
Abstract
Background
Flavokawain B is one of the naturally occurring chalcones in the kava plant (Piper methysticum). It exhibits anticancer, anti-inflammatory and antimalarial properties. Due to its therapeutic potential, flavokawain B holds promise for the treatment of many diseases. However, due to its poor bioavailability and low aqueous solubility, its application remains limited. The attachment of a sugar unit impacts the stability and solubility of flavonoids and often determines their bioavailability and bioactivity. Biotransformation is an environmentally friendly way to improve the properties of compounds, for example, to increase their hydrophilicity and thus affect their bioavailability. Recent studies proved that entomopathogenic filamentous fungi from the genera Isaria and Beauveria can perform O-methylglycosylation of hydroxyflavonoids or O-demethylation and hydroxylation of selected chalcones and flavones.
Results
In the present study, we examined the ability of entomopathogenic filamentous fungal strains of Beauveria bassiana, Beauveria caledonica, Isaria farinosa, Isaria fumosorosea, and Isaria tenuipes to transform flavokawain B into its glycosylated derivatives. The main process occurring during the reaction is O-demethylation and/or hydroxylation followed by 4-O-methylglycosylation. The substrate used was characterized by low susceptibility to transformations compared to our previously described transformations of flavones and chalcones in the cultures of the tested strains. However, in the culture of the B. bassiana KCh J1.5 and BBT, Metarhizium robertsii MU4, and I. tenuipes MU35, the expected methylglycosides were obtained with high yields. Cheminformatic analyses indicated altered physicochemical and pharmacokinetic properties in the derivatives compared to flavokawain B. Pharmacological predictions suggested potential anticarcinogenic activity, caspase 3 stimulation, and antileishmanial effects.
Conclusions
In summary, the study provided valuable insights into the enzymatic transformations of flavokawain B by entomopathogenic filamentous fungi, elucidating the structural modifications and predicting potential pharmacological activities of the obtained derivatives. The findings contribute to the understanding of the biocatalytic capabilities of these microbial cultures and the potential therapeutic applications of the modified flavokawain B derivatives.
Similar content being viewed by others

Background
Flavokawain B (1-(2ʹ-hydroxy-4ʹ,6ʹ-dimethoxyphenyl)-3-phenyl-prop-2-en-1-on) (FB1) is one of the naturally occurring chalcones in the kava plant (Piper methysticum) [1]. Kava is a perennial shrub from the Piperaceae (pepper) family, which is native to the ethnogeographic regions of Melanesia, Micronesia and Polynesia [2]. Since the 1990s, kava has been popularized amongst Western countries due to its sedative, anti-stress and anxiolytic properties [3, 4]. Across European and American markets, kava is sold in tablet or capsule form to treat anxiety disorders [5, 6]. The primary active constituents responsible for the pharmacological effects of kava are known as kavalactones and kavapyrones [7]. Kava chalcone- flavokawain B (FB1) exhibits anti-malarial [8], anti-inflammatory [9] and anti-angiogenic [10] properties; it also weakens the progression of gastric cancer [11]. Compound FB1 substantially slows down the development of colon cancer, inducing mitochondria-dependent apoptosis, which is characterized by the release of cytochrome c [12]. It also plays a crucial role in melanoma cells’ ability to execute and produce ROS-modulated apoptotic and autophagic cell death. Flavokawain B (FB1) shows the potential to inhibit tumor growth in nude mice with xenografts [13] and may represent a novel therapeutic option for patients with synovial sarcoma [14]. This chalcone exhibits toxicity towards various breast cancer cell lines [10, 15] and has been shown to inhibit the growth of human osteosarcoma cells through G2/M cell cycle arrest [16]. However, flavokawain B (FB1), like other chalcones, has poor water solubility and bioavailability, which reduces its in vivo biological effects [17,18,19]. Microbial glycosylation and methylation is a known method of improving the stability, water solubility, and bioavailability of chalcones [20]. In vivo studies have shown that glycosylation may significantly improve the bioavailability of quercetin (flavonol); however, the impact of glycosylation strongly depends on the type of sugar attached [21]. For the two quercetin glycosides isoquercetin (glucoside) and rutin (rutoside), the relative total bioavailability was 148% and 23%, respectively, compared with quercetin aglycone [22]. Certain glycosides, such as quercetin-3-O-glucoside (isoquercetin), are substrates for the small intestinal brush border enzyme—lactase-phlorizin hydrolase (EC 3.2.1.108) [23]. The glucoside form of quercetin can be enzymatically converted to aglycone quercetin, and thus be absorbed mainly in the small intestine. This results in relatively high bioavailability of quercetin after oral administration of isoquercetin [24].
Utilization of whole-cell biocatalysts provides a convenient approach to execute enzymatic cascades encompassing multiple reactions, while simultaneously providing the necessary cofactors required for these intricate biotransformations [25]. Cell coverings protect and stabilize enzymes, enabling them to be used in challenging reaction conditions [26]. Using whole cells in biocatalysis obviates the requirement for cell lysis and the subsequent purification of enzymes, resulting in a significant reduction in catalyst costs. The presence of the intact cell enables the internal supply and regeneration of expensive cofactors, eliminating the need for external supplementation and further reducing costs. Furthermore, whole-cell biocatalysts are generally simpler to prepare, costs of maintaining the cultures are typically feasible, and cells can often be employed repeatedly, enhancing cost-effectiveness [26, 27].
The main aim of this study was to investigate the ability of entomopathogenic filamentous fungi of the genera Beauveria, Isaria and Metarhizium to perform biotransformation of flavokawain B (FB1) and characterize products of this process. Studies have indicated that entomopathogenic fungal strains such as B. bassiana, B. caledonica, Isaria farinosa, and I. fumosorosea may be effectively used as biocatalysts in biotransformations of chalcones [20], flavones [28,29,30], flavanones [29,30,31] and steroids [32,33,34,35]. B. bassiana, an ascomycete fungus, has garnered attention as a versatile pathogenic agent, effectively targeting a wide spectrum of insect species. This remarkable fungus has been harnessed on a commercial scale as an environmentally friendly insecticide. The hallmark of its biological potential lies in the synthesis of a rich array of versatile enzymes and secondary metabolites, including non-peptides and polyketides. Notably, some of these metabolites, such as oxalic acid, demonstrate potent pathogenic and virulent properties, rendering them appealing for applications across diverse sectors, ranging from industrial to pharmaceutical and agricultural realms [36]. B. bassiana exhibits a noteworthy capability for glycosylation of flavonoid compounds such as quercetin [37], warfarin [38], cannflavin B [39] and xanthohumol [40] as well as hydroxylation of steroids such as 1-dehydrotestosterone, testosterone, 19-nortestosterone, 17α-methyltestosterone and progesterone [32]. In addition, I. farinosa and I. fumosorosea strains show the ability to biotransform 2’-hydroxy-2-methylchalcone [41], 2’-methylflavone [30], methoxyflavones [42, 43] and steroids [44, 45]. Metarhizium strains can also be used as biocatalysts in the bioconversion of quercetin [46] and steroids [35, 45].
Results and discussion
In light of the recent findings on the enzymatic modification of flavonoids within the milieu of the entomopathogenic microbial cultures, we decided to employ three distinct strains of Beauveria bassiana (namely KCh J1.5, J1 and BBT) and three strains from the genus Isaria (specifically, I. tenuipes MU35, I. fumosorosea KCh J2 and I. farinosa KCh KW 1.1) as well as a strain of Beauveria caledonica (designated as KCh J3.3) and Metarhizium robertsii (designated as MU4), formerly known as Metarhizium anisopliae, to catalyze biotransformations of (1-(2ʹ-hydroxy-4ʹ,6ʹ-dimethoxyphenyl)-3-phenyl-prop-2-en-1-on) – flavokawain B (FB1) [29,30,31, 47,48,49,50,51]. In this study we decided to determine whether entomopathogenic filamentous fungi transform flavokawain B (FB1) as in case of flavones. Flavokawain B (FB1) was obtained through chemical synthesis, purified by crystallization from ethanol, and its structure was confirmed spectroscopically (Additional file 1: Figures S1–S7) according to the procedure described in earlier publications [42, 52].
Biotransformation of FB1 in the cultures of entomopathogenic filamentous fungi
Biotransformation of the substrate (FB1) in the cultures of selected entomopathogenic, filamentous fungal strains led to obtaining seven identified derivatives.
The products were extracted from the cultures using ethyl acetate then purified with preparative thin-layer chromatography (pTLC). The structures of the purified products were determined using nuclear magnetic resonance spectroscopy (NMR), and retention times (Rt) were determined with ultra-high performance liquid-chromatography (UHPLC) data obtained during the screening procedure. All selected strains except I. fumosorosea KCh J2 and I. farinosa KCh KW1.1 (> 99.9% of the unconverted substrate in the reaction mixture after 10 days of incubation) showed the ability to perform biotransformation of the substrate (FB1). The best conversion rate was observed in B. bassiana KCh BBT and B. bassiana KCh J1.5 (0.5% and 1.6%, respectively, of the remaining substrate in the reaction mixture after 10 days and less than 5% of unreacted substrate after 3 days of biotransformation) (Table 1). Based on TLC and HPLC analyses, it was observed that the resulting products were characterized by significantly higher polarity than the substrate (FB1). This indicated that the main products, akin to the previously described biotransformations of flavonoid compounds [41, 42], are most likely the result of 4-O-methylglycosylation.
