
Abstract
Chronic stress results in disturbances of body hormones through the neuroendocrine system. Cancer patients often experience recurrent anxiety and restlessness during disease progression and treatment, which aggravates disease progression and hinders treatment effects. Recent studies have shown that chronic stress-regulated neuroendocrine systems secret hormones to activate many signaling pathways related to tumor development in tumor cells. The activated neuroendocrine system acts not only on tumor cells but also modulates the survival and metabolic changes of surrounding non-cancerous cells. Current clinical evidences also suggest that chronic stress affects the outcome of cancer treatment. However, in clinic, there is lack of effective treatment for chronic stress in cancer patients. In this review, we discuss the main mechanisms by which chronic stress regulates the tumor microenvironment, including functional regulation of tumor cells by stress hormones (stem cell-like properties, metastasis, angiogenesis, DNA damage accumulation, and apoptotic resistance), metabolic reprogramming and immune escape, and peritumor neuromodulation. Based on the current clinical treatment framework for cancer and chronic stress, we also summarize pharmacological and non-pharmacological therapeutic approaches to provide some directions for cancer therapy.
Similar content being viewed by others

Background
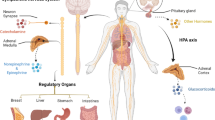
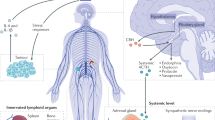
Nowadays, the work environment and economic situation of society, especially with the explosion of COVID-19 worldwide, people are under tremendous psychological pressure [1]. Prolonged and repeated exposure to psychological stress results in a range of endocrine and behavioral responses [2]. The neuroendocrine system, which is activated by chronic stress, consists of the hypothalamic–pituitary–adrenal (HPA) axis and the sympathetic nervous system (SNS). Under chronic stress, the hypothalamus releases corticotropin-releasing hormone (CRH). And then CRH triggers the anterior pituitary to secrete adrenocorticotropic hormone (ACTH) to stimulate the secretion of corticosteroids (e.g., glucocorticoids) by adrenal cortex. Chronic stress also activates the SNS, which responds to sympathetic stimulation and cortisol. The excited SNS promotes the release of norepinephrine from nerve fibers and also promotes the synthesis and secretion of epinephrine from the adrenal medulla. Epinephrine, norepinephrine and dopamine are collectively known as catecholamines. HPA and SNS generally regulate the function of nearly all human organ systems through two signaling pathways: one is the release of the sympathetic neurotransmitter norepinephrine via sympathetic nerve terminals to engage adrenergic receptors (ARs) that are expressed on target organs; the other is the release of epinephrine and corticosteroids from the adrenal glands through the bloodstream to reach target organs, which acts on ARs and glucocorticoid receptors (GRs) expressed on target organs (Fig. 1) [3]. Stress hormones (including epinephrine, norepinephrine and glucocorticoids) that reach target organs effectively promote gluconeogenesis and glycogenolysis, raise blood glucose, and improve body metabolism. Glucocorticoids also have the ability to regulate protein metabolism, fat metabolism and water and salt metabolism. Through the above physiological functions, stress-induced hormones increase brain excitability and sensitivity, relax blood vessels, and accelerate heart rate and cardiac conduction velocity.

Chronic stress regulates the tumor microenvironment through the neuroendocrine system. The nervous system is composed of cranial and spinal nerves. Under chronic stress, the hypothalamus releases CRH, which triggers ACTH secretion from pituitary to stimulate the secretion of glucocorticoids from the adrenal cortex. Chronic stress also activates the SNS, which directly innervates organs through sympathetic neurotransmitter norepinephrine (NE) release from neuro synapse, promoting the synthesis and secretion of epinephrine (E) from the adrenal medulla. NE generally reaches the TME through nerve fibers, and E/glucocorticoids (GCs) reach it through the blood
In acute stress (e.g., acute fight or flight response), elevated cortisol acts via a negative feedback loop and inhibits HPA activity through effects at the pituitary, hypothalamic the paraventricular nucleus and hippocampal levels [2]. However, in chronic stress, cortisol remains elevated, dopamine decreased, negative feedback regulation fails, and cortisol metabolism is reduced, which leads to an increased risk of metabolic syndrome, obesity, cancer, mental health disorders, and cardiovascular disease [2]. In addition, SNS activation does not decay over time and is able to more strongly upregulate norepinephrine levels [1]. Studies in animal models have demonstrated that chronic social stress also increases the growth and distribution of sympathetic nerve fibers in peripheral tissues, thereby upregulating ARs activity in target tissues [4].
Chronic stress affects cancer progression by modulating the tumor microenvironment (TME) through the neuroendocrine system [5]. Recent evidence suggests that epinephrine and norepinephrine and glucocorticoids are major factors in chronic stress-promoted tumor development, which is strongly associated with high malignancy and poor prognosis [6, 7]. Epidemiological studies show that cancer patients have a large burden of mental health disorders and much higher rates of depression and anxiety compared to the general population [8, 9]. Thus, more attention should be paid to the relationship between chronic stress and TME.
