
Abstract
Background
A Phase II, multicentre, randomised, double-blind, placebo-controlled, crossover trial comparing the 24-h forced expiratory volume in 1 s (FEV1) time profile after 3 weeks’ treatment with once-daily (QD) or twice-daily (BID) olodaterol (at the same total daily dose) versus placebo delivered via Respimat® in patients with moderate to severe asthma.
Methods
Patients were randomised to different sequences of olodaterol with 2-week washout, either as a total daily dose of 5 μg (5 μg QD [AM] or 2.5 μg BID) or placebo, or 10 μg (10 μg QD [AM] or 5 μg BID) or placebo. Primary end point was FEV1 area under the curve from 0 to 24 h (AUC0–24) response (defined as change from study baseline FEV1) after 3 weeks. Key secondary end points were FEV1 AUC0–12 and AUC12–24 responses.
Results
Two hundred and six patients received treatment. All olodaterol treatments demonstrated statistically significant improvements in FEV1 AUC0–24 response at 3 weeks versus placebo (p < 0.0001); adjusted mean treatment difference versus placebo was 0.191 L for olodaterol 2.5 μg BID (95 % confidence interval [CI] 0.152, 0.229), 0.150 L for 5 μg QD (95 % CI 0.111, 0.189), 0.228 L for 5 μg BID (95 % CI 0.190, 0.266) and 0.209 L for 10 μg QD (95 % CI 0.170, 0.247). These results were supported by the key secondary end points. Olodaterol 5 μg QD provided numerically lower mean values for 24-h bronchodilation than olodaterol 2.5 μg BID (p = 0.0465), with no statistically significant difference between treatment with olodaterol 10 μg QD and 5 μg BID. No relevant differences in morning and evening peak expiratory flow or Asthma Control Questionnaire scores at 3 weeks were observed between different doses and regimens. Adverse events were generally mild to moderate and comparable between groups.
Conclusions
All doses and dose frequencies provided adequate 24-h bronchodilation superior to placebo. Based on the results of this study, it would be reasonable to include both posologies of 5 μg olodaterol daily (5 μg QD or 2.5 μg BID, both delivered in two puffs per dose from the Respimat® inhaler) in subsequent studies. Further studies are necessary to confirm the optimum dosing regimen in asthma. No safety concerns were identified.
Trial registration
ClinicalTrials.gov NCT01311661
Similar content being viewed by others

Background
Long-acting β2-agonists (LABAs) are used in combination with inhaled corticosteroids (ICS) as controller medication for asthma, and have been shown to improve lung function and symptom scores, and reduce the risk of severe exacerbations [1]. Bronchodilators are also well established as treatment for chronic obstructive pulmonary disease (COPD); LABAs have demonstrated significant improvements in lung function, health-related quality of life and exacerbations in this setting [2, 3].
First-generation LABAs, such as formoterol and salmeterol [4, 5], have a 12-h duration of action that requires a twice-daily (BID) dosing schedule. More recently, research efforts have focused on the development of LABAs with a longer duration of action, which may allow for more convenient once-daily (QD) dosing, potentially improving adherence [6, 7]. Olodaterol is a novel LABA, characterised by high β2 selectivity and a near full-agonist profile at β2 adrenoceptors, with a duration of action over 24 h demonstrated by preclinical studies [8]. Effective 24-h bronchodilation with olodaterol has subsequently been confirmed by single-dose studies in both asthma and COPD [9, 10], along with Phase II studies in COPD investigating four doses of QD olodaterol [11] and a comparison of QD and BID dosing [12] and a large Phase III programme in COPD [13–16]. These data also demonstrated an acceptable tolerability profile and no safety concerns were identified.
Dosing and posology have been the focus of recent discussion in development of new bronchodilators. Firstly, the goal of defining the lowest effective dose to minimise safety risk is of particular importance in view of specific safety considerations for LABA use in asthma [17]. Secondly, while demonstration of 24-h duration of action affords the opportunity to consider QD dosing, assessment of lung function trough measurements alone may not be sufficient to demonstrate that QD dosing is the most appropriate regimen. Specific clinical comparison of different doses and posologies (e.g. QD versus BID) over a 24-h period is the most appropriate method for determining the optimum dose and dosing schedule. The inhaled anticholinergic aclidinium bromide serves as a recent example of a drug developed as a QD treatment [18] but then reassessed at later stages of clinical development and finally approved as a BID drug at a different dose [19].
