
Abstract
Background
Olodaterol is a novel, inhaled long-acting β2-agonist (LABA) with >24-h duration of action investigated in asthma and chronic obstructive pulmonary disease.
Methods
Two multicentre studies examined the efficacy and safety of 4 weeks’ once-daily (QD) olodaterol (2, 5, 10 and 20 μg, with background inhaled corticosteroids) in patients with asthma. One randomised, double-blind, parallel-group study (1222.6; 296 patients) administered treatment in the morning. Pulmonary function tests (PFTs) were performed pre-dose (trough) and ≤3 h post-dose (weeks 1 and 2), and ≤6 h post-dose after 4 weeks; primary end point was trough forced expiratory volume in 1 s (FEV1) response (change from baseline mean FEV1) after 4 weeks. A second randomised, double-blind, placebo- and active-controlled (formoterol 12 μg twice daily) incomplete-block crossover study (1222.27; 198 patients) administered QD treatments in the evening. PFTs were performed over a 24-h dosing interval after 4 weeks; primary end point was FEV1 area under the curve from 0–24 h (AUC0–24) response (change from study baseline [mean FEV1] after 4 weeks).
Results
Study 1222.6 showed a statistically significant increase in trough FEV1 response with olodaterol 20 μg (0.147 L; 95 % confidence interval [CI]: 0.059, 0.234; p = 0.001) versus placebo, with more limited efficacy and no evidence of dose response compared to placebo across the other olodaterol doses (2, 5 and 10 μg). Study 1222.27 demonstrated increases in FEV1 AUC0–24 responses at 4 weeks with all active treatments (p < 0.0001); adjusted mean (95 % CI) differences from placebo were 0.140 (0.097, 0.182), 0.182 (0.140, 0.224), 0.205 (0.163, 0.248) and 0.229 (0.186, 0.272) L for olodaterol 2, 5, 10 and 20 μg, respectively, and 0.169 (0.126, 0.211) for formoterol, providing evidence of increased efficacy with higher olodaterol dose. Olodaterol was generally well tolerated, with a few events associated with known sympathomimetic effects, mainly with 20 μg.
Conclusions
The LABA olodaterol has >24-h duration of action. In patients with asthma, evidence of bronchodilator efficacy was demonstrated with statistically and clinically significant improvements in the primary end point of trough FEV1 response measured in clinics over placebo for the highest administered dose of 20 μg in Study 1222.6, and statistically and clinically significant improvements versus placebo in FEV1 AUC0–24 responses at 4 weeks for all doses tested in Study 1222.27, which also exhibited a dose response. Bronchodilator efficacy was seen over placebo for all olodaterol doses for morning and evening peak expiratory flow in both studies. All doses were well tolerated.
Trial registrations
NCT00467740 (1222.6) and NCT01013753 (1222.27).
Similar content being viewed by others


Background
The use of inhaled long-acting β2-agonists (LABAs) is recommended in asthma guidelines as add-on to inhaled corticosteroids (ICS) and has been shown to improve lung function and reduce symptoms and future risk of severe exacerbations [1–3]. Twice-daily (BID) LABAs such as formoterol and salmeterol are well-established controllers in asthma as add-on to ICS [2] and fixed-dose combination products have been available for a number of years. The development of once-daily (QD) LABAs for the treatment of asthma may simplify the dosing strategy and potentially improve adherence and outcomes [4–6].
Olodaterol is a novel LABA with 24-h bronchodilatory activity and is characterised by high β2 selectivity and almost full agonist activity at β2 adrenoreceptors [7, 8]. Initial single-dose studies in asthma and chronic obstructive pulmonary disease (COPD) have provided clinical evidence of ≥24-h activity. In patients with asthma, olodaterol was evaluated in a single-dose study that showed significant protection against methacholine bronchoconstriction compared to placebo for ≤32 hours [9]. In COPD, a Phase II, clinical, single-dose study showed that QD olodaterol effectively maintained bronchodilation over 24 h [10–12]. Olodaterol has recently been approved at a dose of 5 μg QD for use as maintenance treatment for patients with COPD in Europe and the USA, supported by data from the Phase III olodaterol clinical trial programme that established the long-term efficacy of QD olodaterol for lung-function improvement [13–16].
