
Abstract
Background
Antibiotics biosynthesis is usually regulated by the cluster-situated regulatory gene(s) (CSRG(s)), which directly regulate the genes within the corresponding biosynthetic gene cluster (BGC). Previously, we have demonstrated that LmbU functions as a cluster-situated regulator (CSR) of lincomycin. And it has been found that LmbU regulates twenty non-lmb genes through comparative transcriptomic analysis. However, the regulatory mode of CSRs’ targets outside the BGC remains unknown.
Results
We screened the targets of LmbU in the whole genome of Streptomyces lincolnensis and found fourteen candidate targets, among which, eight targets can bind to LmbU by electrophoretic mobility shift assays (EMSA). Reporter assays in vivo revealed that LmbU repressed the transcription of SLINC_0469 and SLINC_1037 while activating the transcription of SLINC_8097. In addition, disruptions of SLINC_0469, SLINC_1037, and SLINC_8097 promoted the production of lincomycin, and qRT-PCR showed that SLINC_0469, SLINC_1037, and SLINC_8097 inhibited transcription of the lmb genes, indicating that all the three regulators can negatively regulate lincomycin biosynthesis.
Conclusions
LmbU can directly regulate genes outside the lmb cluster, and these genes can affect both lincomycin biosynthesis and the transcription of lmb genes. Our results first erected the cascade regulatory circuit of LmbU and regulators outside lmb cluster, which provides the theoretical basis for the functional research of LmbU family proteins.
Similar content being viewed by others

Introduction
Streptomycetes are high G+C, filamentous Gram-positive bacteria. In order to cope with the complex and changeable living environment, streptomycetes evolved a set of protective mechanisms with competitive advantages [1], producing a large number of secondary metabolites, such as antibiotics with high medical value [2, 3]. Antibiotic biosynthesis is stringently controlled by precise and pyramidal regulatory cascades [4]. Streptomyces can monitor the environmental conditions, growing states, population density, and so on, and then secrete and sense specific signal small molecules named autoregulators, including γ-butyrolactones (GBLs), antibiotics and biosynthetic intermediates [5,6,7]. Then, the receptors of autoregulators respond and transmit these signal inputs to corresponding transcriptional regulators of secondary metabolite, thereby regulating antibiotics biosynthesis. Transcriptional regulators in Streptomyces are usually classified as global/pleiotropic regulators and CSRs [8]. The global/pleiotropic regulators can not only regulate the biosynthesis of secondary metabolites, but also affect the morphological differentiation of Streptomyces [9, 10]. The BGC of each antibiotic usually includes one or more CSRs, which are at the bottom of the secondary metabolic regulatory network, and directly regulate transcription of the corresponding antibiotics biosynthetic genes, thereby regulating antibiotics biosynthesis [11, 12].
Many studies have shown that the targets of CSRs are not limited to the gene cluster in which they are situated, they may also located in disparate antibiotic BGCs, forming cross-regulation [13]. For instance, FscRI regulates candicidin biosynthesis as well as antimycin biosynthesis in Streptomyces albus S4 [14]. GdmRIII up-regulates the production of geldanamycin and down-regulates that of elaiophylin by affecting transcription of the genes in both gene clusters in Streptomyces autolyticus CGMCC0516 [15]. JadR1 can not only activate jadomycin biosynthesis by directly binding to the promoter region of jadJ, but also repress chloramphenicol biosynthesis by directly binding to the promoter region of cmlJ in chloramphenicol BGC in Streptomyces venezuelae [16, 17]. Similar examples are also found in coordinated cephamycin C and clavulanic acid biosynthesis regulated by CcaR in Streptomyces clavuligerus [18], RED, ACT and CDA biosynthesis regulated by RedZ in Streptomyces coelicolor [19], avermectin and oligomycin biosynthesis regulated by AveR in Streptomyces avermitilis [20], and rapamycin and elaiophylin biosynthesis regulated by RapH in Streptomyces rapamycinicus [11]. Though cross-regulation of disparate antibiotics by one CSR has been reported many times, screening of the targets of CSRs outside the BGSs is barely reported. Li et al. [21] showed that NemR functions as a pleiotropic regulator, which not only activates transcription of the genes within nemadectin BGC, but also regulates four targets outside the BGC in Streptomyces cyaneogriseus ssp. noncyanogenus.
Lincomycin, one of the lincosamide antibiotics, was isolated from a soil-derived Gram-positive bacterium Streptomyces lincolnensis in 1962 [22]. The 32-kb BGC of lincomycin (lmb) contains twenty-five structural genes [23, 24], two resistance genes [25], one dual-functional gene [26] and one CSR [27]. Along with the complete elucidation on the biosynthesis route of lincomycin [23, 24], several studies have been reported to explore the regulation mechanism of lincomycin biosynthesis. For the first time, we identified a novel CSR LmbU, which positively regulates lincomycin biosynthesis [27, 42]. Subsequently, several other regulators that are located outside the lmb cluster and modulate lincomycin biosynthesis, including AdpA, BldA, BldD, GlnR, RamR, AflQ1-Q2, Rex, SLCG_2919, SLCG_Lrp, σL and AtrA etc., have been characterized [28,29,30,31,32,33,34,35,36,37,38,39,40].
In our previous study, we characterized LmbU as a CSR of lincomycin, and demonstrated that LmbU homologues are widely found in actinomycetes, indicating LmbU might regulate other target genes except lmb genes. Here, we demonstrated that LmbU functions as a pleiotropic regulator, which negatively regulates transcription of SLINC_0469 and SLINC_1037, while positively regulates transcription of SLINC_8097. In addition, we showed that SLINC_0469, SLINC_1037 and SLINC_8097 can all inhibit the production of lincomycin by repressing transcription of lmb genes.
