
Abstract
Background
A common variant located in the promoter region of MUC5B (rs35705950) is the strongest risk factor for sporadic and familiar IPF, as well as a predictor of outcome. However, there are no data on the effect of MUC5B rs35705950 genotype on the prognosis of IPF patients on antifibrotic treatment. The aim of this study is to determine, in a phenotypically well-characterized population of patients with IPF treated with antifibrotics, the impact of MUC5B rs35705950 genotype on disease progression and survival.
Methods
88 IPF patients on antifibrotic treatment were followed-up from 2014 until transplantation, death or end of follow-up (December 2019). Disease progression was defined as a forced vital capacity (FVC) loss ≥ 5% per year. All patients were genotyped for MUC5B rs35705950 by PCR amplification and Sanger sequencing.
Results
Out of 88 patients, 61 (69%) carried the mutant T allele (TT or TG) and 27 (31%) did not (GG). Carriage of the MUC5B rs35705950 T allele was not associated with a faster decline in FVC. Conversely, at the end of the follow-up, overall survival in carriers of the TT/TG genotype was longer compared to that of the GG genotype carriers. FVC (L) at baseline and time to respiratory failure at rest were independent predictors of worse prognosis.
Conclusions
In IPF patients on antifibrotic treatment, carriage of the MUC5B rs35705950 T allele is associated with longer survival, highlighting the usefulness of MUC5B genetic data in clinical decision making.
Similar content being viewed by others

Background
Idiopathic pulmonary fibrosis (IPF) is a chronic progressive fibrosing interstitial lung disease of unknown origin, characterized by relentless respiratory failure leading to death within 3–5 years from diagnosis [1]. IPF is believed to occur in genetically susceptible individuals because of an aberrant wound-healing response following repetitive alveolar microinjury, resulting in scarring of the lung parenchyma and irreversible loss of function. IPF is likely to result from a complex interaction between environmental and genetic factors; for instance, as many as 20% of affected individuals report to have a family member with pulmonary fibrosis [2].
In 2011, Seibold and colleagues, using a genome-wide linkage analysis, demonstrated that the minor allele (T) of a single nucleotide polymorphism (SNP) located 3 kb upstream of the MUC5B gene transcription start site on 11p15 (rs35705950) was present in 38% of subjects with sporadic IPF and in 34% of subjects with familial interstitial pneumonia [3]. Notably, the risk of disease development increased in a dose-dependent manner, from an odds ratio of 9 for heterozygous carriers of the T allele (i.e., GT) up to 21.8 for the homozygous carriers [3]. The association of MUC5B rs35705950 with IPF has been replicated in several independent cohorts [4,5,6,7,8] and represents the strongest genetic risk factor for sporadic and familial IPF described thus far.
MUC5B encodes a mucin 5B precursor protein that contributes to airway mucus production and homeostasis [9]. Although the precise mechanisms through which MUC5B dysregulation contributes to IPF development are currently unknown, MUC5B overexpression may cause mucociliary dysfunction, retention of particles and disruption of the normal reparative mechanisms in the distal lung, leading to chronic fibroproliferation and regenerative process that results in honeycomb cyst formation [10,11,12,13,14].
MUC5B rs35705950 T allele not only predisposes to IPF but has also been associated with improved survival, although this latter association remains debated and somehow controversial.
With this background, the aim of our study was to evaluate the influence of MUC5B rs35705950 genotype on disease behavior and survival of IPF patients on antifibrotic treatment. To the best of our knowledge, this has never been investigated before.
Methods
Study population and study design
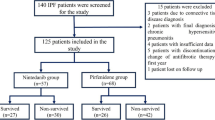
In this longitudinal retrospective study, we analyzed a consecutively collected cohort of well-characterized Caucasian adult patients with sporadic IPF referred to our center between April 2014 and September 2018. Patients were followed-up until transplantation, death or end of follow-up (December 2019), and those who permanently discontinued treatment were excluded from the study. Eighty-eight patients were included in the study (Table 1). The diagnosis of IPF was re-evaluated according to the ATS/ERS/JRS/ALAT guidelines [1]. Occupational or environmental exposure and connective tissue disease were excluded, and only sporadic IPF were considered for the analysis.