To further assess the optimum dose of olodaterol in patients with asthma, two dose-finding studies with different designs were conducted sequentially. Both studies aimed to determine the optimum dose of olodaterol inhalation solution delivered by the Respimat® inhaler QD for 4 weeks in patients with asthma. The first study used a parallel-treatment-group design and the second was conducted using an incomplete-block crossover design. Both studies were randomised and double blind. Data from these studies have previously been presented at the American Thoracic Society Annual Meeting [17, 18].
Methods
Study design
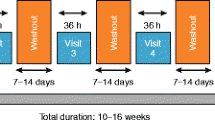
Study 1222.6 was a multicentre, randomised, double-blind, placebo-controlled, parallel-group study to assess the efficacy and safety of 4 weeks of treatment with orally inhaled olodaterol 2, 5, 10 and 20 μg delivered via Respimat® device QD (in the morning) in patients with asthma to determine the optimum dose and confirm 24-h bronchodilation with this posology (Fig. 1a).

Designs of studies: (a) 1222.6 and (b) 1222.27. QD: once daily; BID: twice daily; ICS: inhaled corticosteroid
Study 1222.27 was a multicentre, randomised, double-blind, double-dummy, active- and placebo-controlled, incomplete-block crossover study to determine the efficacy and safety of four different doses of olodaterol (2, 5, 10 and 20 μg) delivered by the Respimat® device QD in the evening versus BID formoterol (12 μg) delivered by the Aerolizer® inhaler and matching placebo for 4 weeks in patients with asthma (Fig. 1b). The 16-week treatment phase comprised four 4-week treatment periods without intervening washout periods, with efficacy end points being assessed after 4 weeks of each treatment at study centre visits.
For Study 1222.6, it was estimated that a sample size of 60 patients per treatment group was required to detect with ≥90 % power a difference of 0.15 L from placebo in the primary end point (trough forced expiratory volume in 1 s [FEV1] response) at the 5 % significance level.
For Study 1222.27, a sample size of approximately 190 patients was planned to detect a difference from placebo of 0.1 L with 95 % power for a single treatment comparison (assuming a standard deviation for the paired differences of 0.25 L), allowing for the loss of efficiency caused by the incomplete-block crossover design, for patient attrition and in order to achieve a balanced design.
Further details of study designs, assessments performed, key inclusion and exclusion criteria, and statistical methodologies are detailed in Additional file 1: Tables S1 and S2.
Both studies were performed in accordance with the principles laid down by the Declaration of Helsinki, International Committee on Harmonisation, Good Clinical Practice Guidelines, European Medical Device Directive and local regulations. Prior to initiation of the studies, the protocols were approved by the ethics research boards of the respective institutions and written, informed consent was obtained from all patients. Both studies were registered with ClinicalTrials.gov (Study 1222.6: NCT00467740; Study 1222.27: NCT01013753).
End points
The primary end point for Study 1222.6 was trough FEV1 response (response being defined as change from study baseline mean FEV1) at 4 weeks. Secondary and other end points included FEV1 area under the curve from 0–3 h (AUC0–3) response (response defined as change from study baseline FEV1) after the first dose and after 1, 2 and 4 weeks of treatment, weekly mean pre-dose morning peak expiratory flow (PEF) response (patient diaries), weekly mean evening PEF response, total score on the Asthma Control Questionnaire (ACQ) and safety.
The primary end point for Study 1222.27 was FEV1 area under the curve from 0–24 h (AUC0–24) response (defined as change from study baseline mean FEV1) after 4 weeks of treatment. Secondary and other end points included FEV1 at individual time points over 24 h post-dose, trough and peak FEV1 responses at the end of each 4-week treatment period, weekly pre-dose PEF (from the patient diaries), control of asthma as assessed by total score on the ACQ and quality of life as assessed by the standardised version of the Asthma Quality of Life Questionnaire (AQLQ[S]) at the end of the treatment period.
For both studies, pulmonary function tests (PFTs) were conducted at screening and baseline. In Study 1222.6 (parallel-group design), PFTs were conducted at 1 h and at 10 min before the morning dose and 30 min, 1, 2 and 3 h post-dosing at the start of randomised treatment and after 1, 2 and 4 weeks of randomised treatment. This was extended to 4, 5 and 6 h post-dosing at the final treatment visit. PFTs were also conducted at follow-up.