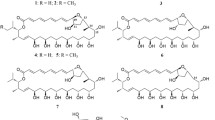
In the case of M. robertsii MU4 several products were obtained (four identified) during the process of biotransformation (Figure 1). The product of demethylation and 4-O-methylglycosydation (FB3) appears to be the main product of the biotransformation (75.8% content in the reaction mixture after 10 days of the biotransformation) with a retention time of about 5.795 min according to UHPLC.

Biotransformation of flavokawain B (FB1) by M. robertsii MU4 strain
The product was identified as 1-(4ʹ-O-β-D-(4ʹʹʹ-O-methylglucopiranosyl)-2ʹ-hydroxy-6ʹ-methoxyphenyl)-3-phenyl-prop-2-en-1-on – 4ʹ-O-β-D-(4ʹʹ-O-methylglucopyranosyl)-cardamonin (FB3) using NMR spectroscopy (Additional file 1: Figures S15–S21). The structure of the chalcone core (FB1) is confirmed by the 1H NMR, 13C NMR, COSY, HSQC and HMBC correlation spectra. However, instead of the signal from the protons of one of the -CH3 groups (visible in the 1H NMR substrate spectrum), signals from the sugar unit are visible. The multiplicities and positions of these signals in both the 1H NMR and 13C NMR spectra indicate that a 4-O-methylglucopyranosyl molecule was introduced in place of this -CH3 group. The HMBC spectrum indicated the coupling of the signal from the C-4ʹ carbon from the chalcone skeleton and H-1ʹʹʹ from the sugar unit, which exactly indicates the attachment position of the sugar unit to the chalcone core. The strains of entomopathogenic filamentous fungi have a unique capacity for 4-O-methylglycosylation of flavonoids. This reaction is most often described for flavonoid compounds containing a free hydroxyl group in their structure [48, 53,54,55,56,57,58]. The ability of entomopathogenic strains to hydroxylate and 4-O-methylglycosylate flavonoid compounds has also been described [31, 42, 59]. In our previous research, we also observed that the fungi perform demethylation and then 4-O-methylglycosylation of methoxyflavones [42, 47]. However, in one of our previous studies, we observed that a series of methoxy derivatives of chrysin, apigenin, and tricetin did not undergo glycosylation. Instead, we observed the tested strains’ ability to selectively demethylate/hydroxylate carbon C-3′ and C-4′ of ring B of the substrates used [43]. Flavokawain B (FB1) also contains two methoxy groups in positions analogous to those in 5,7-dimethoxyflavone. Nevertheless, obtaining 4ʹ-O-β-D-(4″-O-methylglucopyranosyl)-cardamonin (FB3) in the culture of M. robertsii MU4 strain indicates that the test compound (FB1) undergoes progressive demethylation and 4-O-methylglucosylation.
The product of 4ʹ-O-demethylation of the flavokawain B (FB1) known as cardamonin (FB2).
(Rt around 9.075 min) was also observed in the reaction mixture (1.2% of reaction mixture composition after 10 days). Due to the small amount of compound FB2 obtained, its structure was established based only on a comparison of the 1H NMR spectrum obtained for this compound with analogous data previously published in the literature (Additional file 1: Figures S10–S12) [60].
Compound FB4 was characterized by Rt around 4.641 min and it was identified by NMR spectra analysis as 1-(4ʹ-O-β-D-(4ʹʹʹ-O-methylglucopiranosyl)-2ʹ-hydroxy-6ʹ-methoxyphenyl)-3-(3ʹʹ-hydoxyphenyl)-prop-2-en-1-on – 4ʹ-O-β-D-(4″-O-methylglucopyranosyl)-3ʹʹ-hydroxycardamonin (FB4) (Additional file 1: Figures S24–S30). In the 1H NMR spectrum obtained for this compound (similarly to compound FB3), characteristic signals originating from the 4-O-methylglucopyranosyl substituent were observed, along with the observation of 4ʹ-O-demethylation. The position of the sugar unit was identified in a similar way as in the case of product FB3. Based on the 13C NMR spectrum analysis, it was determined that compound FB4 also underwent hydroxylation in ring B. In the HMBC spectrum, couplings were observed between the signal from the introduced –OH group proton and the signals from the C-2ʹʹ and C-4ʹʹ carbons, confirming unequivocally the positioning of the introduced hydroxyl group.
The second most abundant product in the reaction mixture was the compound identified as 1-(4ʹ-O-β-D-(4ʹʹʹ-O-methylglucopiranosyl)-2ʹ-hydroxy-6ʹ-methoxyphenyl)-3-(4ʹʹ-hydoxyphenyl)-prop-2-en-1-on – 4ʹ-O-β-D-(4″-O-methylglucopyranosyl)-4ʹʹ-hydroxycardamonin (FB5), which constituted 16.1% of the reaction mixture (after 10 days, Rt around 4.566 min). The NMR spectra which allowed us to identify product (FB5) (Additional file 1: Figures S33–S39) indicated no changes in the core of the chalcone backbone. Similarly to products FB3 and FB4, the structure of ring A for compound FB5 was determined. In the 1H NMR spectrum obtained for compound FB5, a characteristic signal pattern typical for a para-substituted aromatic ring was observed, unequivocally indicating the introduction of a hydroxyl group at carbon C-4ʹʹ. As a result of enzyme activity produced by the M. robertsii MU4 strain, products generated through cascading transformations were identified: 4ʹ-O-demethylation (compounds FB2-FB5), 4ʹʹ-O-methylglucopyranosylation (compounds FB3-FB5), and hydroxylation of ring B (compounds FB4 and FB5). In the culture of this strain, additional products were also formed, albeit in small quantities that impeded determination of their structure. The reaction mixture contained a decreasing amount of unidentified products between the first and the last day of biotransformation (from 11.5% to 0.1%); it may indicate that these compounds were intermediates in the formation of identified products (FB3–FB5).
Two products were obtained during flavokawain B (FB1) biotransformation in the culture of Isaria tenuipes MU35 (Fig. 2) and Beauveria bassiana KCh J1 (Fig. 3). In both cultures, the formation of cardamonin (FB2) is noticeable at 9.8% and 1.1%, respectively, in the reaction mixture after 10 days of biotransformation). In the culture of I. tenuipes MU35, the main product is 4ʹ-O-β-D-(4″-O-methylglucopyranosyl)-cardamonin (FB3) (36.7% content of the reaction mixture after 10 days of biotransformation).

Biotransformation of flavokawain B (FB1) by I. tenuipes MU35 strain

Biotransformation of flavokawain B (FB1) by B. bassiana KCh J1 strain
The product identified as 1-(2ʹ-hydroxy-4ʹ,6ʹ-dimethoxyphenyl)-3-(3ʹʹ-hydroxyphenyl)-prop-2-en-1-on – 3ʹʹ-hydroxyflavokawain B (FB6) (Additional file 1: Figures S42–S44) appears in the culture of B. bassiana KCh J1 as the main product (11.8% after 10 days, Rt about 8.905 min). However, this substance is also noticeable in the cultures of B. bassiana BBT and KCh J1.5 in decreasing amounts during the biotransformation process (1.8–0.5% and 0.8–0.0%, respectively), which may indicate that this product is an intermediate in the process of formation of the other products with a 4-O-methylglucopyranosyl group. In previous publications, we reported clear differences in the metabolism of the applied substrates in cultures of various strains belonging to the species B. bassiana [34, 42, 46]. We also found that the strain B. bassiana KCh J1 does not possess the ability to glycosylate flavonoid compounds [42, 46], which we also confirmed in the present study.
Biotransformation in the cultures of B. bassiana BBT and KCh J1.5 led to obtaining six identified products (Fig. 4). Products FB3, FB4, FB6 were also identified in the cultures of the strains M. robertsii MU4 and B. bassiana KCh J1, but two main products were obtained: 1-(2ʹ-hydroxy-4ʹ,6ʹ-dimethoxyphenyl)-3-(3ʹʹ-O-β-D-(4ʹʹʹ-O-methylglucopiranosyl)-phenyl)-prop-2-en-1-on – 3ʹ-O-β-D-(4″-O-methylglucopyranosyl)-flavokawain B (FB7) and 1-(2ʹ-hydroxy-4ʹ,6ʹ-dimethoxyphenyl)-3-(3ʹʹ-O-β-D-(4ʹʹʹ-O-methylglucopiranosyl)-4ʹʹ-hydroxyphenyl)-prop-2-en-1-on – 3ʹ-O-β-D-(4″-O-methylglucopyranosyl)-4ʹʹ-hydroxyflavokawain B (FB8) accounted for 32.5% and 41.6%, respectively, after 10 days of the reaction in B. bassiana KCh J1.5 culture.