Patients were followed clinically and functionally for at least one year after initiation of antifibrotic. Patients were treated with pirfenidone or nintedanib according to eligibility criteria and the risk of associated adverse events.
Based on their annual rate of decline in absolute FVC% pred. during the first year of treatment, patients were defined as progressors (≥ 5%pred.) or stable (< 5%pred.), as previously reported [15, 16]. Improvement of FVC was expressed as negative value.
The progression-free survival (PFS) was calculated from the time of treatment initiation until functional progression, which was defined as absolute FVC% pred. loss ≥ 5% compared to the basal FVC% pred.
Based on the level of oxygen in the blood (PaO2), we defined respiratory failure when this value was < 60 mmHg (8.0 kPa).
The time to development of respiratory failure (RF) on exercise and at rest was defined as the time from treatment initiation and development of RF.
The occurrence of acute exacerbation of IPF, defined as an acute worsening of dyspnea with bilateral ground glass opacities superimposed on the UIP pattern not fully explained by fluid overload [17], has been collected.
Blood sample was taken for each patient included in the study for DNA extraction and MUC5B rs35705950 genotyping. Based on their MUC5B genotype, patients were then divided in two groups (TT/TG or GG genotype).
The study was performed in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of the University Hospital of Padova (4280/AO/17). Informed consent was obtained for all study participants.
Sample processing were described in the Additional file 1.
Statistical analysis
Categorical variables are described as absolute (n) and relative values (%) whereas continuous variables are described as median and interquartile range. To compare demographic data and baseline clinical characteristics between TT/TG and GG genotypes, Chi square test and Fisher’s exact test for categorical variables and Mann–Whitney U test for continuous variables were used, as appropriate. Due to the low number of events, in survival analysis, death and death/lung transplantation were combined. Survival was estimated using the Kaplan–Meier method and the p-value of the log-rank test was reported. Analysis on progression was conducted using the Cumulative Incidence Functions (CIF) to account for competing risks.
Clinical characteristics were evaluated to determine their relationship with survival in a univariate analysis of Cox proportional hazards regression testing. The time dependency was evaluated via visual examination of Schoenfeld residuals plot. Variables with a statistically significant association with overall survival on univariate analysis were included in a multivariate Cox proportional hazard regression test to find factors independently associated with disease progression.
All data were analyzed using SPSS Software version 25.0 (New York, NY, US: IBM Corp. USA) and R software. P-values < 0.05 were considered statistically significant.
Results
Clinical and functional characteristics
Clinical and functional characteristics at baseline for the entire study population are shown in Table 1. Most patients were males and former smokers with a median age at diagnosis of 70 years. Almost half of the patients (56%) had a radiological diagnosis, while the remaining required histological confirmation. Mean FVC was 77%, reflecting a mild functional defect, and cardiovascular disease represented the most frequent comorbidity (72%). During their first year of treatment, 63 patients (72%) remained functionally stable while 25 (28%) progressed. Over the entire study period (2014–2019) 27 patients (31%) died, 5 (6%) were transplanted and 5 (6%) experienced an acute exacerbation.
The allele frequency of the MUC5B rs35705950 T was 42% (74/176), while the frequency of the wild type G allele was 58% (102/176). The MUC5B rs35705950 genotype frequencies met the Hardy–Weinberg equilibrium (Additional file 2: Table S1).