Material and methods
Bacterial strains, plasmids and culture conditions
Bacterial strains and plasmids used in this study are listed in Table 1. Escherichia coli strains and S. lincolnensis strains were described in our previous study [27]. Moreover, YEME medium (10 g/L yeast extract, 5 g/L polypeptone, 10 g/L glucose, 3 g/L malt extract, 5 mM MgCl2•2H2O, 340 g/L sucrose) was used for preparation of S. lincolnensis mycelium for conjugation, ISP4 medium (10 g/L soluble starch, 1 g/L K2HPO4, 5 g/L MgSO4•7H2O, 1 g/L NaCl, 2 g/L (NH4)2SO4, 2 g/L CaCO3, 15 g/L Agar, 0.001 g/L FeSO4•7H2O, 0.001 g/L MnCl2•4H2O, 0.001 g/L ZnSO4•7H2O, 0.02 M MgCl2) was used for conjugation of E. coli and S. lincolnensis.
Expression and purification of His6-LmbU
The expression plasmid pLU-1 [27] was transformed into E. coli BL21 (DE3), and used for His6-LmbU expression. The strain was cultivated in 100 mL LB medium at 37 ℃ until OD600 reached about 0.6, then 1 mM Isopropyl β-D-1-thiogalactopyranoside was added. After overnight cultivation at 16 ℃, the cells were washed twice and suspended in PBS buffer (0.1 M phosphate buffer solution, pH 7.5). Total proteins were released by sonication and His6-LmbU was purified using nickeliminodiacetic acid–agarose chromatography (Weishibohui, Beijing, China). After dialysis and concentration, the purified protein was stored in binding buffer (10 mM Tris–HCl [pH 8.0], 1 mM EDTA, 0.2 mM dithiothreitol, 20 μg/mL bovine serum albumin, 1.2% glycerol).
Electrophoretic mobility shift assay (EMSA)
DNA probes of around 200 bp containing the binding sites of LmbU were amplified via two rounds of PCR. Firstly, primer pairs UBS-X-F/R (X indicates the numbers of the 14 putative targets of LmbU) were used to amplify the cold probes without biotin. Then, biotin-labeled primer EMSA-B* was used for the second-round PCR to generate the labeled probes. The probe prepared by primer pair nag-F/R was used as a negative control. EMSAs were performed as described previously [27] using chemiluminescent EMSA kits (Beyotime, Shanghai, China) with some modification in binding buffer, which included 10 mM Tris–HCl (pH 8.0), 1 mM EDTA, 0.2 mM dithiothreitol, 20 g/mL bovine serum albumin, 1.2% glycerol, and 50 μg/mL poly(dI-dC) [27]. EMSAs performed with 200-fold excesses of specific or nonspecific cold probes were added as controls to confirm the specificity of the band shifts.
All primer pairs used in this study are listed in Additional file 1: Table S1.
Construction of lmbU disruption strain ΔlmbU
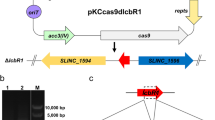
To construct a lmbU disruption strain, the internal region of lmbU (465 bp) was deleted via a CRISPR/Cas9-based genetic editing method [41]. The lmbU-specific single-molecule-guide RNA (sgRNA) was amplified by PCR using the primer pair sgUF/R with pKCcas9dO as template. Upstream (1.2 kb) and downstream (1.2 kb) homologous arms of lmbU were amplified by PCR using primer pairs uU-F/R and dU-F/R, respectively. The lmbU-specific deletion cassette was assembled with the above three DNA fragments by using overlapping PCR. Subsequently, the deletion cassette was digested with SpeI and HindIII (Thermo Fisher, Waltham, MA, USA), and ligated into the corresponding sites of pKCcas9dO. The resulting plasmid pKCcas9dlmbU was introduced into S. lincolnensis NRRL 2936 by conjugation, using E. coli S17-1 as a donor. The conjugants were selected with nalidixic acid and apramycin, and then identified by PCR using the primer pair JDU-F/R and DNA sequencing. The pKCcas9dlmbU plasmid was eliminated through a few rounds of streak cultivation in YEME medium at 37 ℃, which was identified by PCR using the primer pair CR1/2.
Catechol dioxygenase activity analysis
The regions upstream (relative to the translational start site) of SLINC_0469 (−578 to −1), SLINC_1037 (−471 to −1) and SLINC_8097 (−427 to −1) were amplified using primer pairs p0469-F/R, p1037-F/R and p8097-F/R respectively. The reporter genes xylTE were amplified by PCR using primer pair pAxyl-3/4, with pATE152 as a template. Two DNA fragments (promoter region and reporter gene) were cloned into the PvuII (Thermo Fisher) site of the integrative plasmid pSET152 using Super Efficiency Fast Seamless Cloning kits (DoGene, Shanghai, China), resulting in reporter plasmids p0469TE, p1037TE and p8097TE. Then, the reporter plasmids were transferred into the wild-type strain NRRL 2936 and the lmbU disruption strain ΔlmbU, to construct the reporter strains WT/p0469TE, WT/p1037TE, WT/p8097TE, ΔlmbU/p0469TE, ΔlmbU/p1037TE and ΔlmbU/p8097TE.
Catechol dioxygenase activity analysis was performed as described previously [28]. Briefly, the reporter strains were cultivated in YEME medium at 28 ℃ for 1 day, then the cells were harvested and lysed by sonication. An appropriate amount of cell extract was added to the assay buffer (100 mM potassium phosphate [pH 7.5], 1 mM catechol), and the optical density at 375 nm was detected per minute. The rate of change per minute per milligram of absorbance was calculated as catechol dioxygenase activity.
Bioinformatics analysis (Functional domain analysis, sequence alignment and structure modeling)
Functional domain analysis was performed by BlastP in National Center for Biotechnology Information (NCBI) (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastp&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome). Sequence alignment was analyzed using the online software ESPript 3.0 (https://espript.ibcp.fr/ESPript/cgi-bin/ESPript.cgi). Structure modeling was constructed using the online software SWISS MODEL (https://swissmodel.expasy.org/interactive).
Construction of SLINC_0469, SLINC_1037 and SLINC_8097 disruption, complementation and overexpression strains.
