC. We estimated the half-maximal inhibitory concentrations (IC50s) for these drugs using the pRRophetic package in R software.
Statistical analysis
All statistical analyses were performed using SPSS 25 (IBM Corporation, Armonk NY) and R software (version 3.6.1). All statistical results with p < 0.05 were considered significant.
Results
Immune cell-related gene selection, scoring, clustering, and classification
To construct an immune cell gene-based classifier, we performed ssGSEA and consistent cluster analysis. Based on the different gene scores, we obtained four immune cell gene subgroups (Groups 1–4) (Fig. 1A). Advanced-stage HCC was mainly found in Group 4. A total of 16 immune cell-related gene clusters with high scores were also clustered in Group 4, corresponding with the activated immune subgroups. To further explore the prognoses associated with these four different subgroups, we conducted Kaplan–Meier analysis of overall survival, and the results demonstrated that the prognoses of patients in Group 4 were worse than those of patients in the other groups (Fig. 1B). Specifically, the prognoses of patients in Group 1 were better than those of patients in Group 4 (p = 0.00023) (Fig. 1D); Group 2 had more favourable overall survival times than Group 4 (p = 0.00018) (Fig. 1D); and the prognoses of patients in Group 3 were better than those of patients in Group 4 (p = 0.0015) (Fig. 1E). We identified four HCC sample subgroups based on the immune scores, and Group 4 was associated with the most advanced clinical stages and significantly poorer prognoses than the other groups.

ssGSEA based on the immune system-related gene classifier and prognostic analyses. a Immune system-related gene clustering and classification of Groups 1–4. b Overall survival analyses of the four groups, with Group 4 showing the poorest prognoses. c Prognostic analysis comparing Group 1 to Group 4, showing that Group 1 had better prognoses than Group 4. d Group 2 also demonstrated longer overall survival times than Group 4. e Group 3 also demonstrated better overall prognoses than Group 4
Immune cell infiltration in the different subgroups
To further investigate immune cell infiltration in the four subgroups, we applied the CIBERSORT algorithm to the data and confirmed that the different subgroups also had significantly different levels of immune cell infiltration. There were significant differences in the proportions of naïve B cells, plasma cells, M0 macrophages, and resting dendritic cells. Group 4 had the lowest proportions of naïve B cells, plasma cells, and resting dendritic cells but the highest proportion of macrophages (Fig. 2A). The MCP-counter algorithm was used to assess the infiltration levels of immune cells in the HCC samples. The results showed that the infiltration levels of CD8+ T cells and cytotoxic lymphocytes were significantly different. Group 1 had higher degrees of CD8+ T cell and cytotoxic lymphocyte infiltration than the other groups (Fig. 2B). We also used the ESTIMATE method and found that Group 2 had the highest immune purity, with significant differences from that of Group 4 (Fig. 2C). Group 2 also had the lowest immune scores, and these scores were significantly different than those of Group 4 (Fig. 2D). Overall, the four-group classifier of immune system-related genes showed significant differences in immune scores, immune cell infiltration levels, and immune-based prognoses. Group 4 had higher levels of immune cell infiltration, was associated with advanced HCC stages, and had shorter overall survival times. Therefore, tumour immune escape might occur in samples from Group 4, with high immune infiltration, representing disabled and exhausted immune functions.

Immune cell infiltration and differences among the four immune-based subgroups. a Immune cell infiltration levels in the four subgroups. b The levels of CD8+ T cells and cytotoxic lymphocytes were significantly different among the four groups. Group 1 had the highest levels of CD8+ T cell and cytotoxic lymphocyte infiltration. c ESTIMATE analysis indicated that Group 2 had the highest immune purity. Group 2 also had significantly higher immune purity than Group 4. d The immune scores demonstrated that Group 2 had the lowest immune scores and were significantly different from those of Group 4
Correlations between the immune-based classifier and human leukocyte antigen (HLA) and interactions with immune checkpoint molecules
Immune checkpoint blockade (ICB) has been widely applied to treat a variety of tumours and shown significant favourable therapeutic effects [33]. HLA molecules are crucial for immune system function and are very clinically significant in immunotherapy [34]. The functional diversity of HLA genes is closely related to cancer genomics and cancer progression [35]. Importantly, responses to ICIs rely on the evolved efficiency of HLA-mediated immunity [36, 37]. We next investigated the internal relationship between the four groups of immune subgroups and HLA genes and identified several immune regulatory molecules, including HLA-A, HLA-E, and HLA-DRB5 (Fig. 3A). To further support correlations between immune checkpoint molecules and our immune-based classifier, we also determined that CD244, ICOS, ADORA2A, CD70, PD-L1 (CD274), and TIGIT molecules showed significant differences among the different immune-based subgroups (Fig. 3B). These results indicate that HLA genes and immune checkpoint molecules play crucial roles in the different subgroups and that they may be valid therapeutic targets for cancer therapies.