Based on the absence or presence of the minor allele (T) either in homozygosity or in heterozygosity, the population was categorized in two groups: patients with TT/TG genotype (n = 61, 69%) and with GG genotype (n = 27, 31%) (Table 1). The two groups did not differ regarding age, sex, body mass index, comorbidities and antifibrotic treatment. Patients carrying the GG genotype had consistently higher smoking history (30 vs. 10 PY; p < 0.001), lower FVC at treatment start (2.32 vs. 2.86L, p = 0.02; 68 vs. 78%, p = 0.05) and more radiological diagnosis (74 vs. 48%, p = 0.03) compared to TT/TG genotype. However, FVC decline (at the first year) and the percentage of patients with stable disease were similar between the two groups. Respiratory failure (RF) at rest occurred later in patients with the TT/TG genotype (31 vs. 24 months, p = 0.04) (Table 1).
Progression-free survival and survival analysis
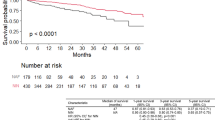
The progression-free survival was similar between patients with the TT/TG and GG genotypes, with a median of 19 months and 20 months, respectively (p = 0.21) (Fig. 1). On univariate analysis earlier occurrence of RF at rest and on exercise and higher levels of neutrophils were associated with disease progression. However, on multivariate analysis, only earlier occurrence of RF at rest (HR 2.36, 95%CI 1.12–4.97; p = 0.02) was independently associated with disease progression in the entire population (Additional file 3: Table S2). Conversely, survival analysis revealed that patients carrying the GG genotype had a significantly worse survival than patients carrying the TT/TG genotypes (42 vs. 74 months, respectively; HR 2.59, 95%CI 1.24–5.40, p = 0.0082) (Fig. 2).

Progression-free survival of TT/TG and GG genotype patients. The red line represents the progression-free survival in the TT/TG group and the green line represents the progression-free survival in the GG group. Kaplan Meier analysis was used with a log-rank test (HR 1.41, 95% CI 0.81–2.44; p = 0.21)

Survival analysis of TT/TG and GG genotype patients. The red line represents the survival in the TT/TG group and the green line represents the survival in the GG group. Kaplan Meier analysis was used with a log-rank test (HR 2.59, 95% CI 1.24–5.40; p = 0.0082)
On multivariate analysis, earlier occurrence of RF at rest (HR 36.7, 95%CI 2.83–47.78; p = 0.006) and lower FVC (L) at treatment initiation (HR 77.2, 95%CI 2.99–199.0; p = 0.009) were significantly associated with mortality (Table 2).
When death is considered together with transplantation, we confirmed that patients carrying the GG genotype had a significantly worse survival than patients carrying the TT/TG genotypes (41 vs. 71 months, respectively; HR 2.73, 95%CI 1.34–5.54, p = 0.0038) (Fig. 3).

Combined survival and transplantation analysis of TT/TG and GG genotype patients. The red line represents the analysis in the TT/TG group and the green line represents the analysis in the GG group. Kaplan Meier analysis was used with a log-rank test (HR 2.73, 95% CI 1.34–5.54; p = 0.0038)
In further analysis, stratifying patients with the TT/TG and GG genotypes by the median time to RF at rest (26 months) and FVC at treatment start (2.6L), a significantly higher percentage of GG genotype carriers had a FVC lower than the median value (67 vs. 41%, p = 0.02), whereas no differences were observed with regard to development of RF at rest (Additional file 4: Table S3).
Discussion
This study shows for the first time that in IPF patients on antifibrotic treatment, survival may be affected by carriage of MUC5B rs35705950 T allele, whether in homozygous or heterozygous form.
MUC5B encodes a major gel-forming mucin that is secreted by proximal submucosal glands and distal airway secretory cells, and plays a key role in mucociliary clearance and host defense [10,11,12,13,14]. A common variant in the promoter region of MUC5B gene has been identified as the strongest genetic risk factor for sporadic and familiar pulmonary fibrosis, although its role in disease development remains speculative. Moreover, mutant T allele has also been associated with pulmonary fibrosis in asbestosis [18], chronic HP [19] and rheumatoid arthritis-ILD [20].