In addition to malignant cells, immune and stromal cells (fibroblasts, adipocytes, endothelial cells, etc.) and extracellular components (cytokines, growth factors, hormones, extracellular matrix, etc.) are present in the TME. They surround the tumor cells and are interspersed with the vascular system, lymphatic network, and peripheral nerves [10]. Stress hormones act on the majority of cells in the TME and ultimately promote tumor progression by activating specific receptors associated with many cancer biological processes, including genomic instability, metabolic disorders, proliferation, angiogenesis, metastasis, and immune evasion. One of the main objectives of this review is to summarize the main mechanisms by which chronic stress-associated hormones regulate tumor development, including the interactions between cancer cells and infiltrating immune and stromal cell populations in the TME. Recent studies have shown that tumors can recruit nerves into the TME and form peritumor nerves, which in turn affects tumorigenesis, angiogenesis, invasion and metastasis [11]. This bidirectional interaction between the nervous system and tumors is now becoming a rapidly growing research topic known as cancer neuroscience [12]. This review will also focus on and summarize the currently reported biological mechanisms of chronic stress with the tumor peripheral nerves.
Clinical and preclinical studies have now reported that stress impairs adjuvant and neoadjuvant treatment, including radiotherapy, immunotherapy, and chemotherapy, through modulation by glucocorticoids or catecholamines [13]. However, clinical studies have not adequately addressed this question. In clinical treatment, the anti-tumor immune responses are indispensable in immunotherapy, chemotherapy, and radiation as well, while chronic stress acting on the immune system impairs the efficacy of clinical therapy [14]. Therefore, the synergistic integration of intervene of chronic stress into existing cancer treatment regimens is a major challenge, based on the various treatment options currently available for cancer. Here, we summarized pharmacological and non-pharmacological therapeutic approaches based on the currently available clinical treatment framework to provide some direction for cancer treatment.
Effect of chronic stress on tumor cells
Chronic stress-induced catecholamines and glucocorticoids directly act on cancer cells and promote tumor development [5]. Stress hormones bind to the corresponding intracellular receptors to promote inflammation, angiogenesis, genomic instability, metastasis, and stem cell-like related genes expression through change of epigenetics or activation of multiple pathways in tumor cells (Fig. 2) [15]. Not only that, chronic stress-induced tumor cells acquire apoptosis resistance and resist cancer therapy [16]. We herein discuss the functional effects of chronic stress on tumor cells and the pathways that impair cancer therapy.

The biological mechanism of chronic stress affecting cancer cells. A Chronic stress promotes the stem cell characteristics in cancer cells. Chronic stress-induced E activates LDHA, promotes glycolysis, and leads to lactate secretion, this enhances the interaction between USP28 and MYC, which promotes stem cell-like-associated genes expression via SLUG. Chronic stress-induced GCs promote β-catenin expression through the interaction of GRP78 with LRP5. GCs also downregulate miR-346 and miR-493, which in turn upregulate Cyclin D1 and accelerate the cell cycle. In addition, GCs promote GRs-dependent nuclear accumulation and TEAD4 transcriptional activation, promoting the maintenance of CSCs. B Chronic stress accelerates cancer cell metastasis. Chronic stress-induced β-ARs signaling activates FAK via cAMP/PKA, which induces extracellular matrix remodeling via Erk1/2-MMP; Src is also activated by cAMP/PKA, and Y419 phosphorylation of Src amplifies HIF1α and MYC, further inducing hTERT overexpression in the nucleus. hTERT activates SLUG and in turn upregulates metastasis-related genes. β-ARs also promotes β-catenin expression and nuclear localization through PI3K/AKT and increases SLUG promoter activity. NE-induced downregulation of miR-337-3p activates STAT3. β-ARs activates MMP7 and releases HB-EGF to activate EGFR, whereas MAOA can target β2-ARs to reverse these processes. In addition, β-ARs promotes neuroendocrine phenotypic transformation and metastasis through MACC1 upregulation, which binds directly to SYP via c-Jun. GRs contribute to increased ROR1. In addition, GRs increase CTGF expression via PI3K/SGK/Nedd4l-Smad2 and promotes lung metastasis. GRs localize to the CDK1 promoter in nucleus and stimulate CDK1 through epigenetic regulation. C Chronic stress promotes angiogenesis. β-ARs activates CREB and STAT3 via cAMP/PKA, STAT3 translocates to the nucleus and stimulates VEGF and MMP2/9 transcription, CREB targets HDAC2 activation, which epistemically represses TSP1. In addition, PPARγ inhibits VEGF by suppressing ROS, while β-ARs signaling reverses the effect of PPARγ on VEGF/FGF2. D Chronic stress promotes DNA damage accumulation and anti-apoptosis. β-ARs mediates Src/FAK and BAD anti-apoptotic pathways by PKA, BAD-S112 phosphorylation and FAK-Y397 phosphorylation gain resistance to apoptosis. GCs stabilize CFLARL through GMEB1-USP40 interaction, inhibit pro-CASP8 activation, thereby inhibiting apoptosis. SGK1, a key downstream effector of Ras, promotes glucose-mediated carbon flux into multiple metabolic pathways to inhibit oxidation by GLUT1 activation; in addition, Ras blocks apoptosis by reducing PHLPP1, which activates p38 MAPK to promote apoptosis. Elevated NE/E and GCs increase MDM2 activity and decrease p53 function via SGK1 and ARRB1, β-ARs and GRs also stimulate the production of ROS and RNS, ultimately leading to DNA damage accumulation