This study has been designed as a posology study for olodaterol in asthma in combination with maintenance therapy with ICS and forms part of a wider series of studies exploring the optimum dose and regimen of olodaterol in both asthma and COPD. One study investigated single-dose olodaterol [9] while two Phase II dose-finding studies were conducted using different designs in moderate asthma. One 4-week, parallel-group study (NCT00467740) demonstrated significant benefits with olodaterol versus placebo, and dose ordering with regards to peak expiratory flow (PEF) but not in forced expiratory volume in 1 s (FEV1), the primary end point of the study [20]. A subsequent incomplete-block crossover study with 4-week treatment periods (NCT01013753) reported significant benefits with olodaterol compared to placebo, and dose ordering observed with FEV1 and PEF [21].
This paper describes the results of our study that was designed to investigate dosing frequencies by comparing the 24-h FEV1 profile of the same olodaterol daily dose administered in either a QD or BID schedule, i.e. 5 μg QD in the morning versus 2.5 μg BID and 10 μg QD in the morning versus 5 μg BID in patients with moderate to severe persistent asthma on maintenance ICS. It is similar in design to a study carried out in patients with COPD (NCT00846768) [12] but with an additional placebo comparison. Total daily doses of 5 and 10 μg were chosen for evaluation as, based on an integrated view of earlier data for QD olodaterol, these doses are approaching the plateau on the dose-response curve [11, 21].
Methods
Patients
Patients were randomised if they met the following main inclusion criteria: aged ≥18 and ≤70 years; a diagnosis of moderate to severe asthma according to the Global Initiative for Asthma [22]; pre-bronchodilator FEV1 ≥60 % and <90 % of predicted FEV1; an increase in FEV1 of ≥12 % and ≥200 mL 15 min after administration of 400 μg salbutamol at screening visit; non-smokers or ex-smokers with a history of <10 pack-years (and smoking cessation ≥1 year prior to enrolment). Patients must have been taking ICS for ≥12 weeks prior to screening and a stable dose for >6 weeks (either medium- to high-dose ICS, or low- to high-dose ICS in fixed-dose combination with a LABA). Patients previously receiving fixed-dose combinations of LABA and ICS were required to demonstrate stability while continuing to receive the equivalent ICS monotherapy for ≥48 h prior to screening visit. Key exclusion criteria included: patients currently diagnosed with a significant disease other than asthma (determined by the investigator as any condition that may put the patient at risk if they entered the study, may influence the results of the study or may cause concern regarding the patient’s ability to participate in the study); patients who had been hospitalised for an asthma exacerbation within 3 months or admitted to an intensive care unit due to asthma within the past 3 years; and patients with a history of myocardial infarction, cor pulmonale or cystic fibrosis. The study was performed in accordance with the Declaration of Helsinki, International Conference on Harmonisation Good Clinical Practice Guidelines and local regulations. Prior to study initiation, the protocol was approved by the ethics research board of the respective institutions and signed consent was obtained from all patients.
The Independent Ethics Committee was Ethikkommittee der Landesärztekammer Hessen, Frankfurt am Main, Germany and the competent authority (Bundesinstitut für Arzneimittel und Medizinprodukte [BfArM], Bonn, Germany) approved the study on 23 Feb 2011.
Study design
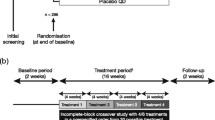
This was a Phase II, multicentre, randomised, placebo-controlled, double-blind, three-period, complete-block, crossover study registered with ClinicalTrials.gov (NCT01311661). After an initial screening visit, patients entered a screening period of 2 weeks to ensure clinical stability. Eligible patients were randomly assigned to receive a three-period treatment sequence comprising 5 μg QD olodaterol (taken in the morning), 2.5 μg BID olodaterol and placebo in a random order, or a three-period treatment sequence comprising 10 μg QD olodaterol (taken in the morning), 5 μg BID olodaterol and placebo in a random order. There were six possible sequences of olodaterol QD, olodaterol BID and placebo for each of the two total daily doses, which ensured that each treatment appeared in each period the same number of times, and each treatment followed every other treatment the same number of times. Patients received each dose regimen for 3 weeks, with a 2-week washout period between regimens (Fig. 1). Treatment was delivered via the Respimat®, with each administration of olodaterol comprising two actuations. All patients were required to take ICS throughout the trial as background medication, as previously instructed by their prescribing physician (BID, QD in the morning or QD in the evening). If administrations coincided, patients were instructed to take the study medications first followed by the ICS.