To construct the SLINC_0469 disruption strain (ΔSLINC_0469), the same CRISPR/Cas9-based genetic editing method was carried out as construction of ΔlmbU with some modification in construction of disruption plasmids. For instance, to construct ΔSLINC_0469, the upstream and downstream homologous arms of SLINC_0469 were amplified by PCR using primer pairs u-0469-F/R and d-0469-F/R, respectively. Specific sgRNA was added to upstream homologous arm by PCR using the primer pair sg-0469/u-0469-R. The above two DNA fragments (sgRNA-containing upstream and downstream homologous arms) were cloned into the SpeI/HindIII (Thermo Fisher) sites of pKCcas9dO using Super Efficiency Fast Seamless Cloning kits (DoGene), resulting in disruption plasmid pKCcas9d0469. The primer pair JD-0469-F/R was used to identify the conjugants selected with nalidixic acid and apramycin, and the primer pair CR1/2 was used to verify the elimination of the disruption plasmid.
To construct SLINC_0469 complementation and overexpression strains, the SLINC_0469 expression cassette containing SLINC_0469p promoter and SLINC_0469 encoding gene was amplified by PCR using the primer pair p0469-F/0469-R. The obtained cassette was cloned into the PvuII (Thermo Fisher) sites of pIB139 by using Super Efficiency Fast Seamless Cloning kits (DoGene). The resulting plasmid pIBpN0469 was introduced into ΔSLINC_0469 by conjugation, resulting in SLINC_0469 complementation strain CSLINC_0469 and ocerexpression strain OSLINC_0469.
Constructions of SLINC_1037 and SLINC_8097 disruption, complementation and overexpression strains were carried out as the similar procedures of SLINC_0469.
Lincomycin bioassay analysis
Lincomycin bioassay analysis was carried out as described in our previous work [27]. FM2 medium (20 g/L lactose, 20 g/L glucose, 10 g/L corn steep liquor, 10 g/L polypeptone, 4 g/L CaCO3, pH 7.0) was used for fermentation cultivation. Micrococcus luteus 28001 was used as an indicator strain, and the concentrations of samples were measured according to the lincomycin standard curves.
Three biological independent experiments were done for the analytical procedures. Error bars indicated means ± standard deviations.
RNA extraction and quantitative real-time PCR (qRT-PCR)
The strains were cultured in FM2 medium for 2 days, and then RNA was extracted by the method using TRIzol (Thermo Fisher) [30]. The trace amount of DNA was removed through incubation with RNase-free DNase I (TaKaRa, Dalian, China) at 28 ℃, and the obtained RNA was analyzed using NanoDrop 2000 (Thermo Fisher). 1 μg RNA was used to synthesize the cDNA using reverse transcription M-MLV (RNase-free) kits (TaKaRa). qRT-PCR was performed with SYBR green PCR master mix (ToYoBo, Shanghai, China) as described previously [27]. PCR was carried out in triplicate for each sample. The transcriptional level of hrdB was used as a positive internal control to normalize the transcriptional levels of target genes, which were measured by the threshold cycle (2−ΔΔCT) method [44].
Results
Potential targets of LmbU were found in the genome of S. lincolnensis
We have previously demonstrated that the conserved palindrome sequence 5ʹ-CGCCGGCG-3ʹ allows LmbU to bind to the promoter regions of lmbA and lmbW [27]. To explore the potential regulatory targets of LmbU and reduce non-specific bindings, we conducted a genome-wide scan of S. lincolnensis using the extended motif (5ʹ-TCGCCGGCGA-3ʹ) derived from the binding motif of the lmbA promoter region. A total of 176 conserved sequences were found throughout the genome, among which 54 were located in the potential regulatory regions (– 600 to + 100 bp relative to the putative translational start site, and not located inside the operon). Subsequently, 14 candidate targets which may be relevant to lincomycin biosynthesis were selected, including 4 regulators, 5 transporters or resistance-related proteins, 2 σ factors, and 3 other functional proteins (Table 2).
LmbU binds to the promoter regions of 8 target genes directly
In order to investigate whether LmbU can bind to the above 14 targets, EMSAs were carried out with purified His6-LmbU and the DNA probes of candidate targets. The results showed that His6-LmbU could obviously bind to the promoter regions of 8 genes in a concentration-dependent manner (Fig. 1). The deduced products of the 8 target genes were as follows: LAL family transcriptional regulator (encoded by SLINC_0469), AcoR family transcriptional regulator (encoded by SLINC_1037), AraC family transcriptional regulator (encoded by SLINC_8097), MFS transporter (encoded by SLINC_0585), ABC transporter permease (encoded by SLINC_6382), MFS transporter (encoded by SLINC_7298), RNA polymerase σ factor (encoded by SLINC_6570) and methyltransferase (encoded by SLINC_1077).

Identification of the binding activities of LmbU to the putative targets. Biotin-labeled probes (5 ng) were incubated with His6-LmbU of increasing concentrations (0, 3.2, 6.4 and 9.6 µM). The free probes and DNA–protein complexes are indicated by filled triangles and hollow triangles respectively
Subsequently, competition experiments were introduced into EMSAs to confirm the binding specificity of His6-LmbU to the above 8 targets. In the presence of 6.4 μM His6-LmbU, the retardant bands of all 8 targets were significantly weakened when 200-fold excesses of unlabeled specific DNA were added, but did not change when 200-fold excesses of unlabeled nonspecific DNA (a negative probe that cannot bind to His6-LmbU, Additional file 1: Fig. S1) were added. These data demonstrated that LmbU can directly and specifically bind to the promoter regions of the above 8 target genes (Fig. 2), including 3 regulators, 3 transporters, 1 σ factor and 1 other functional protein. However, the binding affinities of LmbU with different probes are diverse. LmbU has the highest binding affinity with the probe SLINC_7298p, while the weakest binding affinity with the probe SLINC_6382p. Besides, two retardant bands were observed when His6-LmbU bound to SLINC_1077p, indicating the regulatory models of LmbU to these targets may be different.