Biotransformation of flavokawain B (FB1) by B. bassiana BBT and KCh J1.5 strains
The product (FB7) with Rt about 5.729 min was identified by NMR spectra analysis (Additional file 1: Figures S47–S53). In the 1H NMR spectrum obtained for this compound, it was observed that the modification occurred within the B ring of the chalcone skeleton. Based on chemical shifts, multiplicity of observed signals in the 1H NMR spectrum, and signal positions in the 13C NMR spectrum, it was determined that the functionalization occurred at carbon C-3ʹ. It was also confirmed that the structure of compound FB7 contains a 4-O-methylglucopyranosyl substituent. In the HMBC spectrum, coupling was observed between the signal from proton H-1ʹʹʹ (sugar unit) and the signal from carbon C-3ʹ, unequivocally indicating that the isolated main product formed in the cultures of B. bassiana BBT and KCh J1.5 strains is 3ʹ-O-β-D-(4″-O-methylglucopyranosyl)-flavokawain B (FB7). Product FB8, which was identified as 3ʹ-O-β-D-(4″-O-methylglucopyranosyl)-4ʹʹ-hydroxyflavokawain B, was assigned to Rt around 5.629 min. The product is formed in the cultures of B. bassiana BBT and KCh J1.5 (27.5% and 31.2%, respectively, of the reaction mixture after 10 days). In the 1H NMR spectrum obtained for compound FB8, analogous to the spectra obtained for product FB7, signals originating from protons and carbons in the A ring and propanoic chain were observed at appropriate positions and multiplicities in the 1H NMR spectrum, as well as in the 13C NMR spectrum. The presence of the 4-O-methylglucopyranosyl group in the structure of this compound was also noted. Based on the multiplicity of signals from the B ring protons and the chemical shifts of signals from carbons C-3ʹ and C-4ʹ observed at 147.57 and 146.88, respectively, in the 13C NMR spectrum, it was unequivocally determined that this compound resulted from dihydroxylation. In the HMBC spectrum, coupling between the signal from proton H-1ʹʹʹ (sugar unit) and the signal from carbon C-3ʹ was observed, allowing for the determination of its structure as 3ʹ-O-β-D-(4″-O-methylglucopyranosyl)-4ʹʹ-hydroxyflavokawain B (FB8) (Additional file 1: Figures S56–S62).
In the culture of B. caledonica KCh J3.3 biotransformation of the FKB, three products were obtained (Fig. 5). The main product, which constituted 21.2% of the reaction mixture after 10 days, appeared to be 4ʹ-O-β-D-(4″-O-methylglucopyranosyl)-cardamonin (FB3).

Biotransformation of flavokawain B (FB1) by B. caledonica KCh J3.3
Formation of 4ʹ-O-β-D-(4″-O-methylglucopyranosyl)-3ʹʹ-hydroxycardamonin (FB4) and product FB7 identified as 3ʹ-O-β-D-(4″-O-methylglucopyranosyl)-flavokawain B (FB7) was also observed (9.8% and 14.6% respectively after 10 days).
Based on the conducted experiments, it was observed that the strain M. robertsii MU4 predominantly produces products resulting from modifications of the A ring in the flavokawain B structure; 4-O-methylglucopyranosylation was preceded by 4ʹ-O-demethylation. However, in cultures of B. bassiana BBT and KCh J1.5 strains, transformations within the B ring were observed; the attachment of the sugar unit was preceded by hydroxylation. In our previous publications, through the biotransformation of 2ʹ-hydroxy-2-methylchalcone and 2ʹ-hydroxy-4-methylchalcone in B. bassiana KCH J1.5 culture, we obtained dihydrochalcones and chalcones with a 4-O-methylglucopyranosyl group located in the A ring [20, 41]. In the culture of the strain I. fumosorosea KCh J2, the respective 4″-O-methylglucopyranosyl chalcones were obtained with high efficiency [20, 41]. In this study, we found that the type and positioning of the substituents present in flavokawain B (FB1) hinder its transformation in the culture of the strain I. fumosorosea KCh J2. Simultaneously, none of the obtained biotransformation products of flavokawain B (FB1) resulted from the action of enzymes reducing the double bond of the substrate used. Additionally, in cultures of unconventional yeasts efficiently conducting the hydrogenation reaction, flavokawain B (FB1) remained unhydrogenated [61]. The in vitro metabolism of flavokawain B was examined using human liver microsomes. Major phase I metabolites were generated by demethylation in position C-4ʹʹ, yielding cardamonin, and hydroxylation predominantly in position C-4, yielding flavokawain C as a phase I metabolite [1]. The course of transformations in phase I observed is similar to what we observed in the culture of M. robertsii MU4 (described in this publication). This similarity is also evident in the experiment where flavokawain B was metabolized in the presence of uridine diphosphate (UDP) glucuronic acid by microsomal UDP-glucuronosyl transferases [1]. Flavokawain B in the colorectal cancer cell line LoVo and its doxorubicin-resistant subline LoVo/Dx was converted to the corresponding flavanone 5,7-dimetoxyflavanone. In a previous study it was found that cyclization of chalcone was related to a significant decrease in cytotoxicity [62]. In the cultures of entomopathogenic filamentous fungi described in this publication, we did not observe transformation of flavokawain B (FB1) into the respective flavanone.
Cheminformatics tools such as SwissADME, PreADMET and passOnline were used to compare the physicochemical characteristics, pharmacokinetics, and possible biological activities of flavokawain B (FB1) and its derivatives obtained via biotransformations. Calculations of the physicochemical descriptors for the substrate and the seven products (FB2–FB8) were performed (Additional file 1: Figures S8, S9, S13, S14, S22, S23, S31, S32, S40, S41, S45, S46, S54, S55, S63, S64). Absorption, distribution, metabolism, and excretion (ADME) factors, pharmacokinetic characteristics, and use for medicinal chemistry were all predicted. The Swiss Institute of Bioinformatics (SIB)’s Molecular Modeling Group developed and maintains the online resource SwissADME, which was used to conduct the analysis (http://www.swissadme.ch accessed on September 29, 2023) [63].
Based on the outcomes of this instrument, it was discovered that all of the products indicate lower lipophilicity than flavokawain B (FB1) (Table 2). Moreover, whether there is a hydroxyl group attached to the B ring of the chalcone core or a methylglycoside; the substance is not absorbed from the gastrointestinal tract (FB4, FB5, FB7, FB8). Substances FB1, FB2, FB6, which do not have a sugar unit attached to the chalcone core, exhibit BBB permeability and they cannot be transported by the P-glycoprotein. The simulations are in line with current research on chalcones [64,65,66] and their glycosides [67]. None of the obtained methylglycosides show the ability to inhibit any of the simulated CYP isoforms. This characteristic allows them to be classified as safe in terms of drug-drug interactions [68, 69]. On the other hand, the usage of aglycons and their potential to inhibit the CYP family may have a beneficial effect in terms of lowering treatment costs due to the medications’ reduced dosages with intact drug activity [70, 71]. The Caco-2 permeability assay is considered as the gold standard method to evaluate both the passive and active transport and the absorption of orally administered drugs [72]. Prediction of permeability of substances FB1–FB8 was performed using the online tool PreADMET (https://preadmet.webservice.bmdrc.org/adme/ accessed September 29, 2023). Collected data are presented in Table 2. Studies have indicated that flavonoids with an attached sugar unit are characterized by lower permeability in the Caco-2 model than the unsubstituted ones [73, 74].
Potential bioactivities were calculated using the PassOnline tool (http://www.way2drug.com/passonline/predict.php accessed on September 30, 2023). Table 3 shows predictions of the possible activities of flavokawain B (FB1) and the obtained metabolites (FB2–FB8). In most cases, the activities of metabolites FB2–FB8 are higher than the activities of flavokawain B (FB1) or comparable. Chalcones are generally described as anticarcinogenic [75,76,77] as are their glycosides [78, 79]. A bioinformatics tool predicted that the anticarcinogenic activity of the methylglycosyl residues of FB1 is much more possible than non-sugar metabolites (possibility of 47.6–53.0% to 88.1–90.1%, respectively). According to the predictions, all of substances FB1–FB8 will stimulate caspase 3, leading to cell apoptosis, with a high probability. Bioinformatics projections align with the current state of knowledge [80,81,82,83,84]. It is expected that products FB2–FB5, FB7, and FB8 will indicate antileishmanial activity in accordance with the in silico studies. A great number of flavokawain B (FB1) analogues showed antiprotozoal activity [85], as did other chalcones [86, 87]. Also, some of the aurones synthesized from flavokawain B (FB1) showed toxicity towards Leishmania infantum at a similar level as amphotericin B [88].
Materials and methods
Substrates
The substrates 2-hydroxy-4,6-dimetoxyacetophenone and benzaldehyde were obtained from Sigma-Aldrich (St. Louis, MO, USA). FB1 was synthesized from these substrates. Its NMR spectral data are presented in Additional file Materials.
Synthesis
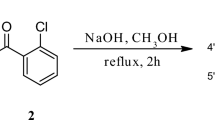
Flavokawain B (FB1) was obtained in the Claisen-Schmidt reaction of benzaldehyde (BA) with 2-hydroxy-4,6-dimetoxyacetophenone (HDMA) in the reaction presented in Fig. 6.

Flavokawain B synthesis
After 2 h of reflux, the product of the Claisen-Schmidt reaction was transferred to an acid environment and filtered using a Buchner funnel. The obtained compound was confirmed by NMR (1H NMR, 13C NMR, COSY, HMBC and HSQC) analysis.
Microorganisms
The microorganisms B. caledonica KCh J3.3, B. bassiana KCh J1.5, KCh J1 and KCh BBT, I. farinosa KCh KW 1.1, I. fumosorosea KCh J2, I. tenuipes MU35 and M. robertsii MU4 were obtained from the collection of the Department of Food Chemistry and Biocatalysis, Wrocław University of Environmental and Life Sciences (Wrocław, Poland).