Study design. All patients received three dose regimens; each was separated by a 2-week washout period. Patients in the 5 μg total daily dose group received one of six possible sequences of olodaterol 2.5 μg BID, 5 μg QD and placebo. Patients in the 10 μg total daily dose group received one of six possible sequences of olodaterol 5 μg BID, 10 μg QD and placebo. Patients continued taking ICS throughout the study, with posology determined by former use. If administration of ICS and study treatment coincided, patients were to take study treatment followed by ICS
Assessments
Spirometry was conducted according to American Thoracic Society and European Respiratory Society recommendations [23]. Identical spirometry equipment was provided to sites for use in clinic measurements. The qualifying pulmonary function test was conducted at screening. Pulmonary function testing (FEV1, forced vital capacity [FVC] and PEF) was performed at 1 h and 10 min prior to trial drug inhalation for the morning dose at all visits, ≤3 h post-dose at the start of each 3-week treatment period (weeks 0, 5 and 10 relative to study baseline) and ≤24 h after inhalation (00:30, 01:00, 02:00, 03:00, 04:00, 06:00, 08:00, 10:00, 11:50, 12:30, 13:00, 14:00, 22:00, 23:00, 23:50 h) at the end of each 3-week treatment period (weeks 3, 8 and 13 relative to study baseline), with a period for night sleep. The primary end point was FEV1 area under the curve from 0 to 24 h (AUC0–24) response at 3 weeks; this was defined as FEV1 AUC0–24 divided by 24 to report in litres minus the study baseline mean FEV1 value. The two key secondary end points were calculated in a similar fashion: FEV1 area under the curve from 0 to 12 h (AUC0–12) response and FEV1 area under the curve from 12 to 24 h (AUC12–24) response, all assessed at the end of each 3-week treatment period. Peak values within 24 h post-dose were defined as the maximum available value between the planned time points 00:30 and 23:50 h after last morning trial-drug inhalation (inclusive), while trough values were defined as the mean of the available values at the planned time points 23:00 and 23:50 h. Similar end points were defined for FVC and PEF.
Daily data on asthma symptoms, night-time awakenings, study medication use during treatment period, ICS medication use, amount of rescue medication (salbutamol) use, and morning and evening PEF and FEV1 were recorded using an electronic peak flow meter with integral patient diary and reviewed by the investigator at each visit. The Asthma Control Questionnaire [24] was completed at screening and weeks 0, 3, 8, 13 and 15, relative to study baseline.
Adverse events (AEs) and serious AEs were monitored throughout the trial at each patient visit. Vital signs, 12-lead electrocardiogram and laboratory tests were also monitored throughout the study, and reported as AEs if they were not associated with a symptom or a diagnosis already reported as an AE.
Statistical analysis
A total sample size of 180 randomised patients was planned to provide ≥90 % power to detect a difference in means of 0.10 L between any one of the active treatments and placebo for the primary end point of FEV1 AUC0–24, assuming a standard deviation for the paired differences of 0.25 L and allowing for approximately 20 % of patients to be non-evaluable.
The full analysis set (FAS) was defined as all patients who received at least one dose of study treatment, for whom a study baseline FEV1 value was available and who had at least one post-dose FEV1 value recorded at an end-of-treatment visit for at least one crossover period. The FAS was used as the basis for the efficacy analyses, including the primary analysis of the primary end point. A per-protocol set was defined (from which patients with important protocol violations were excluded) and was used to perform a sensitivity analysis on the primary end point.
Results for all patients receiving placebo were pooled for the primary analysis, irrespective of whether they received the lower or higher olodaterol daily dose according to the randomised treatment sequence, to use all available data and increase the precision of the estimates. A post hoc sensitivity analysis was also performed on the FAS with placebo split according to treatment sequence (i.e. within each separate complete-block design).
FEV1 AUC0–24 response was analysed in the FAS using a mixed model for crossover studies with ‘treatment’, ‘period’ and ‘study baseline FEV1’ as fixed effects and ‘patient’ as a random effect. The adjusted mean difference between each olodaterol dose/frequency and placebo was calculated using this model, along with the associated p values and 95 % confidence intervals (CIs). Secondary end points were also analysed in the FAS using a mixed model for a crossover study, similar to the primary analysis model. The same statistical model specified above was also used for an exploratory analysis undertaken to compare the different doses and frequencies of olodaterol.
Safety end points were summarised descriptively using the treated set (all randomised patients who were dispensed study medication and were documented to have taken at least one dose of investigational treatment).
Results
Patient population
A total of 206 patients at 36 sites in six countries (Austria, Germany, Hungary, Slovakia, Slovenia and the USA) were randomised (Table 1): the population comprised a relatively even proportion of women to men (53 % versus 47 %), most were aged <50 years and the majority had never smoked. Overall, 199 patients (96.6 %) completed the study (Fig. 2).

CONSORT diagram illustrating participant flow. Since this was a crossover trial and every patient was supposed to receive three treatments, the total number of patients is not the sum of the number of each patient on each treatment. Of the seven patients who discontinued prematurely, the most frequent reason was non-compliance with the trial protocol (three patients). BID: twice daily; QD: once daily; AE: adverse event
Efficacy
Lung function
Highly statistically significant improvements in FEV1 AUC0–24 response were observed with all doses and dose frequencies of olodaterol compared to placebo at week 3 (p < 0.0001) (Table 2), with similar results demonstrated for individual FEV1 values at all individual time points (p < 0.0005) (Fig. 3).