Stem cell-like characteristics
Cancer stem cells (CSCs) have a high self-renewal capacity and are responsible for cancer recurrence and resistance to radiotherapy and chemotherapy [17]. Psychological stress promotes stem-like characteristics and tumorigenic potential in cancer cells, including a significant increase in self-renewal genes expressions, such as CTNNB, OCT4, and NANOG [18, 19]. Chronic stress-induced epinephrine enhances lactate dehydrogenase A (LDHA)-dependent glycolysis, which leads to increased lactate secretion. Excess lactate promotes ubiquitin-specific protease 28 (USP28) to stabilize MYC proteins, which transcriptionally active SLUG expression to enhance breast cancer stem cell-like features (Fig. 2A) [18]. Chronic stress-induced neurotransmitter also activates CSCs through multiple cAMP-mediated pathways (including ERK, AKT, CREB, SHH, and ALDH-1) in non-small cell lung cancer (NSCLC) [20]. Zheng et al. demonstrated that stress-induced excess cortisol increases stem cell properties by enhancing the endoplasmic reticulum stress protein GRP78 (Fig. 2A) [21]. Glucocorticoids have been reported to down-regulate miR-346 and miR-493 levels, which in turn upregulate Cyclin D1 expression and accelerate the cell cycle (Fig. 2A) [22]. In addition, glucocorticoids promote breast cancer cell resistance to cytotoxic compounds like taxanes during cancer treatment through the interaction between GRs and TEA domain transcription factor 4 (TEAD4) [23]. These findings suggest that chronic stress-induced hormones upregulate the expression of proliferation-related genes through multiple pathways, thereby enhancing the stem-like properties of cancer cells.
Metastasis
Cancer metastasis consists of at least two rate-limiting steps, the entry of metastatic tumors into the systemic circulation and the colonization of circulating tumor cells in distant organs [24]. Chronic stress is involved in key steps of tumor metastasis, endows cancer cells with mesenchymal-like characteristics to enhance their capacity of metastasis and invasion [25]. Numerous key molecular switches activated by ARs have been identified to regulate metastasis in solid tumor. In human ovarian cancer samples, norepinephrine correlates strongly with pSrc Y419 and pSrc S17 [26]. Src as a cytoplasmic tyrosine kinase further amplifies the signaling cascade of HIF-1α and c-Myc to induce human telomerase reverse transcriptase (hTERT) overexpression. Subsequently, hTERT induces SLUG expression, upregulates various metastasis-associated genes [27]. β-ARs also promote β-catenin expression and nuclear localization via PI3K/AKT, increasing SLUG promoter activity, thereby promoting epithelial-mesenchymal transformation and invasion of ovarian cancer cells (Fig. 2B) [28]. Additionally, norepinephrine-induced downregulation of miR-337-3p further activates STAT3 for lung metastasis of breast cancer (Fig. 2B) [29]. Focal adhesion kinase (FAK) is increased in prostate cancer patients with metastatic features and high depression scores. Activation of FAK is dependent on the cAMP/PKA signaling pathway and regulates extracellular matrix remodeling via matrix metalloproteinase (MMP) release to promote tumor invasion (Fig. 2B) [30]. Monoamine oxidase A (MAOA), a catecholamine neurotransmitter-degrading enzyme, inhibits metastasis by suppressing β-ARs and ARs-mediated epidermal growth factor receptor (EGFR) signaling (Fig. 2B). MAOA is significantly downregulated in clinical hepatic carcinoma samples [31]. Catecholamines upregulate metastasis‐associated in colon cancer 1 (MACC1), which binds directly to synaptophysin (SYP) via the β2-AR/c-Jun signaling pathway, thereby promoting the neuroendocrine phenotypic transformation of gastric cancer cells and accelerating metastasis (Fig. 2B) [32].
A critical step in metastasis also includes the formation of distant metastatic niches. Studies have shown that adrenergic stimulation upregulates C-C motif chemokine ligand 2 (CCL2) of lung stromal cells and C-C motif chemokine receptor 2 (CCR2) of monocytes, which results in macrophage recruitment and infiltration into the pre-metastatic lung and promote lung metastatic colonization of circulating tumor cells [33, 34]. Obradovic et al. demonstrate that glucocorticoids upregulate the receptor-tyrosine-kinase-like orphan receptor (ROR1), which mediates the lung metastatic colonization process, thus reducing patient survival (Fig. 2B) [35].