Screening procedure
Erlenmeyer flasks (300 mL), each containing 100 mL of the sterile cultivation medium (3% glucose, 1% Aminobac), were inoculated with a suspension of each entomopathogenic strain and then incubated for 4 days at 24 °C on a rotary shaker. After this time, 10 mg of a substrate was dissolved in 1 mL of dimethyl sulfoxide (DMSO) and added to the medium. Samples were collected on the 1st, 3rd, 7th and 10th day of the process. Then, all products were extracted using ethyl acetate, and extracts were dried using MgSO4, concentrated in vacuo and analyzed using TLC and UHPLC methods.
Scale-up biotransformation
For the scale-up process we used Erlenmeyer flasks (2000 mL), each containing 500 mL of the same cultivation medium (3% glucose, 1% Aminobac), which were inoculated in the same way as described above. Three days after inoculation, 100 mg of a substrate was dissolved in 2 mL of DMSO and added to the interior. Samples were collected on the 3–7th day of the process. Products were extracted three times using ethyl acetate and then analyzed using TLC, HPLC and NMR spectroscopy (1H NMR, 13C NMR, COSY, HMBC and HSQC) analysis.
Analysis
Initial tests were carried out using TLC plates (SiO2, DC Alufolien Kieselgel 60 F254 (0.2 mm thick), Merck, Darmstadt, Germany). The mobile phase contained a mixture of chloroform and methanol in a 9:1 (v/v) ratio. The plates were observed using a UV lamp (254 and 365 nm).
The scale-up biotransformation products were separated using 500 µm preparative TLC silica gel plates (Anatech, Gehrden, Germany). The mobile phase contained a mixture of chloroform and methanol in a 9:1 (v/v) ratio. Separation products were scraped out and extracted twice using ethyl acetate.
UHPLC
UHPLC analyses were carried out using a Thermo Scientific Dionex Ultimate 3000 UHPLC + instrument with a photodiode array detector (detection in wavelength: 210–450 nm) and a ZORBAX Eclipse XDB C-18 analytical column (5 m, 4.6 250 mm, Agilent, Santa Clara, CA, USA). Chromatographic separation was achieved using a gradient program as follows: initial conditions–25% B in A, 0.5 min–25% B in A, 4 min–40% B in A, 7.5 min–55% B in A, 8 min–100% B in A, 9.7 mi–25% B in A, 11 min–25% B in A. The flow rate was 0.7 mL min−1, where A: 0.1% HCOOH in H2O; B: 0.1% HCOOH in acetonitrile.
NMR data and isolated yields of biotransformation products
Flavokawain B (FB1): 1H NMR (400 MHz, DMSO-d6) δ (ppm): 13.41 (s, 1H, C-2ʹ-OH); 7.76 (d, 1H, H-3, J = 15.7 Hz); 7.73–7.71 (m, 2H, H-2ʹʹ and H-6ʹʹ); 7.64 (d, 1H, H-2, J = 15.7 Hz); 7.48–7.39 (m, 3H, H-3ʹʹ, H-4ʹʹ and H-5ʹʹ); 6.16 (d, 1H, H-5ʹ, J = 2.3 Hz); 6.13 (d, 1H, H-3ʹ, J = 2.3 Hz); 3.89 (s, 3H, C-6ʹ-OCH3); 3.82 (s, 3H, C-4ʹ-OCH3); 13C NMR (101 MHz, DMSO-d6) δ (ppm): 192.39 (C-1); 165.65 (C-2ʹ); 165.52 (C-4ʹ);161.96 (C-6ʹ); 142.43 (C-3); 134.83 (C-1ʹʹ); 130.54 (C-4ʹʹ); 129.14 (C-3ʹʹ and C-5ʹʹ); 128.54 (C-2ʹʹ and C-6ʹʹ); 127.51 (C-2); 106.38 (C-1ʹ); 93.94 (C-3ʹ); 91.18 (C-5ʹ); 56.29 (C-6ʹ-OCH3); 55.76 (C-4ʹ-OCH3).
Cardamonin (FB2): After 7 days’ transformation of 100 mg of (FB1) in the Isaria tenuipes MU35 culture the isolation yield of (FB2) was 7 mg. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 13.72 (s, 1H, C-2ʹ-OH);10.68 (s, 1H, C-4ʹ-OH); 7.83 (d, 1H, H-3, J = 15.7 Hz); 7.70–7.74 (m, 2H, H-2ʹʹ and H-6ʹʹ); 7.66 (d, 1H, H-2, J = 15.7 Hz); 7.43–7.48 (m, 3H, H-3ʹʹ, H-4ʹʹ and H-5ʹʹ); 6.02 (d, 1H, H-5ʹ, J = 2.3 Hz); 5.93 (d, 1H, H-3ʹ, J = 2.3 Hz); 3.88 (s, 3H, C-6ʹ-OCH3); (400 MHz, CDCl3) δ (ppm): 14.17 (s, 1H, C-2ʹ-OH); 8.63 (s, 1H, C-4ʹ-OH); 7.89 (d, 1H, H-3, J = 15.6 Hz); 7.77 (d, 1H, H-2, J = 15.6 Hz); 7.58–7.62 (m, 2H, H-2ʹʹ and H-6ʹʹ); 7.36–7.43 (m, 3H, H-3ʹʹ, H-4ʹʹ and H-5ʹʹ); 6.05 (d, 1H, H-5ʹ, J = 2.1 Hz); 5.97 (d, 1H, H-3ʹ, J = 2.3 Hz); 3.92 (s, 3H, C-6ʹ-OCH3).
4ʹ-O-β-D-(4″-O-methylglucopyranosyl)-cardamonin (FB3). After 7 days’ transformation of 100 mg of (FB1) in the Metarhizium robertsii MU4 culture the isolation yield of (FB3) was 42 mg. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 7.70–7.76 (m, 2H, H-2ʹʹ and H-6ʹʹ); 7.67 (d, 1H, H-2, J = 15.8 Hz); 7.60 (d, 1H, H-3, J = 15.7 Hz); 7.42–7.48 (m, 3H, H-3ʹʹ, H-4ʹʹ and H-5ʹʹ); 6.25 (d, 1H, H-5ʹ, J = 2.1 Hz); 6.21 (d, 1H, H-3ʹ, J = 2.2 Hz); 5.45 (broad s, 1H, C-2ʹʹʹ-OH); 5.31 (broad s, 1H, C-3ʹʹʹ-OH); 5.02 (d, 1H, H-1ʹʹʹ, J = 7.8 Hz); 4.76 (t,1H, C-6ʹʹʹ-OH, J = 5.4 Hz); 3.87 (s, 3H, C-6ʹ-OCH3); 3.60–3.68 (m, 1H, one of H-6ʹʹʹ); 3,47–3,54 (m, 1H, one of H-6ʹʹʹ); 3.45 (s, 3H, C-4ʹʹʹ-OCH3); 3.42–3.46 (m, 1H, H-3ʹʹʹ); 3,32–3,36 (m, 1H, H-5ʹʹʹ); 3.24 (t, 1H, H-2ʹʹʹ, J = 7.9 Hz); 3.02 (t, 1H, H-4ʹʹʹ, J = 9.2 Hz); 13C NMR (151 MHz, DMSO-d6) δ (ppm): 192.71 (C-1); 163.85 (C-2ʹ); 162.88 (C-4ʹ); 161.45 (C-6ʹ); 142.71 (C-3); 134.72 (C-1ʹʹ); 130.55 (C-4ʹʹ); 129.10 (C-2ʹʹ and C-6ʹʹ); 128.55 (C-3ʹʹ and C-5ʹʹ); 127.64 (C-2); 107.54 (C-1ʹ); 99,36 (C-1ʹʹʹ); 96.33 (C-3ʹ); 92.18 (C-5ʹ); 79.00 (C-4ʹʹʹ); 76.24 (C-3ʹʹʹ); 75.76 (C-5ʹʹʹ); 73.29 (C-2ʹʹʹ); 60.19 (C-6ʹʹʹ); 59.72 (C-4ʹʹʹ-OCH3); 56.20 (C-6ʹ-OCH3).