Adjusted mean FEV1: individual time points from 0 to 24 h at 3 weeks. Analysis with imputation, full analysis set. BID: twice daily; QD: once daily; FEV1: forced expiratory volume in 1 s
For the within-crossover group comparisons of the same total daily dose, the mean FEV1 AUC0–24 response for treatment with olodaterol 2.5 μg BID (0.213 L) was greater than for olodaterol 5 μg QD (0.173 L; p = 0.0465; adjusted mean difference for 5 μg QD versus 2.5 μg BID -0.040 L [95 % CI -0.080, -0.001]). There was no evidence of a difference between treatment with olodaterol 10 μg QD and 5 μg BID (p = 0.3388; adjusted mean difference -0.019 L [95 % CI -0.059, 0.020]).
In the secondary, exploratory analysis of two different total daily doses (i.e. parallel-group comparison), there was no significant difference between mean FEV1 AUC0–24 response with olodaterol 5 μg BID and 2.5 μg BID (p = 0.1631; adjusted mean difference 0.037 L [95 % CI -0.015, 0.090]), while olodaterol 10 μg QD provided a greater mean FEV1 AUC0–24 response than olodaterol 5 μg QD (p = 0.0289; adjusted mean difference 0.059 L [95 % CI 0.006, 0.111]). Mean FEV1 AUC0–24 response was higher with olodaterol 5 μg BID compared to olodaterol 5 μg QD (p = 0.0038; adjusted mean difference 0.078 L [95 % CI 0.025, 0.130]).
The post hoc sensitivity analyses performed with placebo split according to treatment sequence were consistent with the primary analysis (p < 0.0001 for all comparisons with placebo) and gave similar estimates for treatment effect.
Analysis of the key secondary end points of FEV1 AUC0–12 and AUC12–24 responses supported the outcomes of the primary end point and provided additional information from within the 24-h measurement period for olodaterol QD and BID, indicating increased adjusted mean FEV1 AUC12–24 response for BID versus QD of the same daily dose, as would be expected (Table 2). Adjusted mean FEV1 at individual time points are shown in Additional file 1: Table S1.
Increased adjusted mean FEV1 AUC0–12 response with 2.5 μg BID versus 5 μg QD, but not with 5 μg BID versus 10 μg QD, was identified (Fig. 4a; Table 2). The adjusted mean difference from placebo in FEV1 AUC0–12 response ranged from 0.160 L (5 μg olodaterol QD; 95 % CI 0.121, 0.199) to 0.219 L (10 μg olodaterol QD; 95 % CI 0.181, 0.258), while results for FEV1 AUC12–24 response ranged from 0.144 L (5 μg olodaterol QD; 95 % CI 0.102, 0.187) to 0.242 L (5 μg olodaterol BID; 95 % CI 0.200, 0.285) (Fig. 4b). For both end points, an increase in adjusted mean difference from placebo was observed with an increasing total daily dose of olodaterol at both dosing frequencies (p < 0.0001 for all comparisons with placebo).

Difference versus placebo at 3 weeks of adjusted mean FEV1 AUC0–12 (a) and AUC12–24 (b). FEV1: forced expiratory volume in 1 s; AUC0–12: area under the curve from 0 to 12 h; SE: standard error; BID: twice daily; QD: once daily; AUC12–24: area under the curve from 12 to 24 h
The analysis of FEV1 at all individual time points assessed at each visit after 3 weeks of treatment showed strong evidence of efficacy (p < 0.0005 for comparisons with placebo) with all olodaterol doses and dosing frequencies at all post-dosing points on the 24-h curve.
Assessment of mean FVC AUC0–24 response supported the results of the primary end point (Fig. 5), as did peak and trough FEV1 and FVC responses (Table 3 and see Additional file 1: Table S2) and PEF AUC0–24 (see Additional file 1: Table S3), peak and trough responses (data not shown).

Adjusted mean FVC AUC0–24 at 3 weeks. FVC: forced vital capacity; AUC0–24: area under the curve from 0 to 24 h; SE: standard error; BID: twice daily; QD: once daily
Other efficacy variables
All doses and dosing frequencies of olodaterol provided statistically significant improvements in mean morning and evening PEF (from patient diaries) and total Asthma Control Questionnaire scores at 3 weeks compared to placebo (p < 0.0001 for all comparisons with placebo) (see Additional file 1: Table S4). No relevant differences were observed between different doses and regimens.