Notably, glucocorticoids such as dexamethasone (Dex) have been widely used as combination drugs in cancer treatment to combat chemotherapy-induced side effects (e.g. nausea, edema, allergic reactions) [36]. Dex directly inhibits platinum-based chemotherapy-induced 5-hydroxytryptamine 3 (5-HT3) receptors, which mediate the physiological and pathological process of vomiting [37]. Dex also relieves pain in cancer patients by inhibiting the synthesis and release of prostaglandins [38]. However, Dex promotes breast cancer metastasis in the standard hypersensitivity reaction to paclitaxel regimen, which should raise concerns during the treatment of clinical breast cancer. Mechanistically, Dex links with GRs on tumor cells to upregulate serum glucocorticoid-induced kinase 1 (SGK1) expression, which in turn mediates cancer metastasis through connective tissue growth factor (CTGF) (Fig. 2B) [39]. Dex also promotes proliferation and invasion of colon adenocarcinoma by stimulating CDK1 gene expression (Fig. 2B) [40]. Based on these findings, it can be concluded that the effects of Dex are double edged, and the optimal choice is to modify paclitaxel formulations to reduce Dex application.
Given the psychological reactions and physiological stress responses of cancer patients in the perioperative period, surgical excision of primary solid tumors is often accompanied by the development of residual malignant cells metastases [41]. Depression/anxiety is associated with recurrence in breast cancer patients, with recent animal studies providing supportive evidence [42]. Chronic stress impairs TLR-9 immunostimulant (CpG-C) to block cancer metastasis efficacy in the CT-26 metastasis model and the melanoma spontaneous metastasis model. Only the combination of CpG-C and GR and β-AR blockers improve long-term recurrence-free survival after resection of primary metastatic tumors in mice [43].
Angiogenesis
Angiogenesis is a complex process, which usually depends on the balance between activators and inhibitors of angiogenesis [44]. Normal angiogenesis is essential for the development and growth of tissues, but pathological angiogenesis contributes to the spread and growth of cancer by providing nutrients and oxygen [45]. Thaker et al. show that vascularization is markedly increased in stressed animals and is accompanied by upregulation of MMP-2, MMP-9, and vascular endothelial growth factor (VEGF) [46]. This is involved in β-ARs/cAMP/PKA/STAT3 signaling, where activated STAT3 is translocated to the nucleus to bind to specific DNA sites, then stimulates transcription of angiogenesis-related genes (Fig. 2C) [47]. After CREB activation by β-AR, histone deacetylase 2 (HDAC2) epigenetically inhibits thrombospondin-1 (TSP1), a potent angiogenesis inhibitor, thereby promoting angiogenesis and prostate cancer progression (Fig. 2C) [48]. Recent studies have shown that peroxisome proliferator-activated receptor γ (PPARγ) and its agonists induce apoptosis by inhibiting angiogenesis, whereas β2-AR activation reverses this process (Fig. 2C) [49]. Furthermore, in colorectal cancer and melanoma mice, chronic stress impairs the effect of sunitinib, an inhibitor of multiple tyrosine kinase receptors with antiangiogenic and antitumor activity [50].
Cell death and DNA damage
The pathways of cell death are conserved, clinical cancer therapy has aimed to eliminate cancer cells as effectively as possible through programmed cell death or apoptosis. The primary mechanism of radiation therapy is direct damage to tumor cells in the radiation field. The adrenergic response directly increased tumor cell resistance to radiation in vitro and blocked radiation-induced apoptosis [51, 52]. However, it remains to be elucidated how stress affects the radiation resistance of tumor cells and blocks apoptosis.
Chemotherapy kills cancer cells with chemical drugs, blocks DNA synthesis and induces apoptosis. However, in vitro and in xenograft mouse models, chronic stress hormones impair the efficacy of chemotherapy with cisplatin, paclitaxel, and TRAIL in pancreatic cancer [16]. BCL2-associated death promoter (BAD) phosphorylation is necessary to inhibit apoptosis, and epinephrine mediates the β2-AR/PKA/BAD anti-apoptotic signaling pathway, thereby reducing treatment sensitivity and accelerating cancer progression (Fig. 2D) [53]. β2-AR/Src signaling axis not only regulates tumor metastasis but also gains resistance to apoptosis through phosphorylation of FAK Y397 (Fig. 2D) [54]. In addition, β2-AR and human epidermal growth factor receptor-2 (HER2) constitute a positive feedback loop in human breast cancer cells that mediates resistance to trastuzumab-targeted HER2 therapy [55]. In contrast, β2-AR antagonists significantly downregulate Cyclin D1 expression and inhibit the formation of Cyclin D1/CDK4/6 complexes, resulting in G1/S phase arrest [56, 57]. Like epinephrine, stress-induced glucocorticoids enhance cancer cells resistance and reduce the efficacy of chemotherapy agents (such as paclitaxel, doxorubicin, TRAIL, and 5-fluorouracil) by anti-apoptosis mechanisms [58,59,60]. An et al. find that glucocorticoid modulatory element-binding protein 1 (GMEB1) interacts with the deubiquitinase USP40 to stabilize CFLARL and inhibit pro-caspase 8 activation, thereby inhibiting apoptosis in NSCLC cells (Fig. 2D) [61]. While the GRs blocker mifepristone can increase paclitaxel efficacy in triple-negative breast cancer [62].