Correlations between the immunophenotype and expression of HLA genes and immune checkpoint molecules. a HLA genes (HLA-A, HLA-E, HLA-DPB1, and HLA-DRB5) were differentially expressed in the different groups. b Immune checkpoint genes, including BTLA, LAG3, ICOS, ADORA2A, CD70, PD-L1 (CD274), and TIGIT, were differentially expressed in the different groups
Correlations between immune subgroups and interferon-γ (IFN-γ) pathways
The tumour microenvironment is crucial for tumourigenesis and tumour development, and the CD8+ T cell-mediated anti-tumour response has been shown to be significantly increased through the production of cytokines such as INF-γ [38]. Accumulating evidence has shown that immune cells, such as CD8+ T cells, can facilitate the upregulation of immune checkpoints and enhance anti-tumour immune responses [39]. To annotate IFN-γ and T helper cell-related genes, we performed cluster analysis and found that the immune response has a crucial relationship with IFN-γ-related regulatory pathways (Fig. 4A). More importantly, Group 4 had significantly high expression levels of IFN-r-, IFNGR2-, IFNGR1-, JAK1-, and JAK2-related genes, with IFNG showing significant differences among the subgroups (Fig. 4B). Group 4 had higher IFNG expression levels than Group 2 (Fig. 4C).

Associations between the immunophenotype and IFN-γ pathways. a Correlations between immune responses and IFN-γ pathway-related genes. The IFN-γ, IFNGR2, IFNGR1, JAK1, and JAK genes were highly expressed in Group 4. b There were significant differences in IFN-γ expression among the four subgroups. c The expression level of IFN-γ in Group 4 was significantly higher than that in Group 2
Relationship between the immune subgroups and 5-methylcytosine (m5C) regulators
mRNA and its transcription play an important role in immune-related gene regulation [40, 41]. The m5C methylation of RNA modifies the transcription of multiple genes, regulates protein expression, and affects cell phenotypes, and nucleoside modifications have been shown to suppress the potential for RNA to activate innate immune cells [42]. Here, m5C regulators were associated with all four groups. The NSUN7 regulator was expressed at low levels in all of these subgroups (Fig. 5B), and the overall levels of m5C regulator expression showed obvious differences between Group 2 and Group 4 (p < 0.01) and less obvious differences between Group 1 and Group 4 (p < 0.05) (Fig. 5B). The mRNA expression of NSUN4 was remarkably different among the immune subgroups (Fig. 5C).

Relationship between the immune system-based classifier and the expression of m5C mRNA methylation regulators. a All four groups exhibited significant m5C regulator activities. b, c mRNA expression of the m5C regulator NSUN4 was significantly different among the four groups, with Group 4 having higher NSUN4 expression than Groups 1 and 2
Functional annotations of the DEGs
We used the DESeq package in R software to identify and classify immune system-related DEGs. The upregulated DEGs in Group 4 were positively correlated with the immune classifier-based genes, and the upregulated DEGs in Groups 1, 2, and 3 were negatively correlated with the immune classifier-based genes (Fig. 6A). To further explore the regulatory roles of these DEGs, we used the clusterProfiler package in R software to characterize the associated genes. GO pathway analysis indicated that these DEGs are involved in a variety of cell regulation pathways, including the regulation of membrane potentials, neurotransmitter transport, and those for the regulation of blood pressure. CC analysis indicated that these DEGs play crucial roles in synaptic membranes, the presynapse, the ion channel complex, and transmembrane transporter pathways. MF analysis showed that the regulatory mechanisms for the DEGs are mainly enriched in receptor-ligand activity, signalling-receptor activator activity, and other such pathways (Fig. 6B). KEGG pathway annotations showed that Group 4 was enriched in WNT signalling pathway elements. The downregulated DEGs were enriched in immune response-related pathway elements (Fig. 6C).