Safety
The overall proportion of patients who reported at least one AE while on treatment was 35.4 %. Incidence of AEs was highest for 5 μg BID olodaterol and lowest for 10 μg QD, although, in general, AEs were in a similar range (placebo 16.4 %; olodaterol 2.5 μg BID and 5 μg QD 14.9 %; 5 μg BID 18.8 %; 10 μg QD 12.7 %) (Table 4). Overall incidence of AEs by sex was 34.0 % for men and 36.7 % for women, with the largest difference reported with placebo treatment (11.7 % of men versus 20.6 % of women reporting any AE). Serious AEs were reported for four patients in total (1.9 %): two patients (1.0 %) for placebo, one patient in the washout period 4 days after discontinuing 5 μg BID olodaterol treatment and one patient 9 days after starting olodaterol 2.5 μg BID. None was considered to be treatment-related.
Overall, the most frequently reported AEs were infections and infestations (14.1 %), with sinusitis the most frequently reported AE within this category (2.4 % overall). Respiratory, thoracic and mediastinal disorders were the second most frequently reported AE (9.2 %), with the highest rate reported for placebo, at 4.5 %. Within this category, the most common AE reported was asthma (4.4 % overall) with the highest reporting frequency of 2.0 % for placebo and olodaterol 5 μg BID (Table 4).
A total of seven patients (3.4 %) were considered to have had AEs related to study drug by the investigators. Two patients were receiving placebo (1.0 %; headache and cough); two patients who had cough were receiving olodaterol (one receiving 5 μg QD, one receiving 10 μg QD; 1.0 % in both instances). Insomnia, palpitations and asthma were reported by three patients taking 5 μg BID olodaterol (3.0 %). No notable findings were reported via assessment of laboratory parameters, vital signs and electrocardiogram readings.
Discussion
The results of this study add support to the growing evidence base that olodaterol delivered by Respimat® QD provides effective 24-h bronchodilation in both COPD and asthma [9–11, 13–16, 25]. This study was designed to assess the efficacy of different daily doses and dose frequencies of olodaterol versus placebo, and between different doses and frequencies of olodaterol in an exploratory fashion, in patients with moderate to severe persistent asthma after 3 weeks of treatment, thereby providing additional information on the efficacy of QD versus BID dosing.
Both total daily doses and all dosing frequencies of olodaterol tested provided highly significant improvements versus placebo in efficacy variables based on pulmonary function tests, albeit with different 24-h bronchodilatory profiles. Differences were seen between doses and posologies of olodaterol, with a greater mean FEV1 AUC0–24 response for 2.5 μg BID than 5 μg olodaterol QD in the morning but no statistically significant difference was observed between olodaterol 5 μg BID and olodaterol 10 μg QD in the morning.
As expected, the second daily dose in BID schedules gave increased mean FEV1 AUC12–24 response versus QD dosing at the same daily dose after 3 weeks of treatment. However, while a higher mean FEV1 AUC0–12 response might be anticipated for the QD dosing (as was observed for mean area under the curve from 0 to 3 h response at the start of the 3-week treatment period [data not shown]), this was not observed.
Pre-dose morning FEV1 measurements at -1 h and -10 min in this study were numerically higher for both the BID posologies versus QD posologies but notably for 2.5 μg olodaterol BID, hinting at a residual effect; however, further investigation would be required to fully evaluate this potential effect. An inspection of the placebo versus olodaterol time profiles confirms that the reported circadian influence on lung function [26] is not abolished but rather the level of bronchodilation is increased by olodaterol treatment.
Interpretation of the exploratory data in the present study should, however, take into consideration that the study was designed to compare different doses and dosing frequencies of olodaterol with placebo. Consequently, it was not powered to examine statistical differences between active treatments, nor was there any adjustment made for such multiple treatment comparisons. As individual patients only received one total daily dose, inter-patient variability also contributed to increasing uncertainty in the estimation of effect-size comparisons of different daily doses in the parallel arms (wider CIs). Therefore, as is generally true for Phase II studies, any interpretation of the comparisons between different doses and dose frequencies of olodaterol should be undertaken with caution.
Although this study provided some information on the difference in efficacy of different posologies of the same daily dose of olodaterol on top of ICS, along with demonstrating adequate short-term safety, further, longer-term research would be required to fully examine the optimum olodaterol dose regimen in patients with asthma, possibly investigating patient type, combination treatment and the effect of variations in the timing of QD dosing (e.g. morning versus evening dosing).
The lung function efficacy of olodaterol versus placebo has also been demonstrated by other Phase II olodaterol trials carried out in asthma [20, 21]. O’Byrne et al. reported results of their placebo-controlled, parallel-group study in 426 patients (NCT00467740) at the American Thoracic Society meeting in 2012 [20] and demonstrated dose-dependent improvements in lung function with QD olodaterol, although dose ordering was not consistently shown for each of the individual efficacy end points. While the primary end point of trough FEV1 demonstrated numerical improvement for all doses, including 5 and 10 μg olodaterol, only the improvements observed in patients receiving 20 μg olodaterol QD were statistically significant. Evidence of dose ordering was observed in the pre-dose morning PEF response, with dose-related improvements also observed on the Asthma Control Questionnaire [20]. Due to lack of dose ordering seen for the primary end point in this study, Beeh et al. conducted a similar study, but using an incomplete-block crossover design, in 198 patients (NCT01013753) [21]. This study also demonstrated highly statistically significant improvements in FEV1 and PEF response with all doses of olodaterol examined and clear evidence of dose ordering [21].