Whether carriage of the mutant rs35705950 T allele has prognostic implications in patients with IPF is also debated, and conflicting results have been reported. However, these studies were performed before antifibrotics became the standard of care for patients with IPF and the effect of MUC5B rs35705950T on treatment response could not be assessed.
The finding of our study is in line with previous work by Peljto et al. [6], who described the protective effect of the MUC5B rs35705950 T allele in two IPF independent cohorts, one enrolled in the INSPIRE trial and the other recruited at the University of Chicago between 2007 and 2010. Moreover, rs35705950 T was also reported to be independently associated with lower bacterial burden in the bronchoalveolar lavage (p = 0.01), lower lung function decline and mortality [21].
Conversely, Jiang and colleagues showed that T allele was associated to increased mortality in a Chinese population [22]; specifically, T allele carriers had a more severe disease, as assessed by lower FVC and DLCO. One bias that makes it difficult to compare these studies was the T allele frequency of 20%, consistently lower to that reported in previous studies [3, 6, 7] (almost 40%) and replicated in our cohort. Indeed, as with many other genes, the frequency of MUC5B polymorphisms depends on the individual’s ethnic background, with a lower prevalence reported among Asians compared to white non-Hispanics [23].
Nonetheless, the prognostic role of MUC5B polymorphism is under debate, and conflicting results have been recently published as abstracts by two study groups, where no effect of MUC5B variant on survival in IPF patients has been shown [24, 25]. In both cases, it was not clarified whether IPF patients were on antifibrotic treatment or not.
The reason why the T allele may increase the risk of developing IPF in the general population, but confers a survival advantage within the IPF population, can only be speculated upon.
The genetic peculiarity of the MUC5B rs35705950 polymorphism resides in being a common variant with a high effect. Indeed, variants that are common in the general population (i.e., polymorphisms) rarely determine significant clinical or biological effects, except for conferring increased disease susceptibility. Conversely, rare variants (i.e., mutations) tend to be highly penetrant with substantial phenotypic effect. The wild type (G) and mutant (T) allele may interact with distinct environmental factor to determine opposite effect on disease susceptibility and prognosis, but this needs to be explored further. Intuitively, carriers of the T allele may have a better survival than noncarriers as a result of a slower disease progression, but this does not seem to be the case. Indeed, evidence of an association of less severe pathological changes and MUC5B polymorphism is reported, but it is not clear how these changes were defined [26]. Moreover, in a study by Stock [7], it was described only a trend towards a longer time to decline in FVC (HR 0.59, p = 0.052) in those carrying the T allele when multivariate stepwise regression was used.
IPF population in our cohort had a relatively stable disease under antifibrotics, with FVC decline of approximately 50 mL/year, similar in TT/TG and GG carriers. Moreover, the survival rate was very high, up to 70% at 5 years with only 5 cases of acute exacerbations leading the patient to death, confirming the efficacy of antifibrotic treatment in reducing mortality and also acute exacerbations. This rate is higher to that reported in literature; indeed, a recent study described survival rate of the INSIGHT-IPF registry [27] of nearly 60% at 2 years in the treated group, but the disease was more severe compared to our cohort.
Similarly to FVC decline, no between-group difference was observed in progression-free survival, that was nearly two years, supporting the beneficial effect of antifibrotic treatment in IPF, irrespective of MUC5B genotype [28,29,30].
However, at treatment initiation the two groups differed in terms of FVC, which was an independent predictor of mortality. Functional differences between TT and GG genotypes were described also by Peljto and coworkers [6], but authors did not clarify whether the difference was significant; however, in multivariate analysis, MUC5B genotype was associated with survival independently from FVC.
Given the prognostic role of FVC in IPF, it is not surprising that patients with a lower FVC at baseline had a worse survival, and that patients with more preserved lung function at diagnosis live longer [31, 32]. What remains difficult to explain, and somehow counterintuitive, is why patients with more preserved lung function are diagnosed earlier. Answering to this question requires larger prospective studies.