In Study 1222.27 (incomplete-block crossover design), PFTs were performed at screening, at the end of the 2-week baseline period, after 4 weeks of each randomised treatment and at follow-up. At the baseline visit, two PFTs were performed at 1 h and at 10 min prior to the first evening dose of study medication. At the end of each 4-week treatment period, PFTs were performed at 1 h and at 10 min before the evening dose and at defined time points within 24 h post-dosing (30 min, 1, 2, 3, 4 h, 11 h 50 min, 12 h 30 min, 13, 14, 15, 16, 18, 20, 22, 23 h and 23 h 50 min post-dose). A single PFT was performed at the follow-up visit.
Results
In Study 1222.6, a total of 296 patients were randomised to treatment at 37 centres and 289 patients completed the study (Fig. 2a), while in Study 1222.27, 198 patients were randomised at 27 centres and 182 patients completed the study (Fig. 2b). Across the studies, demographic characteristics were generally similar (Tables 1 and 2). Mean ages in Study 1222.6 ranged between 43.6 and 46.3 years across the treatment groups, while in Study 1222.27 the mean age was 45.0 years; across the studies there were slightly more women than men. The majority of patients were never smokers.

Patient disposition in studies: (a) 1222.6 and (b) 1222.27
In Study 1222.6, for the primary end point of trough FEV1 response after 4 weeks of treatment, there was a similar degree of improvement compared with placebo for the 2, 5 and 10 μg doses of olodaterol: mean (95 % confidence interval [CI]) difference of 0.080 (−0.008, 0.167) for the 2 μg olodaterol treatment group, 0.086 (−0.003, 0.174) for the 5 μg treatment group and 0.076 (−0.012, 0.164) for the 10 μg group. The differences between the 20 μg dose and placebo were largest and statistically significant: mean (95 % CI) 0.147 (0.059, 0.234; p = 0.0011) (Fig. 3a and Additional file 1: Table S3). There was no evidence of a dose–response relationship across the 2, 5 and 10 μg doses of olodaterol (Additional file 1: Table S3), with similar results for trough FEV1 response observed at weeks 1 and 2 (Fig. 3a).

FEV1 assessments in Study 1222.6. (a) Trough FEV1 response weeks 1–4 from baseline (±SE), (b) FEV1 profile over time at week 4 from baseline (±SE) and (c) FEV1 AUC0–3 response weeks 1–4 from baseline (±SE). FEV1: forced expiratory volume in 1 s; SE: standard error; AUC0–3: area under the curve from 0–3 h
Figure 3b shows FEV1 at individual time points up to 3 h post-dose at week 4 in Study 1222.6; with olodaterol 20 μg these were statistically significant (p < 0.001) at all time points. Values for FEV1 AUC0–3 response were also statistically significantly (p < 0.05) higher in each of the olodaterol treatment groups compared to placebo at weeks 1, 2 and 4 (Fig. 3c). For both FEV1 over time at week 4 and FEV1 AUC0–3 at weeks 1, 2 and 4, the same pattern was observed as for the primary end point, with little separation between the 2, 5 and 10 μg doses.
A different pattern was observed for other secondary end points, with some evidence of a dose–response relationship apparent for the mean change in weekly morning pre-dose PEF from baseline (Table 3). Similar results were apparent for the evening PEF response (Table 3). After 4 weeks of treatment, mean total ACQ scores also decreased from baseline in each of the active treatment groups, although the changes compared with placebo were only statistically significant (p < 0.05) in the olodaterol 10 and 20 μg dose groups. The mean (95 % CI) differences compared to placebo were −0.142 with 2 μg (−0.368, 0.083; p = 0.216), −0.197 with 5 μg (−0.422, 0.029; p = 0.087), −0.328 with 10 μg (−0.552, −0.103; p = 0.004) and −0.276 with 20 μg (-0.499, −0.052; p = 0.016).
In Study 1222.27, the mean FEV1 AUC0–24 response after 4 weeks of treatment was highly statistically significantly different compared to placebo for all doses of olodaterol and formoterol (p < 0.0001) (Additional file 1: Table S4), with a clear dose–response relationship in the treatment effect for olodaterol. The adjusted mean (95 % CI) FEV1 AUC0–24 response differences from placebo after 4 weeks of treatment were 0.140 (0.097, 0.182), 0.182 (0.140, 0.224), 0.205 (0.163, 0.248) and 0.229 (0.186, 0.272) L for 2, 5, 10 and 20 μg olodaterol, respectively; the adjusted mean difference from placebo with formoterol was 0.169 (0.126, 0.211) L.