The results of our study provide further evidence for the dose ordering seen between 5 and 10 μg total daily doses; the parallel-group comparison of 5 μg QD versus 10 μg QD for several FEV1 response assessments demonstrates a dose response with increasing dose, and further evidence for this relationship is supplied by the comparison of 2.5 μg BID versus 5 μg BID. Indeed, a dose response is also observed when doubling the dose from 5 μg QD to 5 μg BID. When considered as a whole, the weight of the current evidence from several olodaterol trials in asthma indicates a relevant dose-response relationship between a total daily dose of 5 and 10 μg olodaterol.
Increases in FVC observed with all doses of olodaterol were lower than the increases seen with FEV1. This may indicate the effect of olodaterol on peripheral airways, as air trapping is often a feature of small-airway disease in patients with severe asthma [27]. Indeed, bronchodilation using high doses of albuterol has been demonstrated to decrease residual volume:total lung capacity ratio while increasing FVC in parallel, with improvements in FVC partially responsible for increase in FEV1 [27].
A series of clinical studies has examined the dose dependence and posology of olodaterol bronchodilatory efficacy in COPD. A single-dose study [10] and a study of 4-week QD treatment [11] have demonstrated 24-h bronchodilation in COPD. In order to investigate dosing frequency, a complete-block crossover study (NCT00846768) investigated the 24-h bronchodilator profile of different olodaterol regimens in 47 patients with COPD [12]. Although it did not include a placebo treatment, the study demonstrated statistically significant improvements versus baseline for all doses and dose regimens: 5 and 10 μg QD provided near-identical bronchodilation over a 24-h period, while 5 μg QD demonstrated an improved bronchodilation profile versus 2 μg BID [12], thus providing support for the further investigation of 5 and 10 μg QD in COPD [13–16, 28]. Olodaterol 5 μg QD is now approved in several countries for the treatment of COPD. Together with the current study, these data suggest that olodaterol provides effective 24-h bronchodilation in both asthma and COPD. The data in COPD provide a clear rationale for QD schedules; the current data in asthma are limited and less clear, although all doses and schedules provided effective 24-h bronchodilation.
Thorough investigation of new LABAs in asthma is of particular relevance due to the concerns over safety of this class of agent. LABA monotherapy in asthma is contraindicated to minimise the risk of serious exacerbations of asthma and asthma-associated deaths [17]; it is for this reason that patients in the present study were required to continue taking ICS as background medication. To ensure a full picture of the safety of new LABAs, the US Food and Drug Administration also considered asthma safety data when approving the LABA indacaterol in COPD at 75 μg QD (a lower dose than the EU) [29]. With this in mind, the current study demonstrated an acceptable short-term safety profile for olodaterol in asthma on top of maintenance ICS. Incidence of AEs was similar between olodaterol and placebo, and ranged from 12.7 to 18.8 %. All three studies in asthma conducted to date have shown olodaterol to have consistent short-term safety and tolerability profiles, and have raised no safety concerns [20, 21], in line with previous studies in COPD [11, 12].
In conclusion, in this posology study in patients with moderate to severe asthma, while resulting in some differences in 24-h bronchodilation profile, all daily doses (5 and 10 μg) and dose frequencies (2.5 μg BID, 5 μg QD, 5 μg BID, 10 μg QD) of olodaterol demonstrated 24-h bronchodilation superior to placebo in patients with asthma on a background of inhaled steroids. These data should be interpreted with caution, as further studies would be required in asthma before making clinical decisions on optimal dosing and posology. No safety concerns were identified and the short-term safety and tolerability profiles of olodaterol were similar to those seen in previous trials. Further, long-term research would be required in order to fully examine the long-term safety and optimum dose regimen of olodaterol in patients with asthma.
Abbreviations
- AE:
-
Adverse event
- AUC0–12 :
-
Area under the curve from 0 to 12 h
- AUC0–24 :
-
Area under the curve from 0 to 24 h
- AUC12–24 :
-
Area under the curve from 12 to 24 h
- BID:
-
Twice daily
- CI:
-
Confidence interval
- COPD:
-
Chronic obstructive pulmonary disease
- FAS:
-
Full analysis set
- FEV1 :
-
Forced expiratory volume in 1 s
- FVC:
-
Forced vital capacity
- ICS:
-
Inhaled corticosteroids
- LABA:
-
Long-acting β2-agonist
- PEF:
-
Peak expiratory flow
- QD:
-
Once daily
References
Global Initiative for Asthma. Global strategy for asthma management and prevention. Updated 2015. http://www.ginasthma.org/local/uploads/files/GINA_Report_2015_May19.pdf. Accessed 16 Jul 2015.