Anti-anoikis is a marker of malignant transformation. In addition to increasing cancer cell survival in the absence of stromal attachment, anti-anoikis promotes migration, re-attachment, and secondary sites colonization [63]. Overexpression of oncogenes such as Ras, Raf, and Src and tumor suppressor genes downregulation such as phosphatase and tensin homolog and p53 help prevent anoikis [64]. For example, as a key downstream effector of Ras, SGK1 increases glucose uptake via glucose transporter protein 1 (GLUT1), which promotes glucose-mediated carbon flux into multiple metabolic pathways to inhibit anoikis (Fig. 2D) [65, 66]. Ras also reduce the phosphatase PHLPP1 expression, which promotes anoikis by activating p38 MAPK (Fig. 2D) [65].
A hallmark of cancer is genomic instability, which is linked to a greater tendency to DNA damage accumulation. In response to stress signals, the tumor suppressor p53 transcriptionally regulates target genes to initiate apoptosis and cell cycle arrest [67]. Chronic stress elevated glucocorticoids and catecholamines increase MDM2 activity and decreases p53 function by SGK1 [68] and Gs-protein-dependent-PKA/β-arrestin-1(ARRB1) pathways (Fig. 2D) [69], thereby synergistically leading to the DNA damage accumulation. In addition, exposure to cortisol and NE significantly increases the levels of reactive oxygen species (ROS) and reactive nitrogen species (RNS) that interact with DNA, also causing extensive DNA damage (Fig. 2D) [70]. Stress-induced G1 phase arrest significantly reduces the efficacy of paclitaxel in triple-negative breast cancer [71]. Several studies have addressed the effects of chronic stress on DNA integrity, but the specific cellular mechanisms underlying the effects on genome stability remain poorly understood. Moreover, it has not been elucidated whether adrenergic inhibitors of DNA damage repair sufficiently increase spontaneous tumorigenesis in vivo.
It is certain that stress hormones simultaneously influence the various biological mechanisms that promote cancer progression. Some key molecular switches, such as Src and Ras, which regulate important protein families such as MYC and FAK, enhance tumor cells' anti-apoptotic capacity to combat tumor suppression response and cancer therapy in the organism. At the same time, these molecular switches promote tumor cell stemness development and metastasis-related gene expression through transcription factors such as SLUG and β-catenin, accelerating the infiltration of blood vessels and lymphatic vessels around the TME and preparing the tumor for distant metastasis. Intervention in these critical molecular switches may provide new avenues for clinical cancer therapy. It is important that future work should explore additional key molecular switches linking chronic stress to these networks of biological mechanisms.
Effects of chronic stress on immune cells
A growing number of studies suggest that chronic stress can promote an inflammatory TME to activate the interaction between cancer cells and inflammatory immune cells [72]. Immune cells undergo metabolic changes after migrating to TME, including tumor-associated macrophages (TAMs), dendritic cells (DCs), myeloid-derived suppressor cells (MDSCs), and tumor-infiltrating lymphocytes (TILs) [73]. To control tumor growth, various cells communicate through direct contact or producing cytokines and chemokines and act in autocrine and paracrine ways [74].
Stress also modulates immune cell function by affecting tissue blood flow. Chemogenetic activation of the SNS or treatment with adrenergic receptor agonists induces vasoconstriction and reduced local blood flow [75]. This leads to sudden hypoxia in the TME, which triggers rapid calcium signaling in leukocytes, impairs leukocyte migration, and promotes tumor progression [76]. SNS regulating tissue hypoxia may have an impact on antitumor immunity, leading to transient suppression of CD8+ and CD4+ T cell motility in tissues [77].
Natural killer (NK) cells
NK cells are innately cytotoxic cells that play an important role in targeting early and effective immune responses to infection and cancer [78]. NK cell activity suppression in the perioperative period is due to excessive catecholamine secretion, this process is related to immune responses [79]. Epinephrine/norepinephrine inhibits cytotoxicity and cytokine production (including IL-2, IFN-γ, and IL-12) in NK cells by activating DNA-dependent mechanisms and protein synthesis (Fig. 3A) [80, 81]. Compared to catecholamines, glucocorticoids are considered to be secondary mediators of stress-induced NK cell cytotoxicity inhibition [82].