4ʹ-O-β-D-(4″-O-methylglucopyranosyl)-3ʹʹ-hydroxycardamonin (FB4): After 3 days’ transformation of 100 mg of (FB1) in the Beauveria bassiana KCh BBT culture the isolation yield of (FB4) was 12 mg. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 12.94 (s, 1H, C-2ʹ-OH); 9.68 (s, 1H, C-3ʹʹ-OH); 7.60 (d, 1H, H-2, J = 15.5 Hz); 7.52 (d, 1H, H-3, J = 15.7 Hz); 7.25 (t, 1H, H-5ʹʹ, J = 7.8 Hz); 7.13 (d, 1H, H-6ʹʹ, J = 7.8 Hz); 7.07 (broad s, 1H, H-2ʹʹ); 6.85 (dd, 1H, H-4ʹʹ, J = 7.8, 1.8 Hz); 6.25 (d, 1H, H-5ʹ, J = 2.1 Hz); 6.20 (d, 1H, H-3ʹ, J = 2.1 Hz); 5.46 (d, 1H, C-2ʹʹʹ-OH, J = 5.2 Hz); 5.32 (d, 1H, H-3ʹʹʹ-OH, J = 5.5 Hz); 5.02 (d, 1H, H-1ʹʹʹ, J = 7.8 Hz); 4.77 (t, 1H, C-6ʹʹʹ-OH, J = 5.5 Hz); 3.88 (s, 3H, C-6ʹ-OCH3); 3.64 (dd, 1H, one of H-6ʹʹʹ, J = 11.4, 3.6 Hz); 3.45 (s, 3H, C-4ʹʹʹ-OCH3); 3.42–3.54 (m, 2H, H-3ʹʹʹ and one of H-6ʹʹʹ); 3.32–3.36 (m, 1H, H-5ʹʹʹ); 3.23–3.28 (m, 1H, H-2ʹʹʹ); 3.19 (t, 1H, H-4ʹʹʹ, J = 9.2 Hz); 13C NMR (151 MHz, DMSO d6) δ (ppm): 192.62 (C-1); 166.21 (C-2ʹ); 162.98 (C-4ʹ); 161.54 (C-6ʹ); 157.80 (C-3ʹʹ); 142.93 (C-3); 136.00 (C-1ʹʹ); 130.15 (C-5ʹʹ); 127.37 (C-2); 119.78 (C-6ʹʹ); 117.83 (C-4ʹʹ); 114.45 (C-2ʹʹ); 107.43 (C-1ʹ); 99.35 (C-1ʹʹʹ); 96.36 (C-3ʹ); 92.23 (C-5ʹ); 79.02 (C-4ʹʹʹ); 75.78 (C-3ʹʹʹ); 75.59 (C-5ʹʹʹ); 73.30 (C-2ʹʹʹ); 60.21 (C-6ʹʹʹ); 59.75 (C-4ʹʹʹ, -OCH3); 56.22 (C-6ʹ, -OCH3).
4ʹ-O-β-D-(4″-O-methylglucopyranosyl)-4ʹʹ-hydroxycardamonin (FB5): After 7 days’ transformation of 100 mg of (FB1) in the Metarhizium robertsii MU4 culture the isolation yield of (FB5) was 11 mg. 1H NMR (400 MHz, acetone-d6) δ (ppm): 7.87 (d, 1H, H-2, J = 15.5 Hz); 7.76 (d, 1H, H-3, J = 15.5 Hz); 7.60–7.65 (m, 2H, H-2ʹʹ and H-6ʹʹ); 6.88–6.96 (m, 2H, H-3ʹʹ and H-5ʹʹ); 6.27 (d, 1H, H-5ʹ, J = 2.2 Hz); 6.18 (d, 1H, H-3ʹ, J = 2.2 Hz); 5.09 (d, 1H, H-1ʹʹʹ, J = 7.8 Hz); 4.01 (s, 3H, C-6ʹ-OCH3); 3.85 (dd, 1H, one of H-6ʹʹʹ, J = 12.0, 2.0 Hz); 3.69 (dd, 1H, one of H-6ʹʹʹ, J = 11.8, 5.0 Hz); 3.65 (t, 1H, H-3ʹʹʹ, J = 9.0 Hz); 3.57–3.60 (m, 1H, H-5ʹʹʹ); 3.56 (s, 3H, C-4ʹʹʹ-OCH3); 3.47 (dd, 1H, H-2ʹʹʹ, J = 9.1, 7.8 Hz);3.19 (dd, 1H, H-4ʹʹʹ, J = 9.5, 9.1 Hz); 13C NMR (151 MHz, acetone-d6) δ (ppm): 193.55 (C-1); 167.72 (C-2ʹ); 164.81 (C-4ʹ); 163.59 (C-6ʹ); 160.67 (C-4ʹʹ); 143.88 (C-3); 131.39 (C-2ʹʹ and C-6ʹʹ); 127.86 (C-1ʹʹ); 125.02 (C-2); 116.72 (C-3ʹʹ and C-5ʹʹ); 107.62 (C-1ʹ); 100.64 (C-1ʹʹʹ); 97.48 (C-3ʹ); 92.81 (C-5ʹ); 80.04 (C-4ʹʹʹ); 77.77 (C-3ʹʹʹ); 77.19 (C-5ʹʹʹ); 74.62 (C-2ʹʹʹ); 61.91 (C-6ʹʹʹ); 60.53 (C-4ʹʹʹ-OCH3); 56.52 (C-6ʹ-OCH3).
3ʹʹ-hydroxyflavokawain B (FB6): After 3 days’ transformation of 100 mg of (FB1) in the Beauveria caledonica KCh J3.3 culture the isolation yield of (FB6) was 1 mg. 1H NMR (400 MHz, CDCl3) δ (ppm): 14.25 (s, 1H, H-2ʹ, -OH); 7.86 (d, 1H, H-3, J = 15.6 Hz); 7.71 (d, 1H, H-2, J = 15.6 Hz); 7.28 (t, 1H, H-5ʹ, J = 7.9 Hz); 7.18 (d, 1H, H-6ʹ, J = 7.7 Hz); 7.07 (s, 1H, H-2ʹ); 6.87 (dd, 1H, H-4ʹ, J = 8.0, 1.7 Hz); 6.11 (d, 1H, H-5ʹ, J = 2.4 Hz); 5.97 (d, 1H, H-3ʹ, J = 2.4 Hz); 3.92 (s, 3H, C-6ʹ-OCH3); 3.84 (s, 3H, C-4ʹ-OCH3).
3ʹ-O-β-D-(4″-O-methylglucopyranosyl)-flavokawain B (FB7): After 7 days’ transformation of 100 mg of (FB1) in the Beauveria bassiana KCh J1.5 culture the isolation yield of (FB7) was 25 mg. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 13.35 (s, 1H, C-2ʹ-OH); 7.73 (d, 1H, H-2, J = 15.7 Hz); 7.61 (d, 1H, H-3, J = 15.7 Hz); 7.33–7.39 (m, 3H, H-2ʹʹ, 5ʹʹ and H-6ʹʹ); 7.07–7.11 (m, 1H, H-4ʹʹ); 6.17 (d, 1H, H-5ʹ, J = 2.3 Hz); 6.14 (d, 1H, H-3ʹ, J = 2.3 Hz); 5.42 (d, 1H, C-2ʹʹʹ-OH, J = 5.2 Hz); 5.28 (d, 1H, C-3ʹʹʹ-OH, J = 5.2 Hz); 4.99 (d, 1H, H-1ʹʹʹ, J = 7.8 Hz); 4.73 (dd, 1H, C-6ʹʹʹ-OH, J = 6.1, 5.2 Hz); 3.89 (s, 3H, C-6ʹ-OCH3); 3.83 (s, 3H, C-4ʹ-OCH3); 3.64 (ddd, 1H, one of H-6ʹʹʹ, J = 11.8, 5.2, 1.5 Hz); 3.52 (dd, 1H, one of H-6ʹʹʹ, J = 11.7, 5.1 Hz); 3.46 (s, 3H, C-4ʹʹʹ-OCH3); 3.40–4.48 (m, 2H, H-3ʹʹʹ and H-5ʹʹʹ); 3.26 (ddd, 1H, H-2ʹʹʹ, J = 8.9, 8.2, 5.2 Hz); 3.04 (t, 1H, H-4ʹʹʹ, J = 9.3 Hz); 13C NMR (151 MHz, DMSO-d6) δ (ppm): 192.34 (C-1); 165.66 (C-4ʹ); 165.44 (C-2ʹ); 161.95 (C-6ʹ); 157.72 (C-3ʹʹ); 142.10 (C-3); 136.18 (C-1ʹʹ); 130.07 (C-5ʹʹ); 127.86 (C-2); 122.06 (C-6ʹʹ); 118.45 (C-4ʹʹ); 115.68 (C-2ʹʹ); 106.39 (C-1ʹ); 99.89 (C-1ʹʹʹ); 93.88 (C-3ʹ); 91.17 (C-5ʹ); 79.00 (C-4ʹʹʹ); 76.28 (C-3ʹʹʹ); 75.56 (C-5ʹʹʹ); 73.44 (C-2ʹʹʹ); 60.23 (C-6ʹʹʹ); 59.70 (C-4ʹʹʹ-OCH3); 56.28 (C-6ʹ-OCH3); 55.74 (C-4ʹ-OCH3).
