
Abstract
Background
Glucagon-like peptide-1 is an incretin hormone essential for normal human glucose homeostasis. Expression of the glucagon-like peptide-1 receptor in the myocardium has fuelled growing interest in the direct and indirect cardiovascular effects of native glucagon-like peptide-1, its degradation product glucagon-like peptide-1(9-36), and the synthetic glucagon-like peptide-1 receptor agonists. Preclinical studies have demonstrated cardioprotective actions of all three compounds in the setting of experimental myocardial infarction and left ventricular systolic dysfunction. This has led to Phase 2 trials of native glucagon-like peptide-1 and incretin-based therapies in humans with and without Type 2 diabetes mellitus. These studies have demonstrated the ability of glucagon-like peptide-1, independent of glycaemic control, to positively modulate the metabolic and haemodynamic parameters of individuals with coronary artery disease and left ventricular systolic dysfunction. We aim to add to this growing body of evidence by studying the effect of chronic glucagon-like peptide-1 receptor activation on exercise-induced ischaemia in patients with chronic stable angina managed conservatively or awaiting revascularisation. The hypothesis being liraglutide, a subcutaneously injectable glucagon-like peptide-1 receptor agonist, is able to improve exercise haemodynamics in patients with obstructive coronary artery disease when compared with saline placebo.
Methods and design
The Liraglutide to Improve corONary haemodynamics during Exercise streSS (LIONESS) trial is an investigator-initiated single-centre randomised double-blinded placebo-controlled crossover proof-of-principle physiological study. Primary endpoints are change in rate pressure product at 0.1 mV ST-segment depression and change in degree of ST-segment depression at peak exercise during sequential exercise tolerance testing performed over a 6-week study period in which 26 patients will be randomised to either liraglutide or saline with crossover to the opposing regimen at week 3.
Discussion
The study will be conducted in accordance with the principles of Good Clinical Practice and the Declaration of Helsinki. The local Research Ethics Committee and Medicines and Healthcare Products Regulatory Agency have approved the study.
Trial registration
National Institute of Health Research Clinical Research Network (NIHR CRN) Portfolio ID 11112 and ClinicalTrials.gov Identifier NCT02315001.
Similar content being viewed by others

Background
The incretin concept was borne from the observation that an oral load of glucose can provoke a two to three times more potent insulinotropic stimulus than an isoglycaemic intravenous glucose infusion [1,2]. This effect has been attributed to the action of incretin (INtestinal seCRETion of INsulin) hormones, which constitute part of the glucagon superfamily. Glucose-dependent insulinotropic polypeptide (GIP), a 42-amino acid peptide made by duodenal and jejunal enteroendocrine K cells in the proximal small bowel, was the first incretin hormone to be isolated from purified porcine intestinal extracts [3]. More than a decade later, glucagon-like peptide-1 (GLP-1) was identified, a 30-amino acid cleavage product of proglucagon, synthesised predominantly by the enteroendocrine L cells of the distal ileum and colon [4,5]. Despite their site of production, plasma levels of GIP and GLP-1 rise within minutes of enteral nutrition, indicating a combined neural and endocrine signalling axis prompting their secretion [6]. Together, GIP and GLP-1 are fundamental to maintaining normal glucose homeostasis in man. They contribute almost equally to, and have an additive effect on, enhancing glucose-dependent insulin exocytosis after meal ingestion, although GLP-1-mediated effects appear to predominate at higher glucose levels [7].
There is a defective incretin effect in Type 2 diabetes mellitus (T2DM) [8]. Levels of GIP can increase after enteral nutrition but the ability to potentiate postprandial insulin secretion is significantly impaired [9,10]. In contrast, meal-stimulated levels of GLP-1 are severely depressed. A continuous infusion of exogenous GLP-1 can, however, result in a near normal insulin-mediated response to an oral glucose load, suggesting preservation of insulinotropic activity [11-14]. Furthermore the risk of hypoglycaemia with GLP-1 is minimal, as both its stimulatory effect on insulin secretion and its inhibitory action on glucagon release switch off when ambient glucose levels are <4 mmol/L [5,14,15]. Despite these properties, the pharmaco-therapeutic utility of native GLP-1 in T2DM is profoundly limited by its rapid inactivation by the enzyme dipeptidyl dipeptidase-4 (DPP-4), which cleaves 2 amino terminal peptides from GLP-1(7-36) to form the GLP-1(9-36) metabolite [5]. GLP-1 has a half-life of <2 minutes which means only 10-20% of total plasma GLP-1 is biologically active [16]. Novel treatment strategies for T2DM, based on the incretin effect, have been developed to overcome this endogenous cul-de-sac. Subcutaneously injectable GLP-1 receptor (GLP-1R) agonists (DPP-4 mediated degradation-resistant peptides with improved pharmacokinetics that act via the human GLP-1R) and oral DPP-4 inhibitors (small molecules with clinically useful oral bioavailability that shield the endogenous peptide from DPP-4 metabolism and thereby enhance its innate insulinotropic activity) are now licensed and have become part of the standard of care for the management of T2DM [17-19]. In essence GLP-1R agonists provide pharmacologic levels of a GLP-1R stimulus whereas DPP-4 inhibitors preserve physiologic levels of endogenous GLP-1 [6].