The biological mechanism of chronic stress affecting immune cells. A NK cell: E/NE inhibits cytokines production, including IL-12, IFN-γ and IL-2, through DNA-dependent mechanisms activation and protein synthesis. B Dendritic cell: chronic stress downregulates maturation-related markers such as CD40, CD68, and MHC II receptors in DCs. GRs upregulate TSC22D3 expression, mediate immunosuppressive effects through NF-κB, Ras and CVEBP, reduce pro-inflammatory cytokines IL-6, 12, and 23 productions, and block antigen presentation, resulting in the inability of TILs to acquire mature phenotype, as evidenced by low KLRG-1. C TAM & MDSC: chronic stress mediates metabolic reprogramming of macrophages and promotes immunosuppression. β2-ARs increases oxidative phosphorylation and promotes CPT1A expression; β2-ARs also increases FAO, which increases PGE2 production through COX2 overexpression and inhibits IFN-γ production by CD8+ T cells. GCs-GRs inhibit LRP1 and increase SIRPα, leading to an imbalance in the LRP1/SIRPα axis and inhibiting the phagocytosis of tumor cells by macrophages. D TIL: chronic stress reshaped the TIL phenotype in the TME, CD4+ TIL exhibit PD-1+, FOXP3+, CD8+ TIL exhibit PD-1+, TIM3+, Lag3+, IFN-γ−, CD28−. β2-ARs decreased GLUT1 and glucose uptake, resulting in reduced glycolysis and oxidative phosphorylation. GCs-GRs induce GILZ expression, which synergistically induces FoxP3 expression with TGF-β and SMAD2/3/4, further enhancing TGF-β signaling and promoting the conversion of naive T cells to Treg cells. Treg cells can produce immunosuppressive cytokines, including IL-10 and IL-35
Dendritic cells (DCs)
Chronic stress impairs DCs maturation, which is involved in the downregulation of maturation-related factors, such as CD40, CD68, and MHC II receptors, thus reducing antigen presentation and pro-inflammatory cytokines production (including IL-6, IL-12, and IL-23) [83]. Chronic stress also impairs the ability of activated DCs to migrate from the periphery to draining lymph nodes, which directly impairs the initiation of CD8+ T cell responses in vivo [84]. The specific mechanism is that stress-induced glucocorticoid-inducible factor TSC22D3 mediates immunosuppressive effects through homo- and heterodimerization with Ras, NF-κB, and CCAAT enhancer-binding protein (CVEBP), thereby blocking type I interferon response and IFN-γ+ T cell activation in DCs (Fig. 3B) [7]. In mouse melanoma model, the aforementioned functional alterations of DCs prevents cytotoxic T lymphocytes from acquiring a fully mature effector phenotype in vivo, manifested as low expression of KLRG-1 (Fig. 3B) [84]. PLGA-MS vaccination induces prophylactic and therapeutic protection against aggressive melanomas and promotes CD8+ T cell production [85]. However, the reduced tumor protection against immunization in stressed mice after vaccination may suggest that it is important to sufficiently understand the effects of chronic stress on DC function in the therapeutic environment [84].
Tumor-infiltrating lymphocytes (TILs)
T cells are widely classified according to the central markers (CD8 and CD4) and their receptor subunits [86]. T cell receptor-induced activation drives metabolic reprogramming of T cells (including increased glycolysis and oxidative phosphorylation) to meet essential biosynthetic requirements, this process is remodeled by chronic stress. Studies have shown that adrenergic signaling decrease GLUT1 expression in CD8+ T cells, leading to decreased glucose uptake and reduced glycolysis. Not only that, β-ARs-mediated mitochondrial respiration inhibition cause CD8+ T-cell mitochondrial dysfunction during activation (Fig. 3D) [87]. Blocking β-ARs signaling increases the metabolic reprogramming in TILs. This is related to increased costimulatory molecule CD28 and antitumor functions, including IFN-γ production, the proliferation of antigen-specific CD8+ T cells, and cytolytic killing capacity [84, 88]. Chronic stress also contributes to T-cell exhaustion. One possible mechanism is that β-ARs regulate immune checkpoint expression, which strongly suppress anti-tumor immune responses, characterized by increased PD-1, TIM-3, and Lag3 expression [89]. In breast cancer patient samples, the immune checkpoint molecules expression is positively correlated with sympathetic nerve density, and their expression levels correlated with the recurrence rate of breast cancer patients [90]. Blocking β-ARs signaling results in checkpoint genes expression decreased in CD8+ TILs, such as Ceacam1, CD160, Btla, CD274, and Tigit [89].
Immunotherapy aims to enhance the anti-tumor CD8+ T cell response. The most widely used immunotherapy is monoclonal antibodies designed against immune checkpoint molecules, such as PD-1, CTLA4, and 4-1BB, or other stimulatory molecules [91]. However, the effect of chronic stress hormones on the aforementioned T cells leads to the failure of immunotherapy targeting T cells [84, 88, 92]. Notably, positive environmental stimulation (eustress) contributes to reshaping the immunosuppressive microenvironment, enhancing CD8+ T cells antitumor immunity and overcoming resistance to therapeutic PD-L1/PD-1 blockade [93]. The use of corticosteroids is corrected with poorer prognosis in patients with NSCLC, which reduce efficacy of immune checkpoint inhibitor therapy [94, 95]. However, the exact mechanism of how glucocorticoids affect immunotherapy remains elusive.