Differences in adjusted mean FEV1 at each individual time point over the 24-h period also demonstrated a clear dose–response relationship (Fig. 4). There were highly statistically significant (p < 0.0001) improvements in trough and peak FEV1 responses at week 4, again with dose–response relationships apparent with olodaterol (Additional file 1: Table S5).

FEV1 assessments in Study 1222.27: adjusted mean FEV1 trough response at week 4. QD: once daily; BID: twice daily; PFT: pulmonary function test; FEV1: forced expiratory volume in 1 s
While there were significant differences in adjusted mean weekly morning PEF and evening PEF (home-measured) at week 4 for all doses of olodaterol and formoterol compared to placebo (p < 0.0001), the increases with olodaterol did not show the same dose–response relationship as the in-clinic visit FEV1 end points (Additional file 1: Table S6). There were statistically significant reductions in mean total ACQ scores with all active treatments during the study; however, there was no clear dose ordering between the 2, 5 and 10 μg doses. At week 4, the mean (95 % CI) differences compared to placebo were −0.321 (−0.432, −0.210) with the 2 μg dose, −0.293 (−0.403, −0.184) with the 5 μg dose, −0.326 (−0.436, −0.215) with the 10 μg dose and −0.394 (−0.505, −0.282) with the 20 μg dose; all differences were highly statistically significant (p < 0.0001). Compared to placebo, the mean (95 % CI) difference with formoterol was −0.346 (−0.457, −0.235; p < 0.0001). There were also statistically significant increases in AQLQ(S) score at week 4 with all active treatments compared to placebo (p < 0.001). Mean (95 % CI) differences to placebo were 0.289 (0.178, 0.400) with olodaterol 2 μg, 0.209 (0.099, 0.318) with the 5 μg dose, 0.262 (0.151, 0.374) with the 10 μg dose, 0.317 (0.205, 0.429) with the 20 μg dose and 0.315 (0.203, 0.426) with formoterol.
The incidence of adverse events in Study 1222.6 was 37.7 %, 40.0 %, 30.0 %, 39.3 % and 27.8 % in patients receiving olodaterol 2 μg, 5 μg, 10 μg, 20 μg and placebo, respectively (Additional file 1: Table S7). Exacerbation of asthma occurred at a higher incidence in the placebo treatment group (7.4 % compared to 1.7–3.3 % in the olodaterol groups). Only 24 adverse events (total: all treatment groups) were considered drug related, the most common being tremor, headache, dizziness, palpitations and anxiety. However, there was a higher number of patients with drug-related adverse events in the olodaterol 20 μg group compared to the lower-dose groups (16.4 % compared to 3.3–8.3 % in the other groups), the most frequent being tremor.
One patient receiving olodaterol 5 μg was withdrawn from Study 1222.6 due to an adverse event of premature ventricular contractions. Two patients experienced at least one serious adverse event during the study: one in the 10 μg group experienced pneumonia and one in the 20 μg group experienced dizziness, palpitations, hyperhidrosis and chest pain, which were considered drug related. None of the serious adverse events were fatal or life-threatening.
More female patients in the olodaterol 20 μg group experienced drug-related adverse events (seven patients; 21.2 %) than in the placebo group (two patients; 5.1 %) or other active treatment groups (one to three patients; 3.3–8.8 %). No such difference was seen for male patients.
Changes in vital signs, electrocardiogram and laboratory parameters in line with known systemic sympathomimetic effects were observed in Study 1222.6 with olodaterol doses ≥10 μg, with increased heart rate, shortened uncorrected QT interval, increased QT (Bazett corrected) and T-wave abnormalities. Although there was a statistically significant reduction in mean serum potassium levels in patients with the 20 μg olodaterol dose compared to placebo following treatment on day 1 (4.18 versus 4.38 mmol/L; p = 0.0006), no statistically significant treatment differences between placebo and any active treatment dose groups were observed at other time points during the study. Two electrocardiogram results were reported as adverse events: one patient in the placebo group with an atrioventricular block and one in the 5 μg group with ventricular extrasystoles.