MUC5B has an important role in airway immunity, similar to other mucins, by capturing and removing infectious agents through mucociliary clearance [33]. MUC5B rs35705950 T allele is associated with overproduction and accumulation of mucin in distal airspaces and this could lead to an impaired mucociliary activity, that may trigger cough [3]. Interestingly, the mutant MUC5B allele has also been associated with cough severity [34]. Therefore, patients with early cough may seek medical attention when their lung function is still preserved, which may confer a survival benefit.
Another potential consequence of mucociliary dysfunction is the retention of inhaled substances (air pollutants, cigarette smoke, microorganisms, etc.) and endogenous inflammatory debris that over time may result in temporally and spatially distinct areas of microscopic scaring and progressive fibroproliferation in the lung. In this regard, Seibold [3] reported an association between MUC5B gene polymorphism and honeycomb cysts, one of the pathologic hallmarks of IPF. In subjects with IPF, regions of dense accumulation of MUC5B were observed in areas of microscopic honeycombing and involved patchy staining of the metaplastic epithelia lining the honeycomb cysts [35].
These pathological changes are reflected in the radiological abnormalities, characteristic of IPF. Indeed, MUC5B polymorphism is associated with a more typical subpleural distribution of fibrosis and with a greater proportion of confident radiological diagnosis (probable UIP and UIP) [36]. In our cohort, the presence of T allele MUC5B polymorphism was associated with a lower percentage of radiological diagnosis, which implies that carriers of the T allele did not have a CT pattern of UIP and required a histological diagnostic confirmation. In the study by Chung and coworkers [36], no information about functional parameters were given, age was lower compared to our cohort, suggesting a possible more advanced disease.
Our study has some limitation. Firstly, the study population is relatively small and there is no independent validation cohort. Secondly, the retrospective nature of the study might have introduced unintentional biases. However, the study population was carefully characterized and enrolled consecutively, which may have mitigated the selection bias. Finally, although we selected only sporadic cases, three patients were younger than 50 years, which makes one wonder about familiar disease. To the best of our knowledge they are all sporadic cases, although telomere gene mutations screening and monitoring extended to their family members would be needed to detect family aggregation.
Conclusions
In conclusion, we have shown for the first time that MUC5B rs35705950 genotype does not seem to affect response to antifibrotic treatment in patients with IPF. In addition, carriage of the mutant T allele is associated with longer survival in IPF patients on antifibrotic treatment. Larger studies and genotyping of additional genes involved in disease pathogenesis are needed to assess the role of genotype stratification in clinical trial design and in clinical decision making.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- IPF:
-
Idiopathic pulmonary fibrosis
- FVC:
-
Forced vital capacity
- PCR:
-
Polymerase chain reaction
- TTP:
-
Time to progression
- RF:
-
Respiratory failure
References
Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198:e44–68.
Loyd JE. Pulmonary fibrosis in families. Am J Respir Cell Mol Biol. 2003;29:S47-50.
Seibold MA, Wise AL, Speer MC, Steele MP, Brown KK, Loyd JE, et al. A common MUC5B promoter polymorphism and pulmonary fibrosis. N Engl J Med. 2012;364:1503–12. https://doi.org/10.1056/NEJMoa1013660.
Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, Steele MP, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. 2013;45:613–20. https://doi.org/10.1038/ng.2609.
Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med. 2013;1:309–17. https://doi.org/10.1016/S2213-2600(13)70045-6.
Peljto AL, Zhang Y, Fingerlin TE, Ma SF, Garcia JG, Richards TJ, et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA. 2013;309:2232–9. https://doi.org/10.1001/jama.2013.5827.
Stock CJ, Sato H, Fonseca C, Banya WAS, Molyneaux PL, Adamali H, et al. Mucin 5B promoter polymorphism is associated with idiopathic pulmonary fibrosis but not with development of lung fibrosis in systemic sclerosis or sarcoidosis. Thorax. 2013;68:436–41. https://doi.org/10.1136/thoraxjnl-2012-201786.