EMSAs of LmbU with promoter regions of target genes. Biotin-labeled probes (5 ng) were incubated with His6-LmbU of increasing concentrations (0, 6.4 and 9.6 µM). The free probes and DNA–protein complexes are indicated by filled triangles and hollow triangles respectively. 200-fold excess of specific (S) or nonspecific (N) unlabeled probes were used as competitors of the labeled probes
LmbU represses the promoters of SLINC_0469 and SLINC_1037 and activates the promoter of SLINC_8097 in vivo
As we know, antibiotics biosynthesis is strictly controlled by accurate and sophisticated regulatory networks. Through the above studies, we revealed that three regulatory genes may be regulated by LmbU. To investigate the regulation of LmbU to the three regulator genes in vivo, we firstly constructed a lmbU disruption strain ΔlmbU by using a CRISPR/Cas9-based genetic editing method (Additional file 1: Fig. S2). Then, the WT and ΔlmbU strains were chosen for qRT-PCR assays to analyze the effects of LmbU on the transcription of the three target genes. However, transcriptional levels of SLINC_1037 and SLINC_8097 were not enough for quantitative analysis (data not shown).
Therefore, we performed xylTE reporter assays, using the catechol dioxygenase and cofactor genes (xylTE) as a reporter. As results, enzyme activities of XylE controlled by SLINC_0469p and SLINC_1037p exhibited sevenfold and sixfold increase in ΔlmbU compared to that in WT, respectively (Fig. 3a and b), suggesting that LmbU represses the promoters of SLINC_0469 and SLINC_1037 in vivo. In contrast, enzyme activities of XylE controlled by SLINC_8097p showed 19-fold decrease in ΔlmbU compared to that in WT (Fig. 3c), suggesting that LmbU activates the promoter of SLINC_8097 in vivo.

Catechol dioxygenase activity assays of strains WT and ΔlmbU with corresponding reporter plasmids. 3a-3c represent enzyme activities of XylE controlled by SLINC_0469p, SLINC_1037p, and SLINC_8097p respectively. The results were achieved from three independent experiments. **P < 0.01; ***P < 0.001
SLINC_0469 negatively regulates lincomycin biosynthesis
The gene SLINC_0469 is 2,967 bp, and encodes a protein containing 988 amino acids, which belongs to a large ATP-binding regulator of the LuxR family (LAL) transcriptional regulator. Sequence alignment showed that N-terminal of SLINC_0469 has an AAA + (ATPases Associated with a wide variety of Activities) domain with ATPase activity, which contains conserved Walker A motif (A/G-X4-G-K-S/T, X indicates any amino acids) and Walker B motif (hhhhhDD, h indicates hydrophobic amino acids) (Additional file 1: Fig. S3a, b). C-terminal of SLINC_0469 has a DNA-binding domain (DBD) of the helix-turn-helix (HTH) structure of the LuxR family (Additional file 1: Fig. S3c).
To further investigate the function of SLINC_0469 in S. lincolnensis, a SLINC_0469 disruption strain ΔSLINC_0469 was constructed. The mutant ΔSLINC_0469 was confirmed by PCR using the primer pair JD0469-F/R (Fig. 4a). PCR products of WT with intact SLINC_0469 gene and ΔSLINC_0469 with defective SLINC_0469 gene were 4.3 kb (Fig. 4a Lane 1) and 2.4 kb (Fig. 4a Lane 2) respectively. PCR amplification by primer pair CR1/CR2 was used to determine that the disruption plasmid pKCcas9d0469 was eliminated from the mutant ΔSLINC_0469. A 2.6 kb band appeared only using pKCcas9d0469 as template (Lane 6), rather than WT (Lane 4) or ΔSLINC_0469 (Lane 5). Moreover, sequencing analysis verified that the mutant ΔSLINC_0469 was constructed successfully (Fig. 4a). Subsequently, a SLINC_0469 complementation strain CSLINC_0469 was also constructed.

SLINC_0469 suppresses lincomycin biosynthesis. a Identification of ΔSLINC_0469 by PCR and sequencing. Lane M indicated the DNA molecular weight marker. Lanes 1, 2 and 3 indicated PCR products amplified by primer pair JD0469F/R. Lanes 4, 5 and 6 indicated PCR products amplified by primer pair CR1/CR2. 1 and 4, WT; 2 and 5, ΔSLINC_0469; 3 and 6, pKCcas9d0469. b Effect of SLINC_0469 on lincomycin production. c Transcriptional analysis of lincomycin biosynthetic genes in WT and ΔSLINC_0469. The relative expression was normalized using internal reference gene hrdB. The transcriptional level of each gene in WT was set to 1.0. **P < 0.01; ***P < 0.001
The result showed that inactivation of SLINC_0469 had no significant influences on cell growth (Additional file 1: Fig. S4). Then, the WT, ΔSLINC_0469, CSLINC_0469 strains were cultured in FM2 medium to measure the lincomycin production. The results showed that the yield of lincomycin in ΔSLINC_0469 increased 2.6-fold compared to that in WT (Fig. 4b), and complementation of SLINC_0469 restored lincomycin production (Additional file 1: Fig. S7a), indicating that SLINC_0469 negatively regulates lincomycin biosynthesis.
Furthermore, qRT-PCR analysis was carried out to assess the influence of SLINC_0469 on transcription of lmb genes. There are 8 putative operons in the lincomycin cluster, and the first gene of each operon was chosen to perform qRT-PCR, except for lmbK, which could not be detected due to the low transcriptional level according to our previous study [27]. Compared to WT, the transcriptional levels of lmbA, lmbV, lmbW and lmbU were significantly increased in ΔSLINC_0469 with fold changes 3.7, 4.3, 13.0 and 3.3, respectively (Fig. 4c). Similar transcriptional levels of lmbC, lmbD and lmbV were observed in WT and ΔSLINC_0469, suggesting lmbC, lmbD and lmbV were not regulated by SLINC_0469. These data demonstrated that SLINC_0469 can suppress the transcription of lmbA, lmbV, lmbW and lmbU, thereby inhibiting lincomycin biosynthesis.