Identification of differentially expressed genes (DEGs) and pathway enrichment analyses in the four subgroups. a DEGs in Groups 1–3 and in Group 4. b GO analysis indicated that the majority of DEGs (based on different immunotypes) were enriched in cell surface receptor signalling pathways that regulate the immune response and in cell surface receptor signalling pathways that activate the immune response. c KEGG pathway analysis showed that the DEGs in Group 4 were enriched in elements of the WNT signalling pathway, whereas the downregulated DEGs were enriched in elements of immune-related response pathways
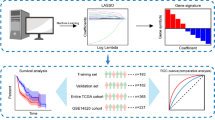
To gain a deeper contextual understanding of their possible interactions, we constructed a PPI network and identified four hub genes (IL6, CCR3, SAA1, and GCG; Fig. 7A). To further identify HCC prognosis-related genes, Cox regression analyses were performed and yielded 45 DEGs. After applying stringent screening conditions (p < 0.01), we identified five immune-related genes that demonstrated significant differences related to prognosis (SEMA3A, TNFRSF11B, GUCA2A, SAA, and CALCR). In addition, the expression levels of IL6, CCR3, SAA1, and GCG in the HCC cell line (Hep3B) and control cell line (L02) were determined by qRT-PCR. As shown in Fig. 7B, C, the expression of IL6 and CCR3 was higher in the HCC cell line (Hep3B) than in the control cell line (L02). The expression level of SAA1 was lower in Hep3B cells than in L02 cells (Fig. 7D). There was no significant difference in GCG expression between the HCC cell line and the control cell line (Fig. 7E). Using multivariate Cox regression for further analysis, we constructed a risk-coefficient model using these five genes. This model demonstrated that the subgroup with the highest risk scores had poorer prognoses than the subgroup with the lowest risk scores (p = 0.014; Fig. 7F).

PPI network analysis, the identification of five key immune system-related genes, and a prognostic model based on these five genes. a The PPI network analysis revealed four hub genes (IL6, CCR3, SAA1, and GCG) that were closely related to the immune classifier-based DEGs. b–e qRT-PCR was performed to determine the relative expression of IL6, CCR3, SAA1, and GCG in the HCC cell line (Hep3B) and the control cell line (L02). f The key immune system-related genes (SEMA3A, TNFRSF11B, GUCA2A, SAA1, and CALCR) and the prognostic model based on these genes demonstrated that the group with high risk scores had shorter overall survival times than the group with low risk scores. g The biomarkers were validated using the ICGC-LIRI-JP dataset. The five-gene-based prognostic model showed that the group with high risk scores had worse prognoses than the group with low risk scores. h Area under the curve (AUC) analyses showed that this predictive model demonstrated good prognostic value
To further validate these five immune gene-based biomarkers, Cox regression analyses were applied to datasets from the International Cancer Genome Consortium (ICGC-LIRI-JP). These alternate independent analyses showed that the group with the highest risk scores also had poorer prognoses than the group with the lowest risk scores (Fig. 7G). Area under the curve (AUC) analyses (Fig. 7H), which were used to quantify and evaluate diagnostic accuracy, showed that the AUC value for the five gene-based prognostic model was 0.71 (95% CI = 61.01–81.94).
Assessment of immunotherapy responses
Further studies were performed to explore the relationship between the four immune subgroups and immunotherapy responses by evaluating the clinical immunotherapy responses to PD-1 and CTLA4. Interestingly, Group 4 was characterized as T cell-deficient and had the highest immune deficiency scores (Fig. 8A). These studies confirmed that immune deficiency is involved in tumour immune escape, especially in Group 4.