Global Initiative for Chronic Obstructive Lung Disease. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. Updated 2014. http://www.goldcopd.org/uploads/users/files/GOLD_Report2014_Feb07.pdf. Accessed 2 Jun 2014.
Hanania NA, Donohue JF. Pharmacologic interventions in chronic obstructive pulmonary disease. Bronchodilators. Proc Am Thorac Soc. 2007;4:526–34.
Anderson GP. Formoterol: pharmacology, molecular basis of agonism, and mechanism of long duration of a highly potent and selective beta 2-adrenoceptor agonist bronchodilator. Life Sci. 1993;52:2145–60.
Ball DI, Brittain RT, Coleman RA, Denyer LH, Jack D, Johnson M, et al. Salmeterol, a novel, long-acting β2-adrenoceptor agonist: characterization of pharmacological activity in vitro and in vivo. Br J Pharmacol. 1991;104:665–71.
Rau JL. Determinants of patient adherence to an aerosol regimen. Respir Care. 2005;50:1346–56.
Rodrigo GJ, Neffen H. Comparison of indacaterol with tiotropium or twice-daily long-acting β-agonists for stable COPD: a systematic review. Chest. 2012;142:1104–10.
Bouyssou T, Casarosa P, Naline E, Pestel S, Konetzki I, Devillier P, et al. Pharmacological characterization of olodaterol, a novel inhaled β 2-adrenoceptor agonist exerting a 24-hour-long duration of action in preclinical models. J Pharmacol Exp Ther. 2010;334:53–62.
O’Byrne PM, van der Linde J, Cockcroft DW, Gauvreau GM, Brannan JD, FitzGerald M, et al. Prolonged bronchoprotection against inhaled methacholine by inhaled BI 1744, a long-acting β2-agonist, in patients with mild asthma. J Allergy Clin Immunol. 2009;124:1217–21.
van Noord JA, Smeets JJ, Drenth BM, Rascher J, Pivovarova A, Hamilton AL, et al. 24-hour bronchodilation following a single dose of the novel β2-agonist olodaterol in COPD. Pulm Pharmacol Ther. 2011;24:666–72.
van Noord JA, Korducki L, Hamilton AL, Koker P. Four weeks once daily treatment with BI 1744 CL, a novel long-acting β2-agonist, is effective in COPD patients [abstract A6183]. Am J Respir Crit Care Med. 2009;179.
Joos G, Aumann JL, Coeck C, Korducki L, Hamilton AL, van Noord JA. Comparison of 24-hour FEV1 profile for once-daily versus twice-daily treatment with olodaterol, a novel long-acting β2 agonist, in patients with COPD [poster A2930]. Presented at the 103rd Annual International Conference of the American Thoracic Society, San Francisco, California, USA, 18–23 May 2012.
Koch A, Pizzichini E, Hamilton A, Hart L, Korducki L, De Salvo MC, et al. Lung function efficacy and symptomatic benefit of olodaterol once daily delivered via Respimat® versus placebo and formoterol twice daily in patients with GOLD 2-4 COPD: results from two replicate 48-week studies. Int J Chron Obstruct Pulmon Dis. 2014;9:697–714.
Ferguson GT, Feldman GJ, Hofbauer P, Hamilton A, Allen L, Korducki L, et al. Efficacy and safety of olodaterol once daily delivered via Respimat® in patients with GOLD 2–4 COPD: results from two replicate 48-week studies. Int J Chron Obstruct Pulmon Dis. 2014;9:629–45.
Lange P, Aumann J-L, Hamilton A, Tetzlaff K, Ting N, Derom E. The 24 hour lung function time profile of olodaterol once daily versus placebo and tiotropium in patients with moderate to very severe chronic obstructive pulmonary disease. J Pulm Respir Med. 2014;4:196.
Feldman GJ, Bernstein JA, Hamilton A, Nivens MC, Korducki L, LaForce C. The 24-h FEV1 time profile of olodaterol once daily via Respimat® and formoterol twice daily via Aerolizer® in patients with GOLD 2–4 COPD: results from two 6-week crossover studies. Springerplus. 2014;3:419.
Chowdhury BA, Dal Pan G. The FDA and safe use of long-acting beta-agonists in the treatment of asthma. N Engl J Med. 2010;362:1169–71.
Jones PW, Rennard SI, Agusti A, Chanez P, Magnussen H, Fabbri L, et al. Efficacy and safety of once-daily aclidinium in chronic obstructive pulmonary disease. Respir Res. 2011;12:55.