The GLP-1R is a heptahelical G-protein coupled receptor found widely in several extra-pancreatic tissue beds, including lung, kidney, central, enteric and peripheral nervous systems, lymphocytes, smooth muscle cells and atrial cardiomyocytes [20-23]. It is this expression of the GLP-1R in the myocardium, and the overall ubiquity of the receptor, that has fuelled growing interest in the direct and indirect cardiovascular (CV) effects of native GLP-1 [24,25], its degradation product GLP-1(9-36) [26,27], and the degradation-resistant GLP-1R agonists [28-30]. Indeed knockout mice lacking the GLP-1R were shown to have a lower resting heart rate, elevated left ventricular (LV) end diastolic pressure and increased LV wall thickness compared to wild type controls [31]. These mice also displayed depressed LV contractility and impaired diastolic function after insulin administration; all suggesting a pivotal role for the GLP-1R in maintaining normal cardiac structure and function, in mice at least [31,32]. It is also important to note that whilst native GLP-1 may exert a regulatory role on CV biology through its interaction with the GLP-1R plus its independently cardioactive metabolite GLP-1(9-36), synthetic GLP-1R agonists are shielded from DPP-4 breakdown and therefore act predominantly via their action on the classical GLP-1R alone [5,20]. Nevertheless, several preclinical studies have demonstrated cardioprotective actions of all three compounds in rodent, canine and porcine hearts in the context of ischaemia/reperfusion (I/R) injury after experimental myocardial infarction (MI) and LV systolic dysfunction [24,26,27,29,30,33-36]. The growing weight of evidence from these animal models has led to several Phase 2 trials of native GLP-1 and incretin-based therapies in humans with and without T2DM (Table 1). Although hypothesis-generating at best, these small, often single-centre, studies have demonstrated the ability of GLP-1, independent of its effect on glycaemic control, to positively modulate the metabolic and haemodynamic parameters of individuals with CAD and cardiomyopathy (Table 1) [37-45].
Our aim is to add to this growing body of evidence by characterising, for the first time, the effect of chronic GLP-1R activation on haemodynamics and exercise-induced ischaemia in a cohort of patients with chronic stable angina managed conservatively or awaiting revascularisation. Rather than study I/R injury following coronary occlusion, we will determine whether the acute anti-ischaemic properties of GLP-1 translate into an anti-anginal action on exertion. Liraglutide (trade name Victoza manufactured by NovoNordisk, Bagsvaerd, Denmark), a synthetic GLP-1R agonist that shares 97% structural homology with native GLP-1, has a half-life of 10–14 hours. It binds to albumin in the circulation, protecting it from DPP-4 degradation and preventing renal elimination. For these reasons liraglutide is a useful surrogate for studying chronic GLP-1R activation in humans. Moreover the need for a continuous GLP-1 infusion is now replaced by a once daily subcutaneous injection, which should help to maximise patient compliance and preclude the risk of fluid overload in those with established coronary heart disease. The relatively small risk of hypoglycaemia associated with liraglutide serves to further support it being used to study the effects of GLP-1 beyond glycaemic control in both diabetics and non-diabetics. Indeed, a trial investigating the treatment of obesity demonstrated significantly greater weight loss achieved with liraglutide versus orlistat or placebo and an absence of hypoglycaemic episodes in a cohort of non-diabetics [46]. Our centre has developed reproducible exercise protocols for the study of angina [47-50], and it is from these that the current trial protocol has been derived.