Potential therapeutic drugs for Group 4 based on T cell expression. a Group 4 had a higher level of T cell dysfunction than the other subgroups. b–s Eighteen drugs with obvious differences in sensitivity in Group 4. These results indicate that Group 4 is more sensitive to EHT 1864, FH535, and lapatinib chemotherapy than the other groups
Chemotherapy responses
To comprehensively examine the responses of the different immune subgroups to chemotherapeutic agents, we used the pRRophetic algorithm in R software. The results indicated that the half-maximal inhibitory concentration (IC50) values of Group 4 were significantly different than those of the other subgroups (the corresponding drugs are listed in Fig. 8B–S), but not all of these drugs exhibited highly sensitive responses. Only EHT 1864, FH535, and lapatinib exhibited higher sensitivities in Group 4, demonstrating the limited selection of drugs for treating advanced-stage HCC tumours.
Discussion
HCC is a very aggressive type of cancer with highly heterogeneous malignancies [43]. The tumour microenvironment plays a crucial role in immune cell regulation and cytokine production and plays a central role in cancer development and progression [44]. The emerging field of immunotherapy has increasingly been applied to HCC [14, 43]. However, compared to other cancers, HCC immunotherapy is still in its infancy, and there are still many challenges that need to be addressed, especially for expanding its indications and increasing its benefits [45, 46]. Therefore, personalized immunotherapy strategies and multiple therapy combinations may be required for effective HCC treatment [16, 47]. In the present study, we comprehensively described HCC subgroups based on the expression of immune system-related genes and identified five genes for a prognostic prediction model. A better understanding is still necessary for the criteria required for patient selection and for optimizing any combination strategies to maximize the potential of these approaches.
Previous studies have shown that the liver is a relatively immune-tolerant organ with a high level of immune cell infiltration [48, 49]. Moreover, a variety of immune system regulators are involved in HCC carcinogenesis and progression [14, 50, 51]. To better elucidate the biological functions of HCC immune system-related genes, we identified four groups (Groups 1–4) based on different degrees of immune system gene expression. Based on this classifier, we demonstrated that Group 4 was closely related to the clinical characteristics of advanced HCC, a high level of macrophage infiltration, low immune purity, and poor overall survival. For this type of HCC, the results indicate high levels of immune cell infiltration with few immune cells contributing to anti-tumour functions and activities.
Recently, immunotherapies that have targeted different immune checkpoints have achieved great successes in treating HCC. Such regulation of the immune system facilitates mast cells and activates HLA-G molecules, which are involved in the pathology of chronic hepatitis [52]. HLA molecules are also crucial for T cell-mediated immune surveillance in HCC patients, and HLA-mediated immunomodulatory functions have been observed in numerous pathological conditions, including cancer [35]. HLA-G has shown both prognostic potential and diagnostic value in HCC patients, and HLA molecules have been reported to play crucial roles in the pathogenesis of HCC [53]; therefore, they may be useful as part of an overall immunotherapy strategy against HCC [54]. Accordingly, we found that the subgroups with high immune scores and Group 4 (with high levels of immune cell infiltration) may have better immunotherapy responses.
Furthermore, we assessed the immune classifier and IFN-γ and m5C regulators in the different groups. Group 4 exhibited both high IFN-related pathway activation and modifications in m5C regulator genes. We also identified four immune system-based hub genes that play crucial roles in immune system-related gene mechanisms that participate as regulators of a variety of gene functions. The five most important immune system DEGs were identified and used as the basis for constructing a prognostic model. This model system was significantly associated with HCC prognoses and may provide better predictions of HCC prognoses when applied clinically.
Conclusions
The gene expression classifier described in the present study provides a comprehensive understanding of immune system-related genes and HCC characteristics. The resulting prognostic model for predicting HCC outcomes may facilitate the determination of clinically relevant and reliable prognostic indicators for immunotherapy responses.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author upon reasonable request.
Abbreviations
- HCC:
-
Hepatocellular carcinoma
- ICB:
-
Immune checkpoint blockade
- PD-1:
-
Programmed cell death ligand 1
- TIM3:
-
T cell immunoglobulin mucin domain-containing-3
- TCGA:
-
The Cancer Genome Atlas
- LIHC:
-
Liver hepatocellular carcinoma
- ICGC:
-
International Cancer Genome Consortium
- ssGSEA:
-
Single-sample gene set enrichment analysis
- GSVA:
-
Gene set variation analysis
- ESTIMATE:
-
Estimation of STromal and Immune cells in MAlignant Tumours using Expression data
- DEGs:
-
Differentially expressed genes
- FDR:
-
False discovery rate
- FC:
-
Fold change
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- GO:
-
Gene Ontology
- BP:
-
Biological process
- MF:
-
Molecular function
- CC:
-
Cellular component
- PPI:
-
Protein–protein interaction
- TIDE:
-
Tumour immune dysfunction and exclusion
- ICIs:
-
Immune checkpoint inhibitors
- GDSC:
-
Genomics of Drug Sensitivity in Cancer
References
Erratum: global cancer statistics 2018. GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2020;70:313.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30.