3ʹ-O-β-D-(4″-O-methylglucopyranosyl)-4ʹʹ-hydroxyflavokawain B (FB8): After 7 days’ transformation of 100 mg of (FB1) in the Beauveria bassiana KCh J1.5 culture the isolation yield of (FB8) was 25 mg. 1H NMR (400 MHz, DMSO-d6) δ (ppm): 13.60 (s, 1H, C-2ʹ-OH); 8.91 (s, 1H, C-4ʹʹ, -OH); 7.62 (d, 1H, H-2, J = 15.6 Hz); 7.56 (d, 1H, H-3, J = 15.6 Hz); 7.18 (d, 1H, H-2ʹʹ, J = 1.5 Hz); 7.14 (dd, 1H, H-6ʹʹ, J = 8.7, 1.5 Hz); 7.11 (d, 1H, H-5ʹʹ, J = 8.6 Hz); 6.16 (d, 1H, H-5ʹ, J = 2.3 Hz); 6.12 (d, 1H, H-3ʹ, J = 2.3 Hz); 5.45 (d, 1H, C-2ʹʹʹ-OH, J = 4.2 Hz); 5.28 (d, 1H, C-3ʹʹʹ-OH, J = 5.4 Hz); 4.99 (d, 1H, H-1ʹʹʹ, J = 7.8 Hz); 4.75 (dd, 1H, C-6ʹʹʹ-OH, J = 6.2, 5.2 Hz); 3.90 (s, 3H, C-6ʹ-OCH3); 3.82 (s, 3H, C-4ʹ-OCH3); 3.64 (ddd, 1H, one of H-6ʹʹʹ, J = 11.1, 4.6, 1.5 Hz); 3.48–3.54 (m, 1H, one of, H-6ʹʹʹ); 3.46 (s, 3H, C-4ʹʹʹ-OCH3); 3.39–4.47 (m, 2H, H-3ʹʹʹ and H-5ʹʹʹ); 3.27–3.33 (m, 1H, H-2ʹʹʹ); 3.05 (t, 1H, H-4ʹʹʹ, J = 9.2 Hz); 13C NMR (151 MHz, DMSO-d6) δ(ppm): 192.17 (C-1); 165.73 (C-4ʹ); 165.51 (C-2ʹ); 161.92 (C-6ʹ); 146.88 (C-4ʹʹ); 142.83 (C-3); 129.29 (C-1ʹʹ); 125.42 (C-2); 121.34 (C-6ʹʹ); 115.93 (C-5ʹʹ); 114.86 (C-2ʹʹ); 106.23 (C-1ʹ); 100.94 (Cʹʹʹ-1); 93.91 (C-3ʹ); 91.15 (C-5ʹ); 79.04 (C-4ʹʹʹ); 75.74 (C-3ʹʹʹ); 75.62 (C-5ʹʹʹ); 73.42 (C-2ʹʹʹ); 60.24 (C-6ʹʹʹ); 59.76 (C-4ʹʹʹ-OCH3); 56.24 (C-6ʹ-OCH3); 55.71 (C-4ʹ-OCH3).
Conclusions
In summary, the study explored the enzymatic modification of flavokawain B (FB1) within the entomopathogenic microbial cultures of B. bassiana, B. caledonica, I. fumosorosea, I. farinosa and M. robertsii strains. The investigation revealed the biotransformation of flavokawain B (FB1) into seven identified derivatives through the catalytic activities of the selected filamentous fungal strains.
The primary findings included the predominant occurrence of 4-O-methylglycosylation in the culture of described strains, each exhibiting distinct patterns of biotransformation. The biotransformation products were characterized using various analytical techniques, including nuclear magnetic resonance (NMR) spectroscopy and high-performance liquid chromatography (HPLC).
M. robertsii MU4, for instance, exhibited the formation of 4ʹ-O-β-D-(4ʹʹ-O-methylglucopyranosyl)-cardamonin (FB3) as the main product, indicating a process of demethylation and 4-O-methylglycosylation. However, in cultures of B. bassiana KCh BBT and KCh J1.5 strains, transformations within the B ring were observed; the attachment of the sugar unit was preceded by hydroxylation.
Cheminformatics tools were employed to compare the physicochemical characteristics, pharmacokinetics, and potential biological activities of flavokawain B (FB1) and its derivatives. The results indicated that the modified derivatives generally displayed lower lipophilicity than the parent compound. Moreover, the introduction of a sugar unit to the chalcone core affected the compoundsʹ absorption from the gastrointestinal tract.
Predictions from ADME factors, pharmacokinetic characteristics, and biological activities suggested that the derivatives, in most cases, exhibited comparable or enhanced activities compared to flavokawain B (FB1). Notably, the products with methylglycosyl residues were predicted to have a higher probability of anticarcinogenic activity, and all derivatives were expected to stimulate caspase 3, leading to cell apoptosis. Furthermore, the in silico studies predicted antileishmanial activity for the derivatives, aligning with the broader knowledge of chalcones and their analogs demonstrating antiprotozoal effects.
In conclusion, this study has provided noteworthy insights into the enzymatic transformations undergone by flavokawain B (FB1) through the actions of entomopathogenic filamentous fungi. It effectively elucidated the structural modifications and furnished predictions regarding the potential pharmacological activities of the resultant compounds. These findings significantly enhance our understanding of the biocatalytic capabilities inherent in these microbial cultures, emphasizing the promising therapeutic applications of the modified flavokawain B derivatives.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its Additional file.
References
Zenger K, Agnolet S, Schneider B, Kraus B. Biotransformation of Flavokawains A, B, and C, Chalcones from Kava ( Piper methysticum ), by Human Liver Microsomes. J Agric Food Chem. 2015;63:6376–85. https://doi.org/10.1021/acs.jafc.5b01858.
Singh YN. Kava: an overview. J Ethnopharmacol. 1992;37:13–45.
LaPorte E, Sarris J, Stough C, Scholey A. Neurocognitive effects of kava (Piper methysticum): a systematic review. Human Psychopharmacol Clin Experim. 2011;26:102–11. https://doi.org/10.1002/hup.1180.
Singh YN, Singh NN. Therapeutic potential of kava in the treatment of anxiety disorders. CNS Drugs. 2002;16:731–43. https://doi.org/10.2165/00023210-200216110-00002.
Sarris J, Stough C, Bousman CA, Wahid ZT, Murray G, Teschke R, et al. Kava in the treatment of generalized anxiety disorder: a double-blind, randomized placebo-controlled study. J Clin Psychopharmacol. 2013. https://doi.org/10.1097/JCP.0b013e318291be67.
Pont-Fernandez S, Kheyfets M, Rogers JM, Smith KE, Epstein DH. Kava (Piper methysticum) in the United States: the quiet rise of a substance with often subtle effects. Am J Drug Alcohol Abuse. 2023;49:85–96. https://doi.org/10.1080/00952990.2022.2140292.
Bilia AR, Gallori S, Vincieri FF. Kava-kava and anxiety: growing knowledge about the efficacy and safety. Life Sci. 2002;70:2581–97.
Hans RH, Guantai EM, Lategan C, Smith PJ, Wan B, Franzblau SG, et al. Synthesis, antimalarial and antitubercular activity of acetylenic chalcones. Bioorg Med Chem Lett. 2010;20:942–4.
Lin C-T, Jayabal Senthil Kumar K, Tseng Y-H, Wang Z-J, Pan M-Y, Xiao J-H, et al. Anti-inflammatory activity of flavokawain B from Alpinia pricei Hayata. J Agric Food Chem. 2009. https://doi.org/10.1021/jf900517d.
Alitheen NB, Abu N, Mohamad NE, Yeap SK, Lim KL, Akhtar MN, et al. In vivo antitumor and antimetastatic effects of flavokawain B in 4T1 breast cancer cell-challenged mice. Drug Des Devel Ther. 2015;9:1401.
Zhu Y, Fan W, Wang Y, Ding H, Yang S, He F, Flavokawain B. Weakens gastric cancer progression via the TGF-β1/SMAD4 pathway and attenuates m2 macrophage polarization. J Immunol Res. 2022;2022:1–22.
Kuo Y-F, Su Y-Z, Tseng Y-H, Wang S-Y, Wang H-M, Chueh PJ, Flavokawain B. Novel chalcone from Alpinia pricei Hayata with potent apoptotic activity: Involvement of ROS and GADD153 upstream of mitochondria-dependent apoptosis in HCT116 cells. Free Radic Biol Med. 2010;49:214–26.
Hseu Y-C, Chiang Y-C, Vudhya Gowrisankar Y, Lin K-Y, Huang S-T, Shrestha S, et al. The in vitro and in vivo anticancer properties of chalcone flavokawain B through Induction of ROS-mediated apoptotic and autophagic cell death in human melanoma cells. Cancers. 2020;12:2936.
Sakai T, Guo Y, Ramez E, Mefford J, Hopkins J, Zi X, et al. Abstract 4241: Flavokawain B, a kava chalcone, induces apoptosis in synovial sarcoma cell lines. Cancer Res. 2011;71:4241–4241.
Abu N, Akhtar MN, Yeap SK, Lim KL, Ho WY, Zulfadli AJ, et al. Induces apoptosis in MCF-7 and MDA-MB231 and inhibits the metastatic process. PLoS One. 2014;9:105244. https://doi.org/10.1371/journal.pone.0105244.
Ji T, Lin C, Krill LS, Eskander R, Guo Y, Zi X, et al. A kava chalcone, inhibits growth of human osteosarcoma cells through G2/M cell cycle arrest and apoptosis. Mol Cancer. 2013;12:55. https://doi.org/10.1186/1476-4598-12-55.
Fei Y, Shao Y, Wang W, Cheng Y, Yu B, He X, et al. Biosynthesis of three chalcone β-D-glucosides by glycosyltransferase from Bacillus subtilis ATCC 6633. Microbiol Biotechnol Lett. 2021;49:174–80. https://doi.org/10.8022/mbl.2012.02010.
Budziak I, Arczewska M, Kamiński DM. Structure and physical properties of cardamonin: a spectroscopic and computational approach. Molecules. 2020;25:4070.
Reddy MR, Aidhen IS. Natural and synthetic glycosylated chalcones as promising bioactive compounds. J Indian Chem Soc. 2021;97:109.