CD4+ helper T cells play a role in antitumor immunity response by inducing CD40 ligand expression to stimulate CD40 on DCs to promote CD8+ T cell initiation [96]. CD4+ T cells differentiate based on various cytokines [97]. It has been shown that β2-AR activation of CD4+ T lymphocytes inhibits Th1-cytokine production and cell proliferation [98]. Interestingly, CD4+ T cell naive and Th1 cells express the β2-AR, while the Th2 cells do not, and the result suggests that β-ARs regulate the CD4+ T cells' Th1/Th2 differentiation, polarizing them toward the Th2 phenotype [99]. Treg cells are usually described as tumor-promoting CD4+ CD25+ FoxP3+ T cells, which produce immunosuppressive cytokines, such as IL-35, IL-10, and TGF-β [100]. Glucocorticoid-induced leucine zipper (GILZ) promotes Treg cells production, which enhances TGF-β signaling by binding to Smad2/3/4 and promoting FoxP3 expression (Fig. 3D) [101]. The balance between Th17 and Treg cells has emerged as an essential factor in the modulation of autoimmunity and cancer [97]. However, the balance of Th17 and Treg cells in tumor progression in the context of chronic stress remains unreported.
TAMs and MDSCs
The macrophages residing within TME are known as TAMs, which are the main infiltrating immune cells in TME. Psychological depression promotes intratumoral infiltration of TAMs and is correlated with poor clinical prognosis and resistance to tumor therapy due to the immune-suppressive and tumor-promoting activity of TAMs [102, 103]. Catecholamines similarly increase the recruitment of MDSCs, which are immature bone marrow mononuclear cells characterized by a CD11b+ Gr1+ phenotype [104]. Chronic stress promotes CXCL2/CXCL3 secretion of tumor cells, induces CXCR2 expression of myeloid cells, and thus facilitating spleen myeloid cells movement into tumor tissue through the CXCL2/CXCL3-CXCR2 axis [105]. Besides, enhanced adrenergic signaling mobilizes tumor cells to secrete monocyte/macrophage chemotactic factor CCL2, which increases CD14+/CD68+ macrophage infiltration in the TME [106]. Glucocorticoids-GRs signal inhibits low-density lipoprotein receptor-related protein-1 (LRP1) expression in tumor-associated macrophages, and increases signal regulatory protein alpha (SIRPα) expression in macrophages, leading to an imbalance in the LRP1/SIRPα axis, which inhibits the phagocytosis of tumor cells by macrophages (Fig. 3C) [107]. Repeated social defeats result in the recruitment of bone marrow-derived monocytes to the brain, where they increase neuroinflammation, leading to prolonged anxiety-like behavior [108], and forming a vicious circle.
Functionally, chronic stress mediates the metabolic reprogramming of macrophages and promotes immunosuppression [109, 110]. β2-ARs signaling in MDSCs or TAMs reduces glycolysis and increases oxidative phosphorylation and fatty acid oxidation (FAO). The latter two are critical metabolic pathways for driving the immune-suppressive function of bone marrow cells [111], and increased FAO promotes prostaglandin E2 (PGE2) production through cytochrome c oxidase subunit 2 (COX2) overexpression (Fig. 3C) [110]. PGE2 is a critical lipid metabolite that effectively recruits neutrophils and macrophages, strongly suppresses both the proliferation and IFN-γ production of CD8+ T cells (Fig. 3C) [112]. Ben-Shaanan et al. show that positive emotions reduce norepinephrine levels in bone marrow, decrease MDSCs production, and reduce the suppressive effects of MDSCs on T cell proliferation and effector phenotypes in tumor-bearing mice [113].
On the other hand, chronic stress-induced catecholamines promote macrophage M2 polarization [114]. M2 macrophages are commonly associated with tumor metastasis and angiogenesis, as evidenced by increased expression of pro-metastatic genes, production of pro-angiogenic molecules and promotion of perivascular extracellular matrix degradation [115]. TAMs signal to tumor cells via COX2/PGE2 in response to β-ARs signaling to generate VEGFC required for lymphatic remodeling [116]. VEGFC is central to lymph angiogenesis, which increases lymphatic tumor cell dissemination, leading to increased lymph node metastasis [117].
It is reasonable that metabolic reprogramming and polarization of macrophages by chronic stress play a key role in tumor development. Understanding the mechanisms by which chronic stress drives their immunosuppressive functions could help improve cancer immunotherapy and contribute to the discovery of new therapeutic approaches.
These data suggest that chronic stress reshapes immune cells in multiple ways, thereby promoting tumor progression. Stress directly inhibits the anti-tumor function of TILs and NK cells through epigenetic or metabolic reprogramming, which also leads to immunotherapy failure. Stress hormones also indirectly alter the immune function of macrophages and DC cells, leaving TILs unsupported and ultimately leading to uncontrolled tumor cell proliferation capacity.
Effects of chronic stress on tumor-associated stromal cells
Cancer-associated fibroblasts (CAFs) are the largest stromal cell population in the TME and play an active role in shaping TME to support tumor cell survival, metastasis, angiogenesis, immunosuppression, and treatment resistance [118]. Nagaraja et al. have shown that epinephrine stimulates tumor cells to produce inhibin-βA, which drives elevated levels of collagen production by CAFs, such as COL3A1, COL5A1, COL5A2, and COL11A1 [119]. Increased collagen expression is associated with cancer progression and poor prognosis in breast cancer patients. Collagen accumulation enhances the stemness of cancer cells, induces apoptosis resistance, and promotes cancer metastasis [120].