Zou RC, Liang Y, Li LL, Tang JZ, Yang YP, Geng YC, et al. Bioinformatics analysis identifies protein tyrosine kinase 7 (PTK7) as a potential prognostic and therapeutic biomarker in stages I to IV hepatocellular carcinoma. Med Sci Monit. 2019;25:8618–27.
Liao X, Bu Y, Jia Q. Traditional Chinese medicine as supportive care for the management of liver cancer: past, present, and future. Genes Dis. 2020;7:370–9.
Liu C, Zha Z, Zhou C, Chen Y, Xia W, Wang YN, et al. Ribonuclease 7-driven activation of ROS1 is a potential therapeutic target in hepatocellular carcinoma. J Hepatol. 2020;S0168–8278:33674–6.
Cerrito L, Ponziani FR, Garcovich M, Tortora A, Annicchiarico BE, Pompili M, et al. Regorafenib: a promising treatment for hepatocellular carcinoma. Expert Opin Pharmacother. 2018;19:1941–8.
Fernandes ESM, Rodrigues PD, Álvares-da-Silva MR, Scaffaro LA, Farenzena M, Teixeira UF, et al. Treatment strategies for locally advanced hepatocellular carcinoma. Transl Gastroenterol Hepatol. 2019;4:12.
Kulik L, El-Serag HB. Epidemiology and management of hepatocellular carcinoma. Gastroenterology. 2019;156:477-491.e1.
Khemlina G, Ikeda S, Kurzrock R. The biology of Hepatocellular carcinoma: implications for genomic and immune therapies. Mol Cancer. 2017;16:149.
Bruix J, Reig M, Sherman M. Evidence-based diagnosis, staging, and treatment of patients with hepatocellular carcinoma. Gastroenterology. 2016;150:835–53.
Gunasekaran G, Bekki Y, Lourdusamy V, Schwartz M. Surgical treatments of hepatobiliary cancers. Hepatology. 2020. https://doi.org/10.1002/hep.31325.
Zhou T, Liang X, Wang P, Hu Y, Qi Y, Jin Y, et al. A hepatocellular carcinoma targeting nanostrategy with hypoxia-ameliorating and photothermal abilities that, combined with immunotherapy, inhibits metastasis and recurrence. ACS Nano. 2020. https://doi.org/10.1021/acsnano.0c01453.
Tseng HC, Xiong W, Badeti S, Yang Y, Ma M, Liu T, et al. Efficacy of anti-CD147 chimeric antigen receptors targeting hepatocellular carcinoma. Nat Commun. 2020;11:4810.
Feng GS, Hanley KL, Liang Y, Lin X. Improving the efficacy of liver cancer immunotherapy: the power of combined preclinical and clinical studies. Hepatology. 2020. https://doi.org/10.1002/hep.31479.
Zhang PF, Gao C, Huang XY, Lu JC, Guo XJ, Shi GM, et al. Cancer cell-derived exosomal circUHRF1 induces natural killer cell exhaustion and may cause resistance to anti-PD1 therapy in hepatocellular carcinoma. Mol Cancer. 2020;19:110.
BoDal M, De Mattia E, Baboci L, Mezzalira S, Cecchin E, Assaraf YG, et al. New insights into the pharmacological, immunological, and CAR-T-cell approaches in the treatment of hepatocellular carcinoma. Drug Resist Updat. 2020;51:100702.
Flynn MJ, Sayed AA, Sharma R, Siddique A, Pinato DJ. Challenges and opportunities in the clinical development of immune checkpoint inhibitors for hepatocellular carcinoma. Hepatology. 2019;69:2258–70.
Brown ZJ, Greten TF, Heinrich B. Adjuvant treatment of hepatocellular carcinoma: prospect of immunotherapy. Hepatology. 2019;70:1437–42.