Aims and objectives
The objective of the present protocol is to evaluate the physiological basis of chronic GLP-1R occupancy on exercise haemodynamics, as manifest by specific electrophysiological parameters, in those patients with known chronic stable angina, a significant coronary stenosis in at least one major epicardial vessel confirmed by coronary angiography and evidence of reversible myocardial ischaemia seen on exercise tolerance testing. The hypothesis being liraglutide, a subcutaneously injectable synthetic analogue of endogenous GLP-1, is able to improve exercise haemodynamics in a cohort of patients with obstructive coronary artery disease (CAD) when compared with saline placebo. Primary and secondary outcome measures are listed in Tables 2 and 3 respectively.
Methods and design
Study design
The Liraglutide to Improve corONary haemodynamics during Exercise streSS (LIONESS) (NIHR CRN Study ID 11112 and clinicaltrials.gov NCT02315001) trial is an investigator-initiated single-centre randomised double-blinded placebo-controlled crossover proof-of-principle physiological study. Patients will be enrolled according to the inclusion and exclusion criteria listed in Tables 4 and 5 respectively.
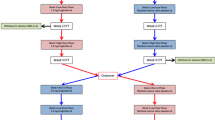
Both investigator and patient will be blinded to the study drug. Patients will be randomised to enter a GLP-1 treatment arm or matched-volume saline placebo arm. Following a 1-week run-in phase with 0.6 mg liraglutide followed by a 1-week course of 1.2 mg liraglutide, patients in the active intervention arm will have their first exercise tolerance test (ETT) at the end of Week 2. Patients will then be up-titrated to high-dose 1.8 mg liraglutide for another week before performing a Week 3 ETT. With a stepwise increase in liraglutide therapy over a 3-week period we hope to observe a dose–response effect on exercise haemodynamics. An initial 1-week run-in phase of once daily 0.6 mg liraglutide should also allow for improved gastrointestinal tolerability. Whilst in the placebo arm, patients will have matched-volume saline injections for the first two weeks before the Week 2 ETT and then another week of saline injections before the Week 3 ETT. Patients will then crossover at this stage so that those in the treatment arm cross to the placebo arm and vice versa (please refer to Figure 1).

The LIONESS Trial study design.
Criteria for terminating an exercise stress test will be: 1) physical exhaustion, 2) severe chest pain, 3) attaining maximal age-related heart rate, 4) ST-segment depression (STD) >0.4 mV, or 5) haemodynamically-significant dysrhythmia.
Sample selection
Potential trial participants fulfilling the inclusion criteria (Table 4) and who consent will be recruited consecutively from the following groups:
-
Patients with known obstructive CAD on angiography and a previously abnormal exercise test, but who are currently stable on conservative medical management, will be enrolled from general cardiology outpatient clinic.
-
Patients with known obstructive CAD on angiography and evidence of reversible myocardial ischaemia on exercise testing, awaiting elective percutaneous coronary intervention (PCI) or coronary artery bypass graft (CABG) surgery.
Sample size calculation
The host department has shown that the time taken to 0.1 mV STD (368 ± 34 → 418 ± 36 seconds) and the rate pressure product at 0.1 mV STD (20500 ± 755 → 21907 ± 764 mmHg/min) is significantly increased during the second of two serial exercise tests separated by 15 minutes in chronic CAD patients with known left anterior descending artery stenoses [47,48]. This effect is attributed to warm-up angina, which is thought to augment the innate resistance of the myocardium to an ischaemic insult [51,52]. We would expect the administration of Liraglutide to mimic the beneficial cardioprotective effects of warm-up angina. A sample size of 26 patients randomised in a 1:1 fashion to each treatment arm (taking into consideration a >10% drop-out rate) followed by crossover would have 90% power to detect a difference between the means of approximately 15% (2-sided α = 0.05).
Randomisation procedure
All randomisations will be performed through a computer-generated randomisation program (www.sealedenvelope.com) in a 1:1 randomisation ratio utilising a study-specific ID assigned to each enrolled patient. Treatment allocation can be unblinded in case of an emergency or serious adverse event if deemed necessary by the investigator.
Study agent and placebo comparator
Liraglutide will be supplied to trial recruits in pre-filled single-use plastic syringes ready for once daily subcutaneous injection at doses of 0.6 mg, 1.2 mg and 1.8 mg. Matched-volume saline placebo will also be supplied in identical pre-filled syringes for subcutaneous injection. Study medication will be produced and dispensed by the Aseptic Services Unit of St Thomas’ Hospital Pharmacy as a set of 7 separate disposable syringes at the beginning of each study week. Patients will be asked to store these in a refrigerator and to dispose of their used syringes in a sharps bin. The sealed bin will be brought to the next study visit to check for compliance with the study agents. Patients will be advised to administer their study agent at approximately the same time each day to reinforce compliance. The recommended injection site will be the abdomen, and that injection site around the abdomen should be varied from day to day.