Normal adipocytes around the tumor are driven into cancer-associated adipocytes (CAAs) by cancer cells. Cancer cells capture metabolites from stromal adipocytes via CAAs and become metabolic parasites [121, 122]. Avril et al. have shown that adrenergic stimulation changes the secretome of CAAs, subsequently promoting cancer cell proliferation [123].
In addition to CAFs and CAAs, endothelial cells are also the main targets of adrenergic nerves in the TME [124]. β-ARs signaling promotes angiogenesis by altering aerobic glycolysis in endothelial cells [125]. In contrast, inhibition of adrenergic neural activity enhances oxidative phosphorylation in endothelial cells through increased cytochrome c oxidase assembly factor 6 (COA6) expression, thereby inhibiting angiogenesis [125].
Effects of chronic stress on the perineural nerve of tumor
Growing pre-clinical and clinical evidence suggests sustained adrenergic signaling in the TME plays a critical role in tumor growth and progression [90, 126]. However, the exact mechanism by which adrenergic neurotransmitters are delivered to the TME is unclear. Neurotrophic factors in the TME promote axonal growth of preexisting nerves. Thereby tumor-associated neural networks are established, which generate neural signals to regulate tumorigenesis and metastasis [127, 128].
Data to date suggest that peripheral nerves are present in a wide range of cancers and influence cancer behavior, closely related to tumorigenesis, angiogenesis, invasion, and metastasis. Patients with densely innervated tumors have increased metastasis rates and decreased survival compared to patients with less innervated tumors [125, 127, 129]. The neuronal organization in tumors has two origins. One is that tumor cells secrete neurotrophic factors that promote the production of new tumor-guided axons from pre-existing local nerves [130,131,132,133]. The other is the transformation of tumor cells into a neuroendocrine phenotype that recruit of neural progenitor cells from the CNS to infiltrate and reside in the TME and metastases [134, 135].
Allen et al. find that norepinephrine causes tumor cells to secrete brain-derived neurotrophic factor (BDNF) in an β3-ARs/cAMP/EPAC/JNK-dependent manner [130]. Similarly, in pancreatic ductal adenocarcinoma, norepinephrine promotes β2-ARs-dependent nerve growth factor (NGF) secretion [131]. Elevated BDNF/NGF levels promote innervation via neurotrophic receptor tyrosine kinase 2 (TrkB) receptors [130, 136], and overexpression of TrkB is related to shorter survival with ovarian cancer patients (Fig. 4) [137]. Interestingly, neurotrophic factors can also be released into the TME in the form of exosomes. As pointed out by Madeo, human head and neck squamous carcinoma cells can induce tumor innervation by secreting exosomes containing neurotrophic factor Ephrin B1 to peripheral sensory nerves (Fig. 4) [133]. Tumor innervation further increases adrenergic input into the TME, forming a feed-forward loop [131, 136]. Sustained accumulation of adrenergic signaling promotes tumor progression [130].

The biological mechanism of chronic stress affecting perineural nerve of tumor. Chronic stress-induced ADRB increases BDNF via ADRB3/cAMP/EpAC/JNK and NGF via ADRB2/CREB or ADRB2/ERK. BDNF and NGF bind to TrkB in peritumor sensory neurons, promoting innervation of the tumor microenvironment and producing a sustained accumulation of adrenergic signals, forming a feed-forward loop. Chronic stress-induced low p53 in tumor cells leads to miR-34a loss in exosomes, resulting in increased ASCL1 in peritumor sensory neurons, which redifferentiates peritumor sensory neurons into adrenergic neurons. In addition, tumor cells secrete exosomes containing EphrinB1 into peritumor sensory nerves, inducing tumor innervation
The sustained accumulation of adrenergic signaling can act not only through nerve growth factor (BDNF, NGF) but also by reprogramming sensory neurons in cancerous tissue to differentiate into new adrenergic neurons. Amit et al. show that in tumorigenesis, TP53 mutations in oral cancer cells leads to miR-34a loss in exosomes, which increases the expression of adrenergic-related mRNAs (e.g. ASCL1) in sensory neurons to promote differentiation of the adrenergic phenotype (Fig. 4). Then adrenergic neurons release local norepinephrine or epinephrine into the TME, resulting in tumor nerve density increased and tumor growth, providing an additional source of catecholaminergic in chronic stress models [132].
Under stressful conditions, CNS activates the autonomic nervous system or the HPA axis, secreting variety of mediators that are beneficial to tumorigenesis and progression. Moreover, CNS also directly establish contact with the distant TME to regulate tumor development. Mauffrey et al. found that neuronal progenitors located in the CNS express dual corticotropin, which is able to infiltrate and reside in prostate tumor tissues as well as their metastases. These neurons are able to differentiate into adrenergic type that contributed to the growth and metastasis of the primary tumor [135]. These data indicate the potential mechanisms by which stress hormones enter the TME, activate β-ARs pathways to promotes tumor development. However, whether neuronal infiltration into tumor tissues and reprogramming of neurons innervating tumor tissues to an adrenergic phenotype are specific phenomena for some cancers requires further investigation.