Fan X, Yuan H, Zhao S, Yang X, Shi R, Wang J, et al. Epigenetic age acceleration of early stage hepatocellular carcinoma tightly associated with hepatitis B virus load, immunoactivation, and improved survival. Cancer Biol Ther. 2020. https://doi.org/10.1080/15384047.2020.1804284.
Xu D, Wang Y, Zhou K, Wu J, Zhang Z, Zhang J, et al. Development and validation of a novel 8 immune gene prognostic signature based on the immune expression profile for hepatocellular carcinoma. Onco Targets Ther. 2020;13:8125–40.
Bhattacharya S, Dunn P, Thomas CG, Smith B, Schaefer H, Chen J, et al. ImmPort, toward repurposing of open access immunological assay data for translational and clinical research. Sci Data. 2018;5:180015.
He Y, Jiang Z, Chen C, Wang X. Classification of triple-negative breast cancers based on Immunogenomic profiling. J Exp Clin Cancer Res. 2018;37:327.
Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 2013;14:7.
Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612.
Le T, Aronow RA, Kirshtein A, Shahriyari L. A review of digital cytometry methods: estimating the relative abundance of cell types in a bulk of cells. Brief Bioinform. 2020. https://doi.org/10.1093/bib/bbaa219.
Dienstmann R, Villacampa G, Sveen A, Mason MJ, Niedzwiecki D, Nesbakken A, et al. Relative contribution of clinicopathological variables, genomic markers, transcriptomic subtyping and microenvironment features for outcome prediction in stage II/III colorectal cancer. Ann Oncol. 2019;30:1622–9.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106.
Yang J, Wang F, Chen B. HLA-DPA1 gene is a potential predictor with prognostic values in multiple myeloma. BMC Cancer. 2020;20:915.
Si C, Wang J, Ma W, Hua H, Zhang M, Qian W, et al. Circular RNA expression profile in human fibroblast premature senescence after repeated ultraviolet B irradiations revealed by microarray. J Cell Physiol. 2019;234:18156–68.
Sanz-Pamplona R, Melas M, Maoz A, Schmit SL, Rennert H, Lejbkowicz F, et al. Lymphocytic infiltration in stage II microsatellite stable colorectal tumors: a retrospective prognosis biomarker analysis. PLoS Med. 2020;17:e1003292.
Wang Q, Li M, Yang M, Yang Y, Song F, Zhang W, et al. Analysis of immune-related signatures of lung adenocarcinoma identified two distinct subtypes: implications for immune checkpoint blockade therapy. Aging. 2020;12:3312–39.
Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, et al. A Landscape of pharmacogenomic interactions in cancer. Cell. 2016;166:740–54.
Xu F, Jin T, Zhu Y, Dai C. Immune checkpoint therapy in liver cancer. J Exp Clin Cancer Res. 2018;37:110.
Singh R, Kaul R, Kaul A, Khan K. A comparative review of HLA associations with hepatitis B and C viral infections across global populations. World J Gastroenterol. 2007;13:1770–87.
Catamo E, Zupin L, Crovella S, Celsi F, Segat L. Non-classical MHC-I human leukocyte antigen (HLA-G) in hepatotropic viral infections and in hepatocellular carcinoma. Hum Immunol. 2014;75:1225–31.
Chowell D, Krishna C, Pierini F, Makarov V, Rizvi NA, Kuo F, et al. Evolutionary divergence of HLA class I genotype impacts efficacy of cancer immunotherapy. Nat Med. 2019;25:1715–20.
Lee MH, Huang YH, Chen HY, Khor SS, Chang YH, Lin YJ, et al. Human leukocyte antigen variants and risk of hepatocellular carcinoma modified by hepatitis C virus genotypes: a genome-wide association study. Hepatology. 2018;67:651–61.
Liao P, Song K, Zhu Z, Liu Z, Zhang W, Li W, et al. Propranolol suppresses the growth of colorectal cancer through simultaneously activating autologous CD8(+) T cells and inhibiting tumor AKT/MAPK pathway. Clin Pharmacol Ther. 2020. https://doi.org/10.1002/cpt.1894.