Concomitant medication
Study participants will have any heart rate-limiting drugs cautiously withdrawn prior to commencing the 6-week trial protocol. These include beta-blockers, rate-limiting calcium antagonists (e.g., diltiazem and verapamil), and ivabradine to ensure a normal chronotropic response to exercise stress testing. Long-acting oral nitrates and nicorandil will also be stopped to avoid any potential interference with haemodynamic physiology. Patients will be allowed to continue their antiplatelet therapy (e.g., aspirin, clopidogrel, prasugrel, and/or ticagrelor), ACE-inhibitors, angiotensin receptor blockers, and statins. Patients will also continue their sublingual glyceryl trinitrate (GTN) spray or buccal GTN to relieve breakthrough angina. Those patients experiencing poor control of pre-existing hypertension following the cessation of the drugs listed previously will be prescribed alternative antihypertensive medication at the discretion of the investigator, or have their current medication they can continue up-titrated, and monitored closely for response.
Study visits and procedures
Screening Visit
Once a patient is deemed to have adequately fulfilled the inclusion and exclusion criteria, informed consent will be obtained. Part of the inclusion criteria includes the demonstration of reversible ischaemia during an ETT. If a recent ETT is not available, the patient will be asked to perform an ETT (free of any heart rate-limiting medication for the past 48 hours) at the screening visit. The result of this ETT will not be used in the final data analysis but will be captured as part of the screening process. Those patients ineligible for trial participation will be registered, and the reason for screening failure will be recorded. Patients entering the trial will receive a tutorial on home blood glucose monitoring and the signs and symptoms of hypoglycaemia. A safety information card will be issued. A full medical history and clinical examination will be performed. Blood tests will be taken (Table 6). Medications that need to be withheld will be identified at this visit and withdrawn cautiously (see Table 7). Those patients successfully recruited will be randomised at this stage.
Trial visits (week 0 to week 6)
Trial participants will perform their first self-administration of study agent and first blood glucose measurement, which will be recorded in their patient diary, at trial commencement (Week 0). Both will be supervised by an investigator. Recruited patients will attend a total of 7 study visits and have procedures performed according to the schedule detailed in Table 7. Patients will be asked to record twice daily home blood glucose measurements at approximately the same time am and pm. Patients will have blood tests performed at every study visit (Table 6).
Expected duration of the study
From the point of administration of study drug on day 1 the total length of the study will be 6 weeks. There will be no follow-up of patients after completion of the 6-week trial protocol, at which point study drugs will be terminated and patients will have their pre-study medication recommenced immediately thereafter.
Procedures for recording and reporting adverse events
All study participants will be issued a patient diary in which they can record any adverse events, side effects, injection site reactions and angina burden. This diary will be inspected at every study visit. Patients will also be able to contact the Main Patient Liaison at all times for general enquiries and advice regarding any signs and symptoms they are experiencing pertaining to the study. The primary care physicians of all study participants will be issued with an information sheet regarding the potential side effects of the GLP-1R agonist Liraglutide and the medication that a patient may have had discontinued prior to commencing the trial. The importance of informing the Main Patient Liaison if any adverse events were to occur will be stressed.
Withdrawal of subjects
Participants will have the right to withdraw from the study at any time for any reason. Should a patient decide to withdraw from the study, all efforts will be made to report the reason for withdrawal as thoroughly as possible. The investigator also has the right to withdraw patients from the study in the event of inter-current illness, adverse events or serious adverse events, protocol violations, administrative reasons or other reasons.
The study drug will be discontinued and the participant withdrawn from the study if:
-
the participant misses 2 consecutive courses of treatment;
-
the participant decides they no longer wish to continue;
-
if the participant experiences an intolerable degree of angina as a result of withdrawing their pre-study heart rate-limiting or anti-anginal medications;
-
if the participant experiences significant side effects from their study medication, in particular excessive or intolerable nausea, vomiting or diarrhoea; or
-
the investigator recommends it.
The Sponsor or Chief Investigator may prematurely discontinue the study on the basis of new safety information or for other reasons given by the Study Steering Committee or Research Ethics Committee concerned.
Data management and analysis
Data will be collected prospectively using a customised paper Case Report Form (CRF). Investigators will record adverse events, injection site reactions, angina episodes and clinical and laboratory parameters on the CRF at each study visit.