Krawczyk-Łebek A, Dymarska M, Janeczko T, Kostrzewa-Susłow E. Glycosylation of methylflavonoids in the cultures of Entomopathogenic Filamentous fungi as a tool for obtaining new biologically active compounds. Int J Mol Sci. 2022;23:5558.
Manach C, Morand C, Crespy V, Demigné C, Texier O, Régérat F, et al. Quercetin is recovered in human plasma as conjugated derivatives which retain antioxidant properties. FEBS Lett. 1998;426:331–6. https://doi.org/10.1016/S0014-5793%2898%2900367-6.
Cermak R, Landgraf S, Wolffram S. The bioavailability of quercetin in pigs depends on the glycoside moiety and on dietary factors. J Nutr. 2003;133:2802–7.
Day AJ, DuPont MS, Ridley S, Rhodes M, Rhodes MJC, Morgan MRA, et al. Deglycosylation of flavonoid and isoflavonoid glycosides by human small intestine and liver β-glucosidase activity. FEBS Lett. 1998;436:71–5. https://doi.org/10.1016/S0014-5793%2898%2901101-6.
Reinboth M, Wolffram S, Abraham G, Ungemach FR, Cermak R. Oral bioavailability of quercetin from different quercetin glycosides in dogs. British Journal of Nutrition. 2010;104:198–203.
Lin B, Tao Y. Whole-cell biocatalysts by design. Microb Cell Fact. 2017;16:106. https://doi.org/10.1186/s12934-017-0724-7.
Ladkau N, Schmid A, Bühler B. The microbial cell—functional unit for energy dependent multistep biocatalysis. Curr Opin Biotechnol. 2014;30:178–89.
Lin B-X, Zhang Z-J, Liu W-F, Dong Z-Y, Tao Y. Enhanced production of N-acetyl-d-neuraminic acid by multi-approach whole-cell biocatalyst. Appl Microbiol Biotechnol. 2013;97:4775–84. https://doi.org/10.1007/s00253-013-4754-8.
Łużny M, Tronina T, Kozłowska E, Kostrzewa-Susłow E, Janeczko T. Biotransformation of 5,7-methoxyflavones by selected Entomopathogenic Filamentous Fungi. J Agric Food Chem. 2021;69:3879–86.
Perz M, Krawczyk-Łebek A, Dymarska M, Janeczko T, Kostrzewa-Susłow E. Biotransformation of Flavonoids with -NO2, -CH3 Groups and -Br, -Cl Atoms by Entomopathogenic Filamentous Fungi. Int J Mol Sci. 2023;2023(24):9500.
Krawczyk-Łebek A, Dymarska M, Janeczko T, Kostrzewa-Susłow E. Fungal biotransformation of 2′-methylflavanone and 2′-methylflavone as a method to obtain glycosylated derivatives. Int J Mol Sci. 2021;22:9617.
Krawczyk-Łebek A, Dymarska M, Janeczko T, Kostrzewa-Susłow E. Entomopathogenic Filamentous Fungi as biocatalysts in glycosylation of methylflavonoids. Catalysts. 2020;10:1148.
Huszcza E, Dmochowska-Gładysz J, Bartmańska A. Transformations of steroids by Beauveria bassiana. Zeitschrift für Naturforschung C. 2005;60:103–8. https://doi.org/10.1515/znc-2005-1-219/html.
Gonzalez R, Nicolau F, Peeples T. N-alkane solvent-enhanced biotransformation of steroid DHEA by Beauveria bassiana as biocatalyst. J Adv Biol Biotechnol. 2015;2:30–7.
Kozłowska E, Urbaniak M, Hoc N, Grzeszczuk J, Dymarska M, Stępień Ł, et al. Cascade biotransformation of dehydroepiandrosterone (DHEA) by Beauveria species. Sci Rep. 2018;8:13449.
Feng M, Liao Z, Han L, Li J, Ye L. Enhancement of microbial hydroxylation of 13-ethyl-gon-4-ene-3,17-dione by Metarhizium anisopliae using nano-liposome technique. J Ind Microbiol Biotechnol. 2014;41:619–27.
Xiao G, Ying S-H, Zheng P, Wang Z-L, Zhang S, Xie X-Q, et al. Genomic perspectives on the evolution of fungal entomopathogenicity in Beauveria bassiana. Sci Rep. 2012;2:483.
Strugała P, Tronina T, Huszcza E, Gabrielska J. Bioactivity in vitro of quercetin glycoside obtained in Beauveria bassiana culture and its interaction with liposome membranes. Molecules. 2017;22:1520.
Cannell RJP, Rashid T, Ismail IM, Sidebottom PJ, Knaggs AR, Marshall PS. Novel metabolites of warfarin produced by Beauveria bassiana and Streptomyces rimosus a novel application of hplc nmr. Xenobiotica. 1997;27(147):57. https://doi.org/10.1080/004982597240659.
Ibrahim AK, Radwan MM, Ahmed SA, Slade D, Ross SA, Elsohly MA, et al. Microbial metabolism of cannflavin A and B isolated from Cannabis sativa. Phytochemistry. 2010;71:1014–9.
Huszcza E, Bartman´ska A, Bartman´ska B, Tronina T. Glycosylation of Xanthohumol by Fungi [Internet]. Z. Naturforsch. 2008.
Krawczyk-Łebek A, Dymarska M, Janeczko T, Kostrzewa-Susłow E. New glycosylated dihydrochalcones obtained by biotransformation of 2′-hydroxy-2-methylchalcone in cultures of Entomopathogenic Filamentous Fungi. Int J Mol Sci. 2021;22:9619.
Łużny M, Tronina T, Kozłowska E, Dymarska M, Popłoński J, Łyczko J, et al. Biotransformation of methoxyflavones by selected Entomopathogenic Filamentous Fungi. Int J Mol Sci. 2020;21:6121.
Łużny M, Tronina T, Kozłowska E, Kostrzewa-Susłow E, Janeczko T. Biotransformation of 5,7-methoxyflavones by selected entomopathogenic Filamentous Fungi. J Agric Food Chem. 2021;69:3879–86. https://doi.org/10.1021/acs.jafc.1c00136.
Kozłowska E, Hoc N, Sycz J, Urbaniak M, Dymarska M, Grzeszczuk J, et al. Biotransformation of steroids by entomopathogenic strains of Isaria farinosa. Microb Cell Fact. 2018;17:71. https://doi.org/10.1186/s12934-018-0920-0.
Panek A, Wójcik P, Świzdor A, Szaleniec M, Janeczko T. Biotransformation of Δ1-progesterone using selected Entomopathogenic Filamentous fungi and prediction of its products’ bioactivity. Int J Mol Sci. 2023;25:508.
Tronina T, Łużny M, Dymarska M, Urbaniak M, Kozłowska E, Piegza M, et al. Glycosylation of quercetin by selected entomopathogenic Filamentous Fungi and prediction of its products’ bioactivity. Int J Mol Sci. 2023;24:11857.
Dymarska M, Janeczko T, Kostrzewa-Susłow E. Glycosylation of methoxylated flavonoids in the cultures of Isaria fumosorosea KCH J2. Molecules. 2018;23:2578.
Dou F, Wang Z, Li G, Dun B. Microbial transformation of flavonoids by Isaria fumosorosea ACCC 37814. Molecules. 2019;24:1028.
Krawczyk-Łebek A, Dymarska M, Janeczko T, Kostrzewa-Susłow E. 4′-Methylflavanone glycosides obtained using biotransformation in the Entomopathogenic Filamentous fungi cultures as potential anticarcinogenic, antimicrobial, and hepatoprotective agents. Int J Mol Sci. 2022;23:5373.
Dymarska M, Janeczko T, Kostrzewa-Susłow E. Glycosylation of 3-hydroxyflavone, 3-methoxyflavone, quercetin and Baicalein in Fungal cultures of the genus Isaria. Molecules. 2018;23:2477.
Tronina T, Bartmańska A, Milczarek M, Wietrzyk J, Popłoński J, Rój E, et al. Antioxidant and antiproliferative activity of glycosides obtained by biotransformation of xanthohumol. Bioorg Med Chem Lett. 2013;23:1957–60.
Łużny M, Kaczanowska D, Gawdzik B, Wzorek A, Pawlak A, Obmińska-Mrukowicz B, et al. Regiospecific hydrogenation of bromochalcone by unconventional yeast strains. Molecules. 2022;27:3681.
Cao H, Chen X, Jassbi AR, Xiao J. Microbial biotransformation of bioactive flavonoids. Biotechnol Adv. 2015;33:214–23.
Dymarska M, Janeczko T, Kostrzewa-Susłow E. Biotransformations of flavones and an Isoflavone (Daidzein) in cultures of Entomopathogenic Filamentous Fungi. Molecules. 2018;23:1356.
Xie L, Zhang L, Wang C, Wang X, Xu Y, Yu H, et al. Methylglucosylation of aromatic amino and phenolic moieties of drug-like biosynthons by combinatorial biosynthesis. Proc Natl Acad Sci. 2018;115:E4980-9. https://doi.org/10.1073/pnas.1716046115.
Zhan J, Leslie Gunatilaka AA. Selective 4′- O -methylglycosylation of the pentahydroxy-flavonoid quercetin by Beauveria bassiana ATCC 7159. Biocatal Biotransformation. 2006;24:396–9. https://doi.org/10.1080/10242420600792169.