SLINC_1037 negatively regulates lincomycin biosynthesis
The 1908-bp SLINC_1037 gene encodes an AcoR family transcriptional regulator. A conserved Walker A motif (GERGTGK) and a HTH motif were found in the internal and the C-terminal of SLINC_1037 respectively (Additional file 1: Fig. S5a, b), indicating SLINC_1037 has putative ATP-binding and DNA-binding activities.
Then, SLINC_1037 disruption strain ΔSLINC_1037, complementation strain CSLINC_1037 was constructed using the method as above. PCR products amplified by the primer pair JD1037-F/R were 3.8 kb and 2.5 kb with WT and ΔSLINC_1037 as templates, respectively (Fig. 5a). Inactivation of SLINC_1037 had no significant influences on cell growth (Additional file 1: Fig. S4). The results of lincomycin bioassays showed that the yield of lincomycin in ΔSLINC_1037 increased 3.1-fold compared to that in WT (Fig. 5b), and complementation of SLINC_1037 restored lincomycin production (Additional file 1: Fig. S7b), indicating that SLINC_1037 negatively regulates lincomycin biosynthesis. qRT-PCR analysis revealed that, compared to WT, the transcriptional levels of lmbA, lmbV, lmbW and lmbU were significantly increased in ΔSLINC_1037 with fold changes 4.9, 7.6, 20.4 and 9.5, respectively (Fig. 5c). These data demonstrated that SLINC_1037 can suppress transcription of lmbA, lmbV, lmbW and lmbU, thereby inhibiting lincomycin biosynthesis.

SLINC_1037 suppresses lincomycin biosynthesis. a Identification of ΔSLINC_1037 by PCR and sequencing. Lane M indicated the DNA molecular weight marker. Lanes 1, 2 and 3 indicated PCR products amplified by primer pair JD1037F/R. Lanes 4, 5 and 6 indicated PCR products amplified by primer pair CR1/CR2. 1 and 4, WT; 2 and 5, ΔSLINC_1037; 3 and 6, pKCcas9d1037. b Effect of SLINC_1037 on lincomycin production. c Transcriptional analysis of lincomycin biosynthetic genes in WT and ΔSLINC_1037. The relative expression was normalized using internal reference gene hrdB. The transcriptional level of each gene in WT was set to 1.0. *P < 0.05; ***P < 0.001
SLINC_8097 negatively regulates lincomycin biosynthesis
The 906-bp SLINC_8097 gene encodes an AraC family transcriptional regulator, the C-terminal of which contains an AraC type DBD with putative DNA-binding activity (Additional file 1: Fig. S6a). Structure modeling and sequence alignment revealed that the DBD of SLINC_8097 includes similar HTH motifs referred to the template structure of AdpA (SMTL ID: 3w6v.1) [45], and the amino acids are conserved in Streptomyces (Additional file 1: Fig. S6B).
Then, SLINC_8097 disruption strain ΔSLINC_8097, complementation strain CSLINC_8097 were constructed. PCR products amplified by the primer pair JD8097-F/R were 2.8 kb and 2.2 kb with WT and ΔSLINC_8097 as templates, respectively (Fig. 6a). Inactivation of SLINC_8097 had no significant influences on cell growth (Additional file 1: Fig. S4). The results of lincomycin bioassays showed that the yield of lincomycin in ΔSLINC_8097 increased 3.2-fold compared to that in WT (Fig. 6b), and complementation of SLINC_8097 restored lincomycin production (Additional file 1: Fig. S7c), indicating that SLINC_8097 negatively regulates lincomycin biosynthesis. Furthermore, qRT-PCR analysis demonstrated that, compared to WT, the transcriptional levels of lmbA, lmbC, lmbD, lmbV, lmbW and lmbU were significantly increased in ΔSLINC_8097 with fold changes 4.2, 3.5, 4.1, 4.7, 13.3 and 5.5, respectively (Fig. 6c). The transcriptional levels of lmbJ were similar in WT and ΔSLINC_8097, suggesting lmbJ was not regulated by SLINC_8097. These data demonstrated that SLINC_8097 can suppress the transcription of lmbA, lmbC, lmbD, lmbV, lmbW and lmbU, thereby inhibiting lincomycin biosynthesis.

SLINC_8097 suppresses lincomycin biosynthesis. a Identification of ΔSLINC_8097 by PCR and sequencing. Lane M indicated the DNA molecular weight marker. Lanes 1, 2 and 3 indicated PCR products amplified by primer pair JD8097F/R. Lanes 4, 5 and 6 indicated PCR products amplified by primer pair CR1/CR2. 1 and 4, WT; 2 and 5, ΔSLINC_8097; 3 and 6, pKCcas9d8097. b Effect of SLINC_8097 on lincomycin production. c Transcriptional analysis of lincomycin biosynthetic genes in WT and ΔSLINC_8097. The relative expression was normalized using internal reference gene hrdB. The transcriptional level of each gene in WT was set to 1.0. *P < 0.05; **P < 0.01; ***P < 0.001
Discussion
Cross-regulation of CSRs among disparate antibiotic biosynthetic pathways has been widely studied in Streptomyces [13]. However, it is rarely reported that CSRs regulate the targets outside the BGCs. Here, we found that the DBSs of LmbU are widely distributed in the genome of S. lincolnensis, suggesting that LmbU is likely to function as a pleiotropic regulator and regulate more targets except the lmb genes (Fig. 7). Furthermore, through comparative transcriptomic analysis, Lin et al. [46] revealed that LmbU could regulate the transcription of 20 non-lmb genes, acting as a pleiotropic transcriptional regulator. It is worth noting that, the three targets of LmbU reported in our study, SLINC_0469, SLINC_1037 and SLINC_8097, were not included in the regulated genes identified by Lin’s RNA-seq data [46]. The S. lincolnensis strain NRRL 2936 used in our study is wild type, while the strain SyBE2901 used in RNA-seq analysis is a high lincomycin producer. Furthermore, we conducted additional studies to provide evidence that the targets of LmbU can regulate the lmb genes and affect the production of lincomycin, which forms a cascade regulatory network.