The fundamental principle underpinning the design of this trial is that the 2-week period between the Week 3 ETT and the Week 5 ETT (see Figure 1), in those patients randomised to the active liraglutide intervention first, should be sufficient to allow adequate washout of the drug and therefore minimise the impact of a potential carryover effect. Given that the half-life of liraglutide is 10–14 hours, this 2-week washout period far exceeds the generally accepted mark of ≥5 half-lives. A washout period for those randomised to the placebo arm first would not apply per se. A preliminary test, such as the Wilcoxon rank sum test, will be applied to ensure there is no carryover effect. Once this has been established, standard two-sample t-tests for independent samples using the intra-individual differences between the outcomes in both study periods as the raw data will be utilised [53].
A multiple regression model will be used to compare treatment, visit number, and patient, using least squares. Linear regression will be used to study the correlation between outcome parameters and baseline patient characteristics. A p value <0.05 will be considered statistically significant.
Discussion
The study will be conducted in compliance with the principles of the Declaration of Helsinki (2008), the principles of Good Clinical Practice and in accordance with all applicable requirements laid down by the Co-Sponsors of the study, namely: King’s College London and Guy’s and St Thomas’ NHS Foundation Trust. The trial protocol has received ethical approval from the National Research Ethics Service (NRES) Committee London – Westminster (Research Ethics Committee reference 11/LO/1564) of the United Kingdom. Annual progress and safety reports and a final report at conclusion of the study will be submitted to the Co-Sponsors and the Research Ethics Committee. The Medicines and Healthcare Products Regulatory Agency (MHRA) has also approved the study.
This trial has been funded by the British Heart Foundation via a Clinical Research Training Fellowship awarded to Dr Aung Myat (grant number FS/11/70/28917) and is supported by the Department of Health via the National Institute of Health Research comprehensive Biomedical Research Centre award to Guy’s and St Thomas’ NHS Foundation Trust in partnership with King’s College London and King’s College Hospital NHS Foundation Trust.
The LIONESS study is a single-centre trial based in the United Kingdom. The trial has, therefore, been registered with our national trials database via the National Institute of Health Research Clinical Research Network (NIHR CRN) portfolio (Study ID 11112) since May 4th 2012. The NIHR CRN trials database is accessible to the public at no charge, open to all prospective registrants, managed by a not-for-profit organization, has a mechanism to ensure the validity of the registration data, and is electronically searchable. The study has also been registered on ClinicalTrials.gov as of November 10th 2014 (NCT02315001). There have been minor amendments to the study protocol since first registration and as such the first patient was recruited in January 2014. The study remains actively recruiting as of February 2015.
It is intended that positive or negative study results will be reported and disseminated at international conferences and in an international peer-reviewed scientific journal.
References
Elrick H, Stimmler L, Hlad CJ, Arai Y. Plasma insulin response to oral and intravenous glucose. J Clin Endocrinol Metab. 1964;24:1076–82.
Perley MJ, Kipnis DM. Plasma insulin responses to oral and intravenous glucose: studies in normal and diabetic subjects. J Clin Invest. 1967;46:1954–62.
Dupre J, Ross S, Watson D, Brown JC. Stimulation of insulin secretion by gastric inhibitory polypeptide in man. J Clin Endocrinol Metab. 1973;37:826–8.
Kreymann B, Ghatei MA, Williams G, Bloom SR. Glucagon-like peptide-1 7–36: a physiological incretin in man. Lancet. 1987;330:1300–4.
Ussher JR, Drucker DJ. Cardiovascular biology of the incretin system. Endocr Rev. 2012;33:187–215.
Myat A, Redwood S, Gersh B, Yellon D, Marber M. Diabetes, incretin hormones and cardioprotection. Heart. 2014;100:1550–61.
Nauck M, Bartels E, Orskov C, Ebert R, Creutzfeldt W. Additive insulinotropic effects of exogenous synthetic human gastric inhibitory polypeptide and glucagon-like peptide-1-(7–36) amide infused at near-physiological insulinotropic hormone and glucose concentrations. J Clin Endocrinol Metab. 1993;76:912–7.
Nauck M, Stöckmann F, Ebert R, Creutzfeldt W. Reduced incretin effect in Type 2 (non-insulin-dependent) diabetes. Diabetologia. 1986;2:46–52.
Vilsbøll T, Krarup T, Madsbad S, Holst JJ. Defective amplification of the late phase insulin response to glucose by GIP in obese Type II diabetic patients. Diabetologia. 2002;45:1111–9.