The overall incidence of adverse events in Study 1222.27 was 9.9 %, 13.8 %, 15.7 %, 12.9 %, 6.4 % and 4.8 % in patients receiving olodaterol 2 μg, 5 μg, 10 μg, 20 μg, formoterol and placebo, respectively. The differences in overall adverse event incidences between treatments could not be explained by more frequent occurrences of any particular preferred term. Only three adverse events were reported by >1 % of patients on any treatment: nasopharyngitis, dyspnoea and headache (Additional file 1: Table S8). Two patients experienced serious adverse events: one patient being treated with 10 μg experienced appendicitis and one patient in the post-treatment period (last treatment olodaterol 2 μg) had a cerebral infarction. Both patients recovered (the patient with cerebral infarction recovered with sequelae) and neither serious adverse event was considered drug related.
Eight adverse events were considered related to study drug in Study 1222.27: four with olodaterol 5 μg (candidiasis, headache, palpitations and muscle spasms), two with olodaterol 10 μg (palpitations and muscle spasms) and one each with olodaterol 2 μg (insomnia) and placebo (metrorrhagia). No drug-related adverse events were reported by patients being treated with 20 μg olodaterol or formoterol.
With the exception of olodaterol 20 μg, there was generally a greater percentage of female patients reporting at least one adverse event with olodaterol treatment: 11.4 % (2 μg), 18.3 % (10 μg), 22.2 % (15 μg) and 12.7 % (20 μg) with olodaterol compared to 4.1 % with placebo and 5.8 % with formoterol. This is in comparison to 7.8 % (2 μg), 12.5 % (10 μg), 3.4 % (15 μg) and 13.2 % (20 μg) of male patients following olodaterol treatment and 5.8 % and 7.1 % following treatment with placebo or formoterol, respectively.
Analysis of the electrocardiogram results showed that, compared to placebo, there was a slight increase (mean change from baseline: 4.23 ms at 1 h) in the mean Fridericia-corrected QT interval with 20 μg olodaterol. There were statistically significant (p < 0.03) reductions in mean blood potassium concentration seen 1 h post-dose: ratios of geometric mean olodaterol:placebo (95 % CI) were 0.979 (0.962, 0.997) with 5 μg, 0.977 (0.960, 0.995) with 10 μg and 0.980 (0.962, 0.998) with 20 μg olodaterol; however, these changes were not considered to be clinically relevant.
One electrocardiogram result was reported as an adverse event: one patient with an atrioventricular block while receiving olodaterol 20 μg and formoterol.
Discussion
These studies have demonstrated that olodaterol is an inhaled LABA with a duration of effect lasting >24 h in patients with asthma when administered in addition to ICS over 4 weeks, as measured by improvements over placebo in FEV1 and home-measured morning and evening PEF in both studies. Olodaterol also improved symptoms as measured by a reduction in ACQ scores. Both studies demonstrated or indicated dose–response effects of olodaterol to a greater (Study 1222.27) or lesser (Study 1222.6) degree across the 2, 5, 10 and 20 μg doses used. The outcome variables that indicated olodaterol dose response in the two studies varied, being indicated only by secondary end points of weekly morning and evening PEF in Study 1222.6, and by the primary end point of FEV1 AUC0–24 response and secondary end points of trough, peak and individual time point FEV1 responses in Study 1222.27 (as well as other end points studied in 1222.27).
Potential reasons for the differences in the outcome variables demonstrating dose response between the studies could include the populations and/or the designs used in the two studies. However, the study populations were similar with regards to patient demographics; all patients were taking ICS and both populations demonstrated a dose response to olodaterol in some outcome variables. Therefore, it is unlikely that an important difference in patient characteristics is an adequate explanation. By contrast, the study designs used were very different. Study 1222.6 used a parallel-group design and Study 1222.27 an incomplete-block crossover design, with each patient receiving four out of a possible six treatments and acting as their own controls for the dose–response evaluations and an active comparator with formoterol. Another important difference in the design of the studies was the time of administration of olodaterol: in the morning in Study 1222.6 and in the evening in Study 1222.27.