Fava A, Buyon J, Mohan C, Zhang T, Belmont HM, Izmirly P, et al. Integrated urine proteomics and renal single-cell genomics identify an IFN-γ response gradient in lupus nephritis. JCI Insight. 2020. https://doi.org/10.1172/jci.insight.138345.
Xue C, Zhao Y, Li L. Advances in RNA cytosine-5 methylation: detection, regulatory mechanisms, biological functions and links to cancer. Biomark Res. 2020;8:43.
Ally A, Balasundaram M, Carlsen R, Chuah E, Clarke A, Dhalla N, Holt RA, Jones SJ, Lee D, Ma Y, Marra MA. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169:1327-1341.e23.
Estibariz I, Overmann A, Ailloud F, Krebes J, Josenhans C, Suerbaum S. The core genome m5C methyltransferase JHP1050 (M.Hpy99III) plays an important role in orchestrating gene expression in Helicobacter pylori. Nucleic Acids Res. 2019;47:2336–48.
Caruso S, O’Brien DR, Cleary SP, Roberts LR, Zucman-Rossi J. Genetics of HCC: novel approaches to explore molecular diversity. Hepatology. 2020. https://doi.org/10.1002/hep.31394.
Nagl L, Horvath L, Pircher A, Wolf D. Tumor endothelial cells (TECs) as potential immune directors of the tumor microenvironment—new findings and future perspectives. Front Cell Dev Biol. 2020;8:766.
Kole C, Charalampakis N, Tsakatikas S, Vailas M, Moris D, Gkotsis E, et al. Immunotherapy for hepatocellular carcinoma: a 2021 update. Cancers. 2020. https://doi.org/10.3390/cancers12102859.
Zhang L, Ding J, Li HY, Wang ZH, Wu J. Immunotherapy for advanced hepatocellular carcinoma, where are we? Biochim Biophys Acta Rev Cancer. 2020. https://doi.org/10.1016/j.bbcan.2020.188441.
Chen J, Gingold JA, Su X. Immunomodulatory TGF-β signaling in hepatocellular carcinoma. Trends Mol Med. 2019;25:1010–23.
Dwyer BJ, Macmillan MT, Brennan PN, Forbes SJ. Cell therapy for advanced liver diseases: repair or rebuild. J Hepatol. 2020;S0168–8278:33626–6.
Ficht X, Iannacone M. Immune surveillance of the liver by T cells. Sci Immunol. 2020. https://doi.org/10.1126/sciimmunol.aba2351.
Rizvi S, Wang J, El-Khoueiry AB. Liver cancer immunity. Hepatology. 2020. https://doi.org/10.1002/hep.31416.
Fabris L, Sato K, Alpini G, Strazzabosco M. The tumor microenvironment in cholangiocarcinoma progression. Hepatology. 2020. https://doi.org/10.1002/hep.31410.
Amiot L, Vu N, Rauch M, L’Helgoualc’h A, Chalmel F, Gascan H, et al. Expression of HLA-G by mast cells is associated with hepatitis C virus-induced liver fibrosis. J Hepatol. 2014;60:245–52.
Fergusson JR, Wallace Z, Connolly MM, Woon AP, Suckling RJ, Hine DW, et al. Immune-mobilising monoclonal T cell receptors mediate specific and rapid elimination of Hepatitis B-infected cells. Hepatology. 2020. https://doi.org/10.1002/hep.31503.
Tamai T, Mizukoshi E, Kumagai M, Terashima T, Iida N, Kitahara M, et al. A novel α-fetoprotein-derived helper T-lymphocyte epitope with strong immunogenicity in patients with hepatocellular carcinoma. Sci Rep. 2020;10:4021.
Acknowledgements
Not applicable.
Funding
This research was funded by the National Natural Science Foundation of China (81790631) and a Zhejiang University Academic Award for Outstanding Doctoral Candidates (2020055).
Author information
Authors and Affiliations
Contributions
LL designed and guided the study. CX wrote and edited the manuscript. XG helped with the statistical analysis and reference collection. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
The primers for the qRT-PCR assay.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xue, C., Gu, X. & Li, L. Immune classifier-based signatures provide good prognostic stratification and predict the clinical benefits of immune-based therapies for hepatocellular carcinoma. Cancer Cell Int 21, 471 (2021). https://doi.org/10.1186/s12935-021-02183-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-021-02183-5