Moore C, Blumhagen RZ, Yang IV, Walts A, Powers J, Walker T, et al. Resequencing study confirms that host defense and cell senescence gene variants contribute to the risk of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2019;200:199–208.
Roy MG, Livraghi-Butrico A, Fletcher AA, McElwee MM, Evans SE, Boerner RM, et al. Muc5b is required for airway defence. Nature. 2014;505:412–6.
Seibold MA, Smith RW, Urbanek C, Groshong SD, Cosgrove GP, Brown KK, et al. The idiopathic pulmonary fibrosis honeycomb cyst contains a mucociliary pseudostratified epithelium. PLoS ONE. 2013;8:e58658.
Helling BA, Gerber AN, Kadiyala V, Sasse SK, Pedersen BS, Sparks L, et al. Regulation of MUC5B expression in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2017;57:91–9.
Evans CM, Fingerlin TE, Schwarz MI, Lynch D, Kurche J, Warg L, et al. Idiopathic pulmonary fibrosis: a genetic disease that involves mucociliary dysfunction of the peripheral airways. Physiol Rev. 2016;96:1567–91.
Hancock LA, Hennessy CE, Solomon GM, Dobrinskikh E, Estrella A, Hara N, et al. Muc5b overexpression causes mucociliary dysfunction and enhances lung fibrosis in mice. Nat Commun. 2018;9:5363.
Nakano Y, Yang IV, Walts AD, Watson AM, Helling BA, Fletcher AA, et al. MUC5B promoter variant rs35705950 affects MUC5B expression in the distal airways in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2016;193:464–6.
Kreuter M, Costabel U, Richeldi L, Cottin V, Wijsenbeek M, Bonella F, et al. Statin therapy and outcomes in trials of nintedanib in idiopathic pulmonary fibrosis. Respiration. 2018;95:317–26.
Balestro E, Cocconcelli E, Giraudo C, Polverosi R, Biondini D, Lacedonia D, et al. High-resolution CT change over time in patients with idiopathic pulmonary fibrosis on antifibrotic treatment. J Clin Med. 2019;8(9):1469.
Collard HR, Ryerson CJ, Corte TJ, Jenkins G, Kondoh Y, Lederer DJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. an international working group report. Am J Respir Crit Care Med. 2016;194(3):265–75.
Platenburg MGJP, Wiertz IA, van der Vis JJ, Crestani B, Borie R, Dieude P, et al. The MUC5B promoter risk allele for idiopathic pulmonary fibrosis predisposes to asbestosis. Eur Respir J. 2020;55(4):1902361.
Ley B, Newton CA, Arnould I, Elicker BM, Henry TS, Vittinghoff E, et al. The MUC5B promoter polymorphism and telomere length in patients with chronic hypersensitivity pneumonitis: an observational cohort-control study. Lancet Respir Med. 2017;5(8):639–47.
Juge PA, Lee JS, Ebstein E, Furukawa H, Dobrinskikh E, Gazal S, et al. MUC5B promoter variant and rheumatoid arthritis with interstitial lung disease. N Engl J Med. 2018;379(23):2209–19.
Molyneaux PL, Cox MJ, Willis-Owen SA, Mallia P, Russell KE, Russell AM, et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2014;190:906–13.
Jiang H, Hu Y, Shang L, Li Y, Yang L, Chen Y. Association between MUC5B polymorphism and susceptibility and severity of idiopathic pulmonary fibrosis. Int J Clin Exp Pathol. 2015;8:14953–8.
Wang C, Zhuang Y, Guo W, Cao L, Zhang H, Xu L, et al. Mucin 5B promoter polymorphism is associated with susceptibility to interstitial lung diseases in Chinese males. PLoS ONE. 2014;9:e104919.