Elahi D, McAloon-Dyke M, Fukagawa NK, Meneilly GS, Sclater AL, Minaker KL, et al. The insulinotropic actions of glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (7–37) in normal and diabetic subjects. Regul Pept. 1994;51:63–74.
Nauck M, Heimesaat MM, Orskov C, Holst JJ, Ebert R, Creutzfeldt W. Preserved incretin activity of glucagon-like peptide 1 [7–36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J Clin Invest. 1993;91:301–7.
Zander M, Madsbad S, Madsen JL, Holst JJ. Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes: a parallel-group study. Lancet. 2002;359:824–30.
Rachman J, Gribble F, Barrow B, Levy J, Buchanan K, Turner R. Normalization of insulin responses to glucose by overnight infusion of glucagon-like peptide 1 (7–36) amide in patients with NIDDM. Diabetes. 1996;45:1524–30.
Egan J, Meneilly G, Habener JF, Elahi D. Glucagon-like peptide-1 augments insulin-mediated glucose uptake in the obese state. J Clin Endocrinol Metab. 2002;87:3768–73.
Holst JJ. The physiology of glucagon-like peptide 1. Physiol Rev. 2007;225:1409–39.
Deacon CF, Nauck MA, Toft-Nielsen M, Pridal L, Willms B, Holst JJ. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes. 1995;44:1126–31.
Drucker DJ, Nauck M. The incretin system: glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368:1696–705.
Scirica BM, Bhatt DL, Braunwald E, Steg PG, Davidson J, Hirshberg B, et al. SAVOR-TIMI 53 Steering committee and investigators. saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N Engl J Med. 2013;369:1317–26.
White WB, Cannon CP, Heller SR, Nissen SE, Bergenstal RM, Bakris GL, et al. EXAMINE Investigators. Alogliptin after acute coronary syndrome in patients with type 2 diabetes. N Engl J Med. 2013;369:1327–35.
Ussher JR, Drucker DJ. Cardiovascular actions of incretin-based therapies. Circ Res. 2014;114:1788–803.
Pyke C, Knudsen LB. The glucagon-like peptide-1 receptor–or not? Endocrinology. 2013;154:4–8.
Kim M, Platt MJ, Shibasaki T, Quaggin SE, Backx PH, Seino S, et al. GLP-1 receptor activation and Epac2 link atrial natriuretic peptide secretion to control of blood pressure. Nat Med. 2013;19:567–75.
Richards P, Parker H, Adriaenssens A, Hodgson J, Cork S, Trapp S, et al. Identification and characterisation of glucagon-like peptide-1 receptor expressing cells using a new transgenic mouse model. Diabetes. 2014;63:1224–33.
Bose AK, Mm M, Carr RD, Cl B, Yellon DM. Glucagon-like peptide 1 can directly protect the heart against ischemia/reperfusion injury. Diabetes. 2005;54:146–51.
Poornima I, Brown SB, Bhashyam S, Parikh P, Bolukoglu H, Shannon RP. Chronic glucagon-like peptide-1 infusion sustains left ventricular systolic function and prolongs survival in the spontaneously hypertensive, heart failure-prone rat. Circ Heart Fail. 2008;1:153–60.
Ban K, Noyan-Ashraf MH, Hoefer J, Bolz S-S, Drucker DJ, Husain M. Cardioprotective and vasodilatory actions of glucagon-like peptide 1 receptor are mediated through both glucagon-like peptide 1 receptor-dependent and -independent pathways. Circulation. 2008;117:2340–50.
Sonne DP, Engstrøm T, Treiman M. Protective effects of GLP-1 analogues exendin-4 and GLP-1 (9–36) amide against ischemia-reperfusion injury in rat heart. Regul Pept. 2008;146:243–9.
Noyan-Ashraf MH, Shikatani EA, Schuiki I, Mukovozov I, Wu J, Li RK, et al. A glucagon-like peptide-1 analog reverses the molecular pathology and cardiac dysfunction of a mouse model of obesity. Circulation. 2013;127:74–85.
Noyan-Ashraf M, Momen M, Ban K, Al-Muktafi S, Zhou Y, Riazi AM, et al. GLP-1R agonist liraglutide activates cytoprotective pathways and improves outcomes after experimental myocardial infarction in mice. Diabetes. 2009;58:975–83.
Timmers L, Henriques JPS, De Kleijn DPV, Devries JH, Kemperman H, Steendijk P, et al. Exenatide reduces infarct size and improves cardiac function in a porcine model of ischemia and reperfusion injury. J Am Coll Cardiol. 2009;53:501–10.