Another study that examined the dose–response characteristics of olodaterol was also a crossover design and included patients with milder asthma, not taking ICS. The primary outcome variable was change in methacholine provocative concentration causing a 20 % fall in FEV1 (PC20). Olodaterol demonstrated a clear dose response for the changes in methacholine PC20 [9].
There are challenges in conducting β2-agonist dose–response studies in asthma. Studies examining the dose response of β2-agonists using bronchoprovocation models in asthma are inherently easier to conduct, as they involve mild, stable patients with close to normal baseline FEV1 values. When indices of bronchodilation are used to measure effect, the patients need to have baseline airflow obstruction in order to demonstrate a response and in most countries such patients would be treated with ICS. These patients may also be previously treated with LABA and washed out from this medication (for a minimum of 2 weeks in Study 1222.6 and 48 h in Study 1222.27).
Other LABAs with a duration of action of >24 h have been studied in patients with asthma. Beeh et al. demonstrated that indacaterol had a duration of bronchodilation of 24 h for the two highest doses but did not demonstrate a significant dose response for FEV1 across four doses administered [19]. Another study that included patients taking ICS and which evaluated four doses of indacaterol treatment for 7 days also did not demonstrate a dose response when FEV1 was measured [20]. The dose–response characteristics of vilanterol, another LABA, have been evaluated in patients with asthma taking ICS measured by treating with five different doses for 28 days and measuring both FEV1 and PEF [21]. Once again, while prolonged bronchodilation was observed for the three highest doses used, a significant dose response was not seen for either outcome variable. These studies demonstrate the challenges in documenting a clear dose response for a LABA seen in a primary end-point measure (as in Study 1222.27), although evidence for a dose response may be apparent in secondary end-point measures (as in Study 1222.6).
The safety profile of olodaterol was as expected for all β2-agonists, with tremor and changes in serum potassium and the QT interval in the electrocardiogram at the highest dose. There was no evidence of an increase of severe adverse events with olodaterol when compared to placebo in Study 1222.6 or formoterol in Study 1222.27.
These studies did have some limitations in that neither study was adequately powered to identify differences between the different doses of active treatments (both studies were powered for comparisons between olodaterol and placebo), though this is not unusual for Phase II dose-finding studies.
When taken together, the weight of the evidence from several olodaterol trials in asthma indicates a relevant dose–response relationship between a total daily dose of 5 and 10 μg olodaterol. In addition to the results from these present studies, further evidence has been provided by a study examining the 24-h FEV1 time profile after 3 weeks of treatment with QD or BID regimens of olodaterol (at the same total daily dose) versus placebo (NCT01311661; 1222.29). Dose responses, although not the primary objective of the studies, were consistently observed for a total daily dose of 5 versus 10 μg olodaterol (Beeh et al., manuscript in preparation).
In conclusion, treatment with olodaterol QD for 4 weeks was well tolerated at all doses and no new safety concerns were identified, although some sympathomimetic effects were observed, mainly at the 20 μg dose. Parallel, as well as incomplete-block crossover, designs may be suitable for Phase II dose-ranging studies and more than one study may be necessary to strengthen the evidence for a dose response.
Abbreviations
- ACQ:
-
Asthma Control Questionnaire
- AQLQ(S):
-
Asthma Quality of Life Questionnaire, standardised version
- AUC0–3 :
-
Area under the curve from 0–3 h
- AUC0–24 :
-
Area under the curve from 0–24 h
- BID:
-
Twice daily
- CI:
-
Confidence interval
- FEV1 :
-
Forced expiratory volume in 1 s
- ICS:
-
Inhaled corticosteroids
- LABA:
-
Long-acting β2-agonist
- PEF:
-
Peak expiratory flow
- PFT:
-
Pulmonary function test
- QD:
-
Once daily
References
Fuso L, Mores N, Valente S, Malerba M, Montuschi P. Long-acting beta-agonists and their association with inhaled corticosteroids in COPD. Curr Med Chem. 2013;20:1477–95.
Global Initiative for Asthma. Global strategy for asthma management and prevention. Updated 2015. http://www.ginasthma.org/local/uploads/files/GINA_Report_2015_May19.pdf. Accessed 29 Jul 2015.
Montuschi P, Ciabattoni G. Bronchodilating drugs for chronic obstructive pulmonary disease: current status and future trends. J Med Chem. 2015;58:4131–64.