Stock C, Molyneaux P, Saunders P, Kokosi M, George P, Kouranos V, et al. MUC2MUC5B and TOLLIP variants: no association with disease progression and survival in an IPF cohort [abstract]. Eur Respir J. 2020;56:736.
Bonella F, Campo I, Boerner E, Theegarten D, Guzman J, Costabel U, et al. Potential clinical utility of MUC5B and TOLLIP single nucleotide polymorphisms (SNP) in in the management of patients with IPF [abstract]. Eur Respir J. 2019;54:5370.
Cosgrove GP, Groshong SD, Peljto AL, Talbert J, McKean D, Markin C, et al. The MUC5B promoter polymorphism is associated with a less severe pathological form of familial interstitial pneumonia (FIP) [abstract]. Am J Respir Crit Care Med. 2012;185:A6865.
Behr J, Prasse A, Wirtz H, Koschel D, Pittrow D, Held M, et al. Survival and course of lung function in the presence or absence of antifibrotic treatment in patients with idiopathic pulmonary fibrosis: long-term results of the INSIGHTSIPF registry. Eur Respir J. 2020;56:1902279.
King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2083–92.
Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–82.
Biondini D, Balestro E, Lacedonia D, Cerri S, Milaneschi R, Luppi F, et al. Pretreatment rate of decay in forced vital capacity predicts long-term response to pirfenidone in patients with idiopathic pulmonary fibrosis. Sci Rep. 2018;8:5961. https://doi.org/10.1038/s41598-018-24303-4.
Ley B, Collard HR, King TE Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2011;183:431–40.
Erbes R, Schaberg T, Loddenkemper R. Lung function tests in patients with idiopathic pulmonary fibrosis Are they helpful for predicting outcome? Chest. 1997;111:51–7.
Schwartz DA. Idiopathic pulmonary fibrosis is a genetic disease involving mucus and the peripheral airways. Ann Am Thorac Soc. 2018;15:S192–7.
Scholand MB, Wolff R, Crossno PF, Sundar K, Winegar M, Whipple S, et al. Severity of cough in idiopathic pulmonary fibrosis is associated with MUC5 B genotype. Cough. 2014;10:3.
Yang IV, Fingerlin TE, Evans CM, Schwarz MI, Schwartz DA. MUC5B and idiopathic pulmonary fibrosis. Ann Am Thorac Soc. 2015;12:S193–9.
Chung JH, Peljto AL, Chawla A, Talbert JL, McKean DF, Rho BH, et al. CT imaging phenotypes of pulmonary fibrosis in the MUC5B promoter site polymorphism. Chest. 2016;149:1215–22.
Acknowledgements
Not applicable.
Funding
There is no source of funding to declare.
Author information
Authors and Affiliations
Contributions
DB and EC conceived this research, performed the statistical analysis and the investigation, and drafted the original manuscript. NB and GC performed data curation. SL performed data curation and the statistical analysis. CR performed data curation and the investigation. GL and DG performed the statistical analysis. SB, MGC and MS supervised and reviewed the final manuscript. EB and PS conceived this research, supervised and reviewed the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the ethics committee of the University Hospital of Padova (4280/AO/17).
Consent for publication.
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Sample processing.
Additional file 2: Table S1.
MUC5B rs35705950 genotype frequency.
Additional file 3: Table S2.
Predictive factors of progression in the entire population of IPF patients treated with antifibrotics.
Additional file 4: Table S3.
Occurrence of respiratory failure (RF) at rest and FVC (L) at treatment initiation according to MUC5B genotype (TT/TG vs. GG patients).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Biondini, D., Cocconcelli, E., Bernardinello, N. et al. Prognostic role of MUC5B rs35705950 genotype in patients with idiopathic pulmonary fibrosis (IPF) on antifibrotic treatment. Respir Res 22, 98 (2021). https://doi.org/10.1186/s12931-021-01694-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-021-01694-z