Gros R, You X, Baggio L, Golam Kabir M, Sadi A, Mungrue I. Cardiac function in mice lacking the glucagon-like peptide-1 receptor. Endocrinology. 2003;144:2242–52.
Kavianipour M, Ehlers MR, Malmberg K, Ronquist G, Ryden L, Wikstrom G, et al. Glucagon-like peptide-1 (7–36) amide prevents the accumulation of pyruvate and lactate in the ischemic and non-ischemic porcine myocardium. Peptides. 2003;24:569–78.
Zhao T, Parikh P, Bhashyam S. Direct effects of glucagon-like peptide-1 on myocardial contractility and glucose uptake in normal and postischemic isolated rat hearts. J Pharmacol Exp Ther. 2006;317:1106–13.
Nikolaidis L, Doverspike A, Hentosz T. Glucagon-like peptide-1 limits myocardial stunning following brief coronary occlusion and reperfusion in conscious canines. J Pharmacol Exp Ther. 2005;312:303–8.
Nikolaidis LA, Elahi D, Shen Y-T, Shannon RP. Active metabolite of GLP-1 mediates myocardial glucose uptake and improves left ventricular performance in conscious dogs with dilated cardiomyopathy. Am J Physiol Heart Circ Physiol. 2005;289:H2401–8.
Ban K, Kim K-H, Cho C-K, Sauvé M, Diamandis EP, Backx PH, et al. Glucagon-like peptide (GLP)-1 (9–36) amide-mediated cytoprotection is blocked by exendin(9–39) yet does not require the known GLP-1 receptor. Endocrinology. 2010;151:1520–31.
Nikolaidis LA, Mankad S, Sokos GG, Miske G, Shah A, Elahi D, et al. Effects of glucagon-like peptide-1 in patients with acute myocardial infarction and left ventricular dysfunction after successful reperfusion. Circulation. 2004;109:962–5.
Sokos GG, Nikolaidis LA, Mankad S, Elahi D, Shannon RP. Glucagon-like peptide-1 infusion improves left ventricular ejection fraction and functional status in patients with chronic heart failure. J Card Fail. 2006;12:694–9.
Sokos GG, Bolukoglu H, German J, Hentosz T, Magovern GJ, Maher TD, et al. Effect of glucagon-like peptide-1 (GLP-1) on glycemic control and left ventricular function in patients undergoing coronary artery bypass grafting. Am J Cardiol. 2007;100:824–9.
Halbirk M, Nørrelund H, Møller N, Holst JJ, Schmitz O, Nielsen R, et al. Cardiovascular and metabolic effects of 48-h glucagon-like peptide-1 infusion in compensated chronic patients with heart failure. Am J Physiol Heart Circ Physiol. 2010;298:H1096–102.
Read PA, Khan FZ, Heck PM, Hoole SP, Dutka DP. DPP-4 inhibition by sitagliptin improves the myocardial response to dobutamine stress and mitigates stunning in a pilot study of patients with coronary artery disease. Circ Cardiovasc Imaging. 2010;3:195–201.
Read PA, Hoole SP, White PA, Khan FZ, O'Sullivan M, West NE, et al. A pilot study to assess whether glucagon-like peptide-1 protects the heart from ischemic dysfunction and attenuates stunning after coronary balloon occlusion in humans. Circ Cardiovasc Interv. 2011;4:266–72.
Read PA, Khan FZ, Dutka DP. Cardioprotection against ischaemia induced by dobutamine stress using glucagon-like peptide-1 in patients with coronary artery disease. Heart. 2012;98:408–13.
Lønborg J, Vejlstrup N, Kelbæk H, Bøtker HE, Kim WY, Mathiasen AB, et al. Exenatide reduces reperfusion injury in patients with ST-segment elevation myocardial infarction. Eur Heart J. 2012;33:1491–9.
McCormick LM, Kydd AC, Read PA, Ring LS, Bond SJ, Hoole SP, et al. Chronic dipeptidyl peptidase-4 inhibition with sitagliptin is associated with sustained protection against ischemic left ventricular dysfunction in a pilot study of patients with type 2 diabetes mellitus and coronary artery disease. Circ Cardiovasc Imaging. 2014;7:274–81.
Astrup A, Rössner S, Van Gaal L, Rissanen A, Niskanen L, Al Hakim M, et al. NN8022-1807 Study Group. Effects of liraglutide in the treatment of obesity: a randomised, double-blind, placebo-controlled study. Lancet. 2009;374:1606–16.