Cazzola M, Segreti A, Matera MG. Novel bronchodilators in asthma. Curr Opin Pulm Med. 2010;16:6–12.
Cazzola M, Calzetta L, Matera MG. β2-adrenoceptor agonists: current and future direction. Br J Pharmacol. 2011;163:4–17.
Cazzola M, Matera MG. Novel long-acting bronchodilators for COPD and asthma. Br J Pharmacol. 2008;155:291–9.
Bouyssou T, Casarosa P, Naline E, Pestel S, Konetzki I, Devillier P, et al. Pharmacological characterization of olodaterol, a novel inhaled β 2-adrenoceptor agonist exerting a 24-hour-long duration of action in preclinical models. J Pharmacol Exp Ther. 2010;334:53–62.
Casarosa P, Kollak I, Kiechle T, Ostermann A, Schnapp A, Kiesling R, et al. Functional and biochemical rationales for the 24-hour-long duration of action of olodaterol. J Pharmacol Exp Ther. 2011;337:600–9.
O’Byrne PM, van der Linde J, Cockcroft DW, Gauvreau GM, Brannan JD, FitzGerald M, et al. Prolonged bronchoprotection against inhaled methacholine by inhaled BI 1744, a long-acting β2-agonist, in patients with mild asthma. J Allergy Clin Immunol. 2009;124:1217–21.
Ichinose M, Takizawa A, Izumoto T, Fukuchi Y. Efficacy of 4 weeks’ once-daily treatment with olodaterol (BI 1744), a novel long-acting β2-agonist, in Japanese patients with COPD [abstract A2931]. Am J Respir Crit Care Med. 2012;185.
van Noord JA, Smeets JJ, Drenth BM, Rascher J, Pivovarova A, Hamilton AL, et al. 24-hour bronchodilation following a single dose of the novel β2-agonist olodaterol in COPD. Pulm Pharmacol Ther. 2011;24:666–72.
Joos G, Aumann JL, Coeck C, Korducki L, Hamilton AL, van Noord JA. Comparison of 24-hour FEV1 profile for once-daily versus twice-daily treatment with olodaterol, a novel long-acting β2 agonist, in patients with COPD [poster A2930]. Presented at the 103rd Annual International Conference of the American Thoracic Society, San Francisco, California, USA, 18–23 May 2012.
Ferguson GT, Feldman GJ, Hofbauer P, Hamilton A, Allen L, Korducki L, et al. Efficacy and safety of olodaterol once daily delivered via Respimat® in patients with GOLD 2–4 COPD: results from two replicate 48-week studies. Int J Chron Obstruct Pulmon Dis. 2014;9:629–45.
Feldman GJ, Bernstein JA, Hamilton A, Nivens MC, Korducki L, LaForce C. The 24-h FEV1 time profile of olodaterol once daily via Respimat® and formoterol twice daily via Aerolizer® in patients with GOLD 2–4 COPD: results from two 6-week crossover studies. Springerplus. 2014;3:419.
Koch A, Pizzichini E, Hamilton A, Hart L, Korducki L, De Salvo MC, et al. Lung function efficacy and symptomatic benefit of olodaterol once daily delivered via Respimat® versus placebo and formoterol twice daily in patients with GOLD 2–4 COPD: results from two replicate 48-week studies. Int J Chron Obstruct Pulmon Dis. 2014;9:697–714.
Lange P, Aumann J-L, Hamilton A, Tetzlaff K, Ting N, Derom E. The 24 hour lung function time profile of olodaterol once daily versus placebo and tiotropium in patients with moderate to very severe chronic obstructive pulmonary disease. J Pulm Respir Med. 2014;4:196.
Beeh K-M, Beck E, Gahlemann M, Blahova Z, Toorawa R, Flezar M. Dose-finding study of 4-week, once-daily treatment with olodaterol, a novel long-acting β2-agonist, in patients with asthma [abstract A2764]. Am J Respir Crit Care Med. 2012;185.
O’Byrne PM, D’Urzo T, Gahlemann M, Hart L, Wang F, Beck E. Dose-finding study of once-daily treatment with olodaterol, a novel long-acting β2-agonist, in patients with asthma [abstract A3963]. Am J Respir Crit Care Med. 2012;185.