Jones PW, Singh D, Bateman ED, Agusti A, Lamarca R, de Miquel G, et al. Efficacy and safety of twice-daily aclidinium bromide in COPD patients: the ATTAIN study. Eur Respir J. 2012;40:830–6.
O’Byrne PM, D’Urzo T, Gahlemann M, Hart L, Wang F, Beck E. Dose-finding study of once-daily treatment with olodaterol, a novel long-acting β2-agonist, in patients with asthma [abstract A3963]. Am J Respir Crit Care Med. 2012;185.
Beeh K-M, Beck E, Gahlemann M, Blahova Z, Toorawa R, Flezar M. Dose-finding study of 4-week, once-daily treatment with olodaterol, a novel long-acting β2-agonist, in patients with asthma [abstract A2764]. Am J Respir Crit Care Med. 2012;185.
Global Initiative for Asthma. Global strategy for asthma management and prevention. Updated 2012. Global Initiative for Asthma, 2012.
Miller MR, Hankinson J, Brusasco V, Burgos F, Casaburi R, Coates A, et al. Standardisation of spirometry. Eur Respir J. 2005;26:319–38.
Juniper EF, O’Byrne PM, Guyatt GH, Ferrie PJ, King DR. Development and validation of a questionnaire to measure asthma control. Eur Respir J. 1999;14:902–7.
Joos G, Aumann JL, Coeck C, Korducki L, Hamilton AL, van Noord J. Comparison of 24-hour FEV1 profile for once-daily versus twice-daily treatment with olodaterol, a novel long-acting B2-agonist, in patients with COPD [abstract A2930]. Am J Respir Crit Care Med. 2012;185.
Smolensky MH, Barnes PJ, Reinberg A, McGovern JP. Chronobiology and asthma. I. Day-night differences in bronchial patency and dyspnea and circadian rhythm dependencies. J Asthma. 1986;23:321–43.
Sorkness RL, Bleecker ER, Busse WW, Calhoun WJ, Castro M, Chung KF, et al. Lung function in adults with stable but severe asthma: air trapping and incomplete reversal of obstruction with bronchodilation. J Appl Physiol. 2008;104:394–403.
McGarvey L, Niewoehner D, Magder S, Sachs P, Tetzlaff K, Hamilton A et al. One-year safety of olodaterol once daily via Respimat® in patients with GOLD 2-4 chronic obstructive pulmonary disease: results of a pre-specified pooled analysis. COPD 2015; Feb 18 [Epub ahead of print].
Chowdhury BA, Seymour SM, Michele TM, Durmowicz AG, Liu D, Rosebraugh CJ. The risks and benefits of indacaterol — the FDA’s review. N Engl J Med. 2011;365:2247–9.
Acknowledgements
The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors. They take full responsibility for the scope, direction, content of, and editorial decisions relating to, the manuscript, were involved at all stages of development and have approved the submitted manuscript. The authors received no compensation related to the development of the manuscript. This work was supported by Boehringer Ingelheim Pharma GmbH & Co. KG. Medical writing assistance was provided by Caitlin Watson, PhD, of Complete HealthVizion, which was contracted and compensated by Boehringer Ingelheim Pharma GmbH & Co. KG.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
KMB: received funding from Almirall, Boehringer Ingelheim, GlaxoSmithKline, Infinity, Mundipharma, Novartis, Sterna AG and TEVA, and received consultant and lecture fees from Cytos, Boehringer Ingelheim, AstraZeneca, Mundipharma, Novartis, Pfizer and Chiesi. CL: has nothing to declare. MG and AW: are employees of Boehringer Ingelheim. RT: is a contract statistician working for Boehringer Ingelheim. MF: received consultant and expert testimony fees from Boehringer Ingelheim, Novartis and AstraZeneca and is a member of the board for the European Board for Accreditation in Pneumology and the European Union for Medical Specialists.
Authors’ contributions
All authors contributed to the study conception and design, provided oversight of the study, and were responsible for the drafting, review and final approval of the manuscript.
Additional file
Additional file 1: Table S1.
Adjusted mean FEV1 and comparison with placebo at individual time points at 3 weeks. Table S2. Adjusted mean FVC AUC0–24 response and comparison with placebo at 3 weeks. Table S3. Adjusted mean PEF AUC0–24 response and comparison with placebo at 3 weeks. Table S4. Overall adjusted mean PEF and total Asthma Control Questionnaire score after 3 weeks.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Beeh, KM., LaForce, C., Gahlemann, M. et al. Randomised, double-blind, placebo-controlled crossover study to investigate different dosing regimens of olodaterol delivered via Respimat® in patients with moderate to severe persistent asthma. Respir Res 16, 87 (2015). https://doi.org/10.1186/s12931-015-0243-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-015-0243-1