Sordon S, Popłoński J, Tronina T, Huszcza E. Microbial glycosylation of daidzein, genistein and biochanin a: two new glucosides of biochanin A. Molecules. 2017;22:81.
Sordon S, Popłoński J, Tronina T, Huszcza E. Regioselective O-glycosylation of flavonoids by fungi Beauveria bassiana, Absidia coerulea and Absidia glauca. Bioorg Chem. 2019;93:102750.
Dymarska M, Grzeszczuk J, Urbaniak M, Janeczko T, Pląskowska E, Stępień Ł, et al. Glycosylation of 6-methylflavone by the strain Isaria fumosorosea. PLoS One. 2017;12:0184885. https://doi.org/10.1371/journal.pone.0184885.
Yap Li Ching A, Sook Wah T, Aspollah Sukari M, Ee Cheng Lian G, Rahmani M, Khalid K. Characterization of flavonoid derivatives from Boesenbergia rotunda (L). The Malaysian Journal of Analytical Sciences. 2007.
Łużny M, Kozłowska E, Kostrzewa-Susłow E, Janeczko T. Highly effective, regiospecific hydrogenation of methoxychalcone by Yarrowia lipolytica enables production of food Sweeteners. Catalysts. 2020;10:1135.
Palko-Łabuz A, Kostrzewa-Susłow E, Janeczko T, Środa-Pomianek K, Poła A, Uryga A, et al. Cyclization of flavokawain B reduces its activity against human colon cancer cells. Hum Exp Toxicol. 2020;39:262–75. https://doi.org/10.1177/0960327119882986.
Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717.
Higgs J, Wasowski C, Marcos A, Juki M, Pav CH, Gobec S, et al. Chalcone derivatives: synthesis, in vitro and in vivo evaluation of their anti-anxiety, anti-depression and analgesic effects. Heliyon. 2019. https://doi.org/10.1016/j.heliyon.2019.e01376.
Sudevan ST, Oh JM, Abdelgawad MA, Abourehab MAS, Rangarajan TM, Kumar S, et al. Introduction of benzyloxy pharmacophore into aryl/heteroaryl chalcone motifs as a new class of monoamine oxidase B inhibitors. Sci Rep. 2022;12:22404.
Hara H, Ikeda R, Ninomiya M, Kamiya T, Koketsu M, Adachi T. Newly synthesized ‘Hidabeni’ Chalcone derivatives potently suppress LPS-induced no production <i>via</i> Inhibition of STAT1, but Not NF-κB, JNK, and p38, Pathways in Microglia. Biol Pharm Bull. 2014;37:1042–9.
Petit C, Ceccarelli M, Cretton S, Houriet J, Skalicka-Woźniak K, Christen P, et al. Passive intestinal absorption of representative plant secondary metabolites: a physicochemical study. Planta Med. 2017;83:718–26. https://doi.org/10.1055/s-0043-101915.
Deodhar M, Al Rihani SB, Arwood MJ, Darakjian L, Dow P, Turgeon J, et al. Mechanisms of CYP450 inhibition: understanding drug-drug interactions due to mechanism-based inhibition in clinical practice. Pharmaceutics. 2020;12:846.
Ogu CC, Maxa JL. Drug interactions due to cytochrome P450. Baylor Univ Med Center Proc. 2000;13:421–3. https://doi.org/10.1080/08998280.2000.11927719.
De la Garza-Salazar F, Colunga-Pedraza PR, Gómez-Almaguer D. Cytochrome P450 inhibition to decrease dosage and costs of venetoclax and ibrutinib: a proof-of-concept case study. Br J Clin Pharmacol. 2023;89:898–902. https://doi.org/10.1111/bcp.15590.
Lynch T, Price A. The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician. 2007;76:391–6.
Skolnik S, Lin X, Wang J, Chen X-H, He T, Zhang B. Towards prediction of in vivo intestinal absorption using a 96-well caco-2 assay. J Pharm Sci. 2010;99:3246–65.
Wang X-X, Liu G-Y, Yang Y-F, Wu X-W, Xu W, Yang X-W. Intestinal absorption of triterpenoids and flavonoids from Glycyrrhizae radix et rhizoma in the human caco-2 monolayer cell model. Molecules. 2017;22:1627.
Tian X-J, Yang X-W, Yang X, Wang K. Studies of intestinal permeability of 36 flavonoids using Caco-2 cell monolayer model. Int J Pharm. 2009;367:58–64.
Michalkova R, Mirossay L, Kello M, Mojzisova G, Baloghova J, Podracka A, et al. Anticancer potential of natural chalcones in vitro and in vivo evidence. Int J Mol Sci. 2023. https://doi.org/10.3390/ijms241210354.
Leite FF, de Sousa NF, de Oliveira BHM, Duarte GD, Ferreira MDL, Scotti MT, et al. Anticancer activity of chalcones and its derivatives: review and in silico studies. Molecules. 2023;28:4009.
Michalkova R, Kello M, Cizmarikova M, Bardelcikova A, Mirossay L, Mojzis J. Chalcones and gastrointestinal cancers: experimental evidence. Int J Mol Sci. 2023;24:5964.
Yagura T, Motomiya T, Ito M, Honda G, Iida A, Kiuchi F, et al. Anticarcinogenic compounds in the Uzbek medicinal plant Helichrysum maracandicum. J Nat Med. 2008;62:174–8. https://doi.org/10.1007/s11418-007-0223-y.
Orlikova B, Schnekenburger M, Zloh M, Golais F, Diederich M, Tasdemir D. Natural chalcones as dual inhibitors of HDACs and NF-κB. Oncol Rep. 2012;28:797–805. https://doi.org/10.3892/or.2012.1870.
Suzuki K, Hyun Seung B, Soon Sung L, Sanghyun L, Sang Hoon J, Yeon Sil L, et al. The mechanism of anti-inflammatory activity of 2’-hydroxychalcone derivatives. Ensho Saisei. 2005;25:130–6.
Kim N, Pae H, Oh G, Kang T, Kim Y, Rhew H, et al. Butein, a plant polyphenol, induces apoptosis concomitant with increased caspase-3 activity, decreased Bcl-2 expression and increased bax expression in HL-60 cells. Pharmacol Toxicol. 2001;88:261–6. https://doi.org/10.1111/j.1600-0773.2001.880507.x.
Kim H-G, Oh H-J, Ko J-H, Song HS, Lee Y-G, Kang SC, et al. Lanceoleins A-G, hydroxychalcones, from the flowers of Coreopsis lanceolata and their chemopreventive effects against human colon cancer cells. Bioorg Chem. 2019;85:274–81.
Das M, Manna K. Chalcone scaffold in anticancer armamentarium: a molecular insight. J Toxicol. 2016;2016:1–14.
Kobori M, Iwashita K, Shinmoto H, Tsushida T. Phloretin-induced apoptosis in B16 melanoma 4A5 cells and HL60 human leukemia cells. Biosci Biotechnol Biochem. 1999;63:719–25. https://doi.org/10.1271/bbb.63.719.
Boeck P, Bandeira Falcão CA, Leal PC, Yunes RA, Filho VC, Torres-Santos EC, et al. Synthesis of chalcone analogues with increased antileishmanial activity. Bioorg Med Chem. 2006;14:1538–45.
Zheoat AM, Alenezi S, Kotb Elmahallawy E, Ungogo MA, Alghamdi AH, Watson DG, et al. Antitrypanosomal and Antileishmanial activity of chalcones and flavanones from polygonum salicifolium. Pathogens. 2021. https://doi.org/10.3390/pathogens10020175.
Osman MS, Awad TA, Shantier SW, Garelnabi EA, Osman W, Mothana RA, et al. Identification of some chalcone analogues as potential antileishmanial agents: an integrated in vitro and in silico evaluation. Arabian J Chem. 2022;15:103717.
Roussaki M, Costa Lima S, Kypreou A-M, Kefalas P, Cordeiro da Silva A, Aurones DA. A promising heterocyclic scaffold for the development of potent antileishmanial agents. Int J Med Chem. 2012;2012:1–8.
Acknowledgements
The article is part of a PhD dissertation titled: “Chemo-enzymatic synthesis of dihydrochalcones glycosides and their health promoting activity”, prepared during Doctoral School at the Wrocław University of Environmental and Life Sciences.
Funding
This work was financed by the (Polish) National Science Centre, Grant No.2021/43/O/NZ7/01517. The APC is financed by Wrocław University of Environmental and Life Sciences.
Author information
Authors and Affiliations
Contributions
PC, MU, EK and TJ conceived and designed the experiments; MU, EK, MD, ŁS and TJ performed microbiological tests; PC, TT and TJ performed the chemical synthesis; PC performed the biotransformations; PC and TT performed quantification of biotransformation; TJ and EKS analyzed the spectral data; PC, TT, MU and TJ interpreted the results; PC, MD and TJ wrote and edited the manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Supplementary Figures S1–S64.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chlipała, P., Tronina, T., Dymarska, M. et al. Multienzymatic biotransformation of flavokawain B by entomopathogenic filamentous fungi: structural modifications and pharmacological predictions. Microb Cell Fact 23, 65 (2024). https://doi.org/10.1186/s12934-024-02338-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-024-02338-9