Beeh KM, Derom E, Kanniess F, Cameron R, Higgins M, van As A. Indacaterol, a novel inhaled β2-agonist, provides sustained 24-h bronchodilation in asthma. Eur Respir J. 2007;29:871–8.
LaForce C, Alexander M, Deckelmann R, Fabbri LM, Aisanov Z, Cameron R, et al. Indacaterol provides sustained 24 h bronchodilation on once-daily dosing in asthma: a 7-day dose-ranging study. Allergy. 2008;63:103–11.
Lötvall J, Bateman ED, Bleecker ER, Busse WW, Woodcock A, Follows R, et al. 24-h duration of the novel LABA vilanterol trifenatate in asthma patients treated with inhaled corticosteroids. Eur Respir J. 2012;40:570–9.
Acknowledgements
The authors would like to thank Fei Wang and Florian Voß for their assistance in preparing this manuscript. Fei Wang provided statistical support for Study 1222.6. The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors; RT provided statistical input and meets the criteria for Study 1222.27 only. Medical writing assistance was provided by Rob Kite, BSc, of Complete HealthVizion, which was contracted and compensated by Boehringer Ingelheim Pharma GmbH & Co. KG.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
This work was supported by Boehringer Ingelheim Pharma GmbH & Co. KG. PO’B has received research funding from AstraZeneca, Novartis, Amgen and Genentech, consultancy fees from AstraZeneca and Chiesi, and participated in advisory boards for Boehringer Ingelheim, GlaxoSmithKline, MedImmune, Merck, Takeda and Abbott. TD’U has received grants from Boehringer Ingelheim. K-MB has received lecture fees from Almirall Hermal, AstraZeneca, Cytos AG, Boehringer Ingelheim, Novartis, TEVA, Mundipharma, Pfizer and GlaxoSmithKline, compensation for clinical trial conduct from Almirall Hermal, AstraZeneca, Boehringer Ingelheim, Novartis, TEVA, Mundipharma, Sterna AG and GlaxoSmithKline, consultation fees from Ablynx and participated in advisory boards for Almirall Hermal, AstraZeneca, Cytos AG, Boehringer Ingelheim, Chiesi, Novartis, TEVA, Mundipharma and Pfizer. MG, LH and ZB are employees of Boehringer Ingelheim. RT is a contract statistician working for Boehringer Ingelheim and was compensated for all work on Study 1222.27, including this manuscript.
Authors’ contributions
PO’B and K-MB were the coordinating investigators for Studies 1222.6 and 1222.27, respectively. LH and ZB were the Boehringer Ingelheim trial monitors for Studies 1222.6 and 1222.27, respectively. MG was the Boehringer Ingelheim team member medicine for both studies. RT was a contract statistician for Study 1222.27. TD’U was involved in patient recruitment, data interpretation and analysis. All authors were involved in the development and review of the manuscript. All authors take full responsibility for the scope, direction, content of, and editorial decisions relating to, the manuscript, were involved at all stages of development and have approved the submitted manuscript. The authors received no compensation related to the development of the manuscript, except RT (as noted above). All authors read and approved the final manuscript.
Additional file
Additional file 1: Table S1.
Study methodology. Table S2. Key inclusion and exclusion criteria. Table S3. Adjusted mean (95 % CI) FEV1 trough and peak responses compared to placebo after 4 weeks of treatment in Study 1222.6. Table S4. Adjusted mean (95 % CI) FEV1 AUC0–24 responses compared to placebo after 4 weeks of treatment in Study 1222.27. Table S5. Adjusted mean (95 % CI) FEV1 responses compared to placebo after 4 weeks of treatment in Study 1222.27. Table S6. Adjusted mean (95 % CI) morning and evening PEF compared to placebo after 4 weeks of treatment in Study 1222.27. Table S7. Adverse events reported in Study 1222.6 occurring in more than one patient in any treatment group. Table S8. Adverse events reported in Study 1222.27 with an incidence of >1 % on any treatment on any system class or individual adverse event.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
O’Byrne, P.M., D’Urzo, T., Beck, E. et al. Dose-finding evaluation of once-daily treatment with olodaterol, a novel long-acting β2-agonist, in patients with asthma: results of a parallel-group study and a crossover study. Respir Res 16, 97 (2015). https://doi.org/10.1186/s12931-015-0249-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-015-0249-8