Proposed model of LmbU mediating regulation network to lincomycin biosynthesis. The locations of the lmb cluster and the three target genes on the chromosome were indicated. The arrows indicate activation, and the vertical virgules indicate inhibition. The solid lines indicate direct actions; the dotted lines indicate unknown mechanisms
Here, we used the conserved DBS of LmbU to screen the targets of LmbU, and performed EMSAs to investigate whether LmbU can bind to the targets directly. Though the sequence of DBS of LmbU within each target is perfectly matched, the binding affinity is not similar, indicating that the flanking sequence of DBS or the structure of the DNA may be also important for DNA-binding of LmbU. As reported, two ways have been found to be involved in DNA sequences recognition of proteins [47]. One way is directly based on contacts of amino acids and bases, and the flanking sequences are also important [48]. For example, the first and second flanking positions 5ʹ to the consensus DBS play important roles in DNA-binding affinity for E12 homodimer and E12-TAL1 heterodimer [49]. The other way is indirectly mediated by the conformation of the DNA [50,51,52]. Thus, the structures of DNA, including bendability, stability, groove shape, flexibility and so on, rather than the simple sequence are more appropriate to determine the DNA-binding affinities of proteins.
According to the study, LmbU inhibited the transcription of SLINC_0469 and SLINC_1037, which negatively regulate the production of lincomycin, suggesting that LmbU affect the production of lincomycin not only by activating the lmb genes, but also by suppressing the genes against lincomycin biosynthesis. On the contrary, LmbU activates the transcription of SLINC_8097, which negatively regulates the production of lincomycin. This may be conducive to maintain the level of lincomycin within a certain range in vivo. These data revealed that the regulatory network of LmbU on lincomycin biosynthesis is complex and accurate. In addition, LmbU can bind to the promoter regions of SLINC_0585 (encode an MFS transporter), SLINC_6382 (encode an ABC transporter permease), SLINC_7298 (encode an MFS transporter), SLINC_6570 (encode an RNA polymerase sigma factor) and SLINC_1077 (encode a methyltransferase). The studies about the regulatory mechanisms of LmbU to these targets and the effects of these target genes on lincomycin biosynthesis are ongoing.
Conclusion
In our previous studies, we have demonstrated that LmbU functions as a CSR of lincomycin, and LmbU homologues are widely found in actinomycetes, but their positions on the chromosome are not limited to the antibiotic BGCs [27, 44]. Based on this, we screened and identified the targets of LmbU which are located outside the lmb cluster, and showed the effect of these targets on production of lincomycin. The data elucidated that LmbU promotes lincomycin biosynthesis not only through regulating transcription of the lmb genes, but also by regulating three target genes outside the lmb cluster. In addition, the three targets SLINC_0469, SLINC_1037 and SLINC_8097 have been found negatively regulated lincomycin biosynthesis via regulating transcription of the lmb genes including lmbU, forming cross-regulation. This study can further illuminate the regulatory network of lincomycin biosynthesis, and will bring light to the functional analysis of LmbU family regulators.
Availability of data and materials
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- CSRG(s):
-
Cluster-situated regulatory gene(s)
- CSR:
-
Cluster-situated regulator
- BGC:
-
Biosynthetic gene cluster
- lmb :
-
Lincomycin biosynthetic gene
- LB:
-
Luria–Bertani media
- MS:
-
Mannitol-soya flour
- YEME:
-
Yeast extract-malt extract
- PCR:
-
Polymerase chain reaction
- bp:
-
Base pair
- Cas9:
-
CRISPR-associated protein 9
- CRISPR:
-
Clustered Regularly Interspaced Short Palindromic Repeats
- qRT-PCR:
-
Quantitative real-time PCR
- EMSA:
-
Electrophoretic mobility shift assay
- HTH:
-
Helix-turn-helix
- DBD:
-
DNA-binding domain
- DBS:
-
DNA-binding site
References
van der Heul HU, Bilyk BL, McDowall KJ, Seipke RF, van Wezel GP. Regulation of antibiotic production in Actinobacteria: new perspectives from the post-genomic era. Nat Prod Rep. 2018;35(6):575–604.
Ward AC, Allenby NEE. Genome mining for the search and discovery of bioactive compounds: the Streptomyces paradigm. FEMS Microbiol Lett. 2018;365(24):fny240.
Carroll AR, Copp BR, Davis RA, Keyzers RA, Prinsep MR. Marine natural products. Nat Prod Rep. 2022;39(6):1122–71.
Xia H, Li X, Li Z, Zhan X, Mao X, Li Y. The application of regulatory cascades in Streptomyces: yield enhancement and metabolite mining. Front Microbiol. 2020;11:406.
Kong DK, Wang X, Nie J, Niu GQ. Regulation of antibiotic production by signaling molecules in Streptomyces. Front Microbiol. 2019;10:2927.
Li J, Li Y, Niu G, Guo H, Qiu Y, Lin Z, et al. NosP-regulated nosiheptide production responds to both peptidyl and small-molecule ligands derived from the precursor peptide. Cell Chem Biol. 2018;25(2):143–53.
Gou L, Han T, Wang X, Ge J, Liu W, Hu F, et al. A novel TetR family transcriptional regulator, CalR3, negatively controls calcimycin biosynthesis in Streptomyces chartreusis NRRL 3882. Front Microbiol. 2017;8:2371.
Liu G, Chater KF, Chandra G, Niu GQ, Tan HR. Molecular regulation of antibiotic biosynthesis in Streptomyces. Microbiol Mol Biol Rev. 2013;77(1):112–43.
Lu X, Liu X, Chen Z, Li J, van Wezel GP, Chen W, et al. The ROK-family regulator Rok7B7 directly controls carbon catabolite repression, antibiotic biosynthesis, and morphological development in Streptomyces avermitilis. Environ Microbiol. 2020;22(12):5090–108.