Edwards RJ, Redwood SR, Lambiase PD, Tomset E, Rakhit RD, Marber MS. Antiarrhythmic and anti-ischaemic effects of angina in patients with and without coronary collaterals. Heart. 2002;88:604–10.
Edwards RJ, Redwood SR, Lambiase PD, Marber MS. The effect of an angiotensin-converting enzyme inhibitor and a K+(ATP) channel opener on warm up angina. Eur Heart J. 2005;26:598–606.
Lambiase PD, Edwards RJ, Cusack MR, Bucknall CA, Redwood SR, Marber MS. Exercise-induced ischemia initiates the second window of protection in humans independent of collateral recruitment. J Am Coll Cardiol. 2003;41:1174–82.
Lockie TPE, Rolandi MC, Guilcher A, Perera D, De Silva K, Williams R, et al. Synergistic adaptations to exercise in the systemic and coronary circulations that underlie the warm-up angina phenomenon. Circulation. 2012;126:2565–74.
Williams RP, Manou-Stathopoulou V, Redwood SR, Marber MS. “Warm-up Angina”: harnessing the benefits of exercise and myocardial ischaemia. Heart. 2014;100:106–14.
Saha M, Redwood SR, Marber MS. Exercise training with ischaemia: is warming up the key? Eur Heart J. 2007;28:1543–4.
Wellek S, Blettner M. Vom richtigen Umgang mit dem Crossover-Design in klinischen Studien: Teil 18 der Serie zur Bewertung wissenschaftlicher Publikationen. Dtsch Arztebl Int. 2012;109:276–81.
Funding statement
The trial is supported by the Department of Health via the National Institute of Health Research comprehensive Biomedical Research Centre award to Guy’s and St Thomas’ NHS Foundation Trust in partnership with King’s College London and King’s College Hospital NHS Foundation Trust. The trial is also supported by the British Heart Foundation via a Clinical Research Training Fellowship awarded to AM (Grant Number FS/11/70/28917).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
AM, SA, BJG, SRR and MSM report no conflicts of interest pertaining to this manuscript or the trial in general.
DLB discloses the following relationships - Advisory Board: Cardax, Elsevier Practice Update Cardiology, Medscape Cardiology, Regado Biosciences; Board of Directors: Boston VA Research Institute, Society of Cardiovascular Patient Care; Chair: American Heart Association Get With The Guidelines Steering Committee; Data Monitoring Committees: Duke Clinical Research Institute, Harvard Clinical Research Institute, Mayo Clinic, Population Health Research Institute; Honoraria: American College of Cardiology (Senior Associate Editor, Clinical Trials and News, ACC.org), Belvoir Publications (Editor in Chief, Harvard Heart Letter), Duke Clinical Research Institute (clinical trial steering committees), Harvard Clinical Research Institute (clinical trial steering committee), HMP Communications (Editor in Chief, Journal of Invasive Cardiology), Journal of the American College of Cardiology (Associate Editor; Section Editor, Pharmacology), Population Health Research Institute (clinical trial steering committee), Slack Publications (Chief Medical Editor, Cardiology Today’s Intervention), WebMD (CME steering committees); Other: Clinical Cardiology (Deputy Editor); Research Funding: Amarin, AstraZeneca, Bristol-Myers Squibb, Eisai, Ethicon, Forest Laboratories, Ischemix, Medtronic, Pfizer, Roche, Sanofi Aventis, The Medicines Company; Unfunded Research: FlowCo, PLx Pharma, Takeda. The authors declare that they have no competing interests.
Authors’ contributions
AM and MSM conceived the study and participated in its design, coordination, registration and acquisition of ethical approval. AM and SA are responsible for patient screening, recruitment and examination of patients during the study. MSM is the principal investigator. SRR is the co-principal investigator and will contribute to the analysis and interpretation of data. DLB and BJG will also contribute to the analysis and interpretation of data. All authors have been involved in drafting and revising the manuscript for important intellectual content. All authors have read and approved the final manuscript prior to submission.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Myat, A., Arri, S., Bhatt, D.L. et al. Design and rationale for the randomised, double-blinded, placebo-controlled Liraglutide to Improve corONary haemodynamics during Exercise streSS (LIONESS) crossover study. Cardiovasc Diabetol 14, 27 (2015). https://doi.org/10.1186/s12933-015-0193-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-015-0193-4