Zhu YP, Wang XY, Zhang J, Ni X, Zhang X, Tao MF, et al. The regulatory gene wblA is a target of the orphan response regulator OrrA in Streptomyces coelicolor. Environ Microbiol. 2022;24(7):3081–96.
He WY, Wang WF, Ma JX, Zheng GS, Zimin AA, Jiang WH, et al. Crossregulation of rapamycin and elaiophylin biosynthesis by RapH in Streptomyces rapamycinicus. Appl Microbiol Biotechnol. 2022;106(5–6):2147–59.
Liu K, Hu XR, Zhao LX, Wang YM, Deng ZX, Tao MF. Enhancing ristomycin a production by overexpression of ParB-like StrR family regulators controlling the biosynthesis genes. Appl Environ Microbiol. 2021;87(19):e01066-e1121.
McLean TC, Wilkinson B, Hutchings MI, Devine R. Dissolution of the disparate: co-ordinate regulation in antibiotic biosynthesis. Antibiotics. 2019;8(2):83.
McLean TC, Hoskisson PA, Seipke RF. Coordinate regulation of antimycin and candicidin biosynthesis. Msphere. 2016;1(6):e00305-e316.
Jiang M, Yin M, Wu S, Han X, Ji K, Wen M, et al. GdmRIII, a TetR family transcriptional regulator, controls geldanamycin and elaiophylin biosynthesis in Streptomyces autolyticus CGMCC0516. Sci Rep. 2017;7(1):4803.
Wang L, Tian X, Wang J, Yang H, Fan K, Xu G, et al. Autoregulation of antibiotic biosynthesis by binding of the end product to an atypical response regulator. Proc Natl Acad Sci U S A. 2009;106(21):8617–22.
Xu G, Wang J, Wang L, Tian X, Yang H, Fan K, et al. “Pseudo” γ-butyrolactone receptors respond to antibiotic signals to coordinate antibiotic biosynthesis. J Biol Chem. 2010;285(35):27440–8.
Santamarta I, Rodriguez-Garcia A, Perez-Redondo R, Martin JF, Liras P. CcaR is an autoregulatory protein that binds to the ccaR and cefD-cmcI promoters of the cephamycin C-clavulanic acid cluster in Streptomyces clavuligerus. J Bacteriol. 2002;184(11):3106–13.
Huang J, Shi J, Molle V, Sohlberg B, Weaver D, Bibb MJ, et al. Cross-regulation among disparate antibiotic biosynthetic pathways of Streptomyces coelicolor. Mol Microbiol. 2005;58(5):1276–87.
Guo J, Zhao JL, Li LL, Chen Z, Wen Y, Li JL. The pathway-specific regulator AveR from Streptomyces avermitilis positively regulates avermectin production while it negatively affects oligomycin biosynthesis. Mol Genet Genomics. 2010;283(2):123–33.
Li C, He HR, Wang JB, Liu H, Wang HY, Zhu YJ, et al. Characterization of a LAL-type regulator NemR in nemadectin biosynthesis and its application for increasing nemadectin production in Streptomyces cyaneogriseus. Sci China Life Sci. 2019;62(3):394–405.
Pížek J, Řezanka T. Lincosamides: chemical structure, biosynthesis, mechanism of action, resistance, and applications. Biochem Pharmacol. 2017;133:20–8.
Wang SA, Lin CI, Zhang JW, Ushimaru R, Sasaki E, Liu HW. Studies of lincosamide formation complete the biosynthetic pathway for lincomycin A. Proc Natl Acad Sci U S A. 2020;117(40):24794–801.
Zhao Q, Wang M, Xu D, Zhang Q, Liu W. Metabolic coupling of two small-molecule thiols programs the biosynthesis of lincomycin A. Nature. 2015;518(7537):115–9.
Zhang HZ, Schmidt H, Piepersberg W. Molecular cloning and characterization of two lincomycin-resistance genes, lmrA and lmrB, from Streptomyces lincolnensis 78–11. Mol Microbiol. 1992;6(15):2147–57.
Koberska M, Vesela L, Vimberg V, Lenart J, Vesela J, Kamenik Z, et al. Beyond self-resistance: ABCF ATPase LmrC is a signal-transducing component of an antibiotic-driven signaling cascade accelerating the onset of lincomycin biosynthesis. MBio. 2021;12(5):e01731-e1821.
Hou B, Lin Y, Wu H, Guo M, Petkovic H, Tao L, et al. The novel transcriptional regulator LmbU promotes lincomycin biosynthesis through regulating expression of its target genes in Streptomyces lincolnensis. J Bacteriol. 2018;200(2):e00777-e817.
Hou B, Tao L, Zhu X, Wu W, Guo M, Ye J, et al. Global regulator BldA regulates morphological differentiation and lincomycin production in Streptomyces lincolnensis. Appl Microbiol Biotechnol. 2018;102(9):4101–15.
Hou B, Wang R, Zou J, Zhang F, Wu H, Ye J, et al. A putative redox-sensing regulator Rex regulates lincomycin biosynthesis in Streptomyces lincolnensis. J Basic Microbiol. 2021;61(9):772–81.
Kang Y, Wang Y, Hou B, Wang R, Ye J, Zhu X, et al. AdpAlin, a pleiotropic transcriptional regulator, is involved in the cascade regulation of lincomycin biosynthesis in Streptomyces lincolnensis. Front Microbiol. 2019;10:2428.
Kang Y, Wu W, Zhang F, Chen L, Wang R, Ye J, et al. AdpAlin regulates lincomycin and melanin biosynthesis by modulating precursors flux in Streptomyces lincolnensis. J Basic Microbiol. 2023;63(6):622–31.
Li J, Wang N, Tang Y, Cai X, Xu Y, Liu R, et al. Developmental regulator BldD directly regulates lincomycin biosynthesis in Streptomyces lincolnensis. Biochem Biophys Res Commun. 2019;518(3):548–53.
Meng S, Wu H, Wang L, Zhang B, Bai L. Enhancement of antibiotic productions by engineered nitrate utilization in actinomycetes. Appl Microbiol Biotechnol. 2017;101(13):5341–52.
Xu Y, Ke M, Li J, Tang Y, Wang N, Tan G, et al. TetR-Type regulator SLCG_2919 is a negative regulator of lincomycin biosynthesis in Streptomyces lincolnensis. Appl Environ Microbiol. 2019;85(1):e02091-e2118.
Xu Y, Tang Y, Wang N, Liu J, Cai X, Cai H, et al. Transcriptional regulation of a leucine-responsive regulatory protein for directly controlling lincomycin biosynthesis in Streptomyces lincolnensis. Appl Microbiol Biotechnol. 2020;104(6):2575–87.
Wang R, Cao Y, Kong F, Hou B, Zhao J, Kang Y, et al. Developmental regulator RamRsl controls both morphological development and lincomycin biosynthesis in Streptomyces lincolnensis. J Appl Microbiol. 2022;133(2):400–9.
Wang R, Kong F, Wu H, Hou B, Kang Y, Cao Y, et al. Complete genome sequence of high-yield strain S. lincolnensis B48 and identification of crucial mutations contributing to lincomycin overproduction. Synth Syst Biotechnol. 2020;5(2):37–48.
Tu B, Mao Y, Wang R, Kang Y, Ye J, Zhang H, et al. An alternative sigma factor σLsl regulates lincomycin production in Streptomyces lincolnensis. J Basic Microbiol. 2023;63(2):190–9.
Wu W, Kang Y, Hou B, Ye J, Wang R, Wu H, et al. Characterization of a TetR-type positive regulator AtrA for lincomycin production in Streptomyces lincolnensis. Biosci Biotechnol Biochem. 2023;87(7):786–95.
Wang R, Zhou T, Kong F, Hou B, Ye J, Wu H, et al. AflQ1-Q2 represses lincomycin biosynthesis via multiple cascades in Streptomyces lincolnensis. Appl Microbiol Biotechnol. 2023;107(9):2933–45.
Huang H, Zheng G, Jiang W, Hu H, Lu Y. One-step high-efficiency CRISPR/Cas9-mediated genome editing in Streptomyces. Acta Biochim Biophys Sin. 2015;47(4):231–43.
Hou B, Zhu X, Kang Y, Wang R, Wu H, Ye J, et al. LmbU, a cluster-situated regulator for lincomycin, consists of a DNA-binding domain, an auto-inhibitory domain, and forms homodimer. Front Microbiol. 2019;10:989.
Bierman M, Logan R, O’Brien K, Seno ET, Rao RN, Schoner BE. Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene. 1992;116(1):43–9.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25(4):402–8.
Yao MD, Ohtsuka J, Nagata K, Miyazono K, Zhi Y, Ohnishi Y, et al. Complex structure of the DNA-binding domain of AdpA, the global transcription factor in Streptomyces griseus, and a target duplex DNA reveals the structural basis of its tolerant DNA sequence specificity. J Biol Chem. 2013;288(43):31019–29.
Lin CY, Pang AP, Zhang Y, Qiao J, Zhao GR. Comparative transcriptomic analysis reveals the significant pleiotropic regulatory effects of LmbU on lincomycin biosynthesis. Microb Cell Fact. 2020;19(1):30.
Rohs R, West SM, Sosinsky A, Liu P, Mann RS, Honig B. The role of DNA shape in protein-DNA recognition. Nature. 2009;461(7268):1248–53.
Gordân R, Shen N, Dror I, Zhou TY, Horton J, Rohs R, et al. Genomic regions flanking E-box binding sites influence DNA binding specificity of bHLH transcription factors through DNA shape. Cell Rep. 2013;3(4):1093–104.
Beltran AC, Dawson PE, Gottesfeld JM. Role of DNA sequence in the binding specificity of synthetic basic-helix-loop-helix domains. ChemBioChem. 2005;6(1):104–13.
Bansal M, Kumar A, Yella VR. Role of DNA sequence based structural features of promoters in transcription initiation and gene expression. Curr Opin Struct Biol. 2014;25:77–85.
Fujii S, Kono H, Takenaka S, Go N, Sarai A. Sequence-dependent DNA deformability studied using molecular dynamics simulations. Nucleic Acids Res. 2007;35(18):6063–74.
Rube HT, Rastogi C, Kribelbauer JF, Bussemaker HJ. A unified approach for quantifying and interpreting DNA shape readout by transcription factors. Mol Syst Biol. 2018;14(2):e7902.
Acknowledgements
Not applicable.
Funding
This work was supported by Ministry of Science and Technology (MOST) of China (2021YFC2100600) and the National Natural Science Foundation of China (NSFC) (31900059).
Author information
Authors and Affiliations
Contributions
WH and ZH supervised the experiments. HB, WR, YJ and ZX conceived and designed research. MY and ZX conducted experiments. MY, ZX, WR, and ZT analyzed the data. HB and WR wrote the original manuscript. MY, WH and WR edited the manuscript. All authors read and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1:
Fig. S1. EMSA of His6-LmbU (0, 3.2, 6.4 and 9.6 µM) with the negative probe. Fig. S2. Construction and identification of S. lincolnensis lmbU disruption mutant ΔlmbU. Fig. S3. Functional domains and sequence alignment of SLINC_0469. Fig. S4. Growth curves of S. lincolnensis strains NRRL 2936, ΔSLINC_0469, ΔSLINC_1037, and ΔSLINC_8097. Fig. S5. Functional domains and sequence alignment of SLINC_1037. Fig. S6. Functional domains and sequence alignment of SLINC_8097. Fig. S7. Effect of three new LmbU targets towards lincomycin production. Table S1. Primers used in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mao, Y., Zhang, X., Zhou, T. et al. Three new LmbU targets outside lmb cluster inhibit lincomycin biosynthesis in Streptomyces lincolnensis. Microb Cell Fact 23, 3 (2024). https://doi.org/10.1186/s12934-023-02284-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-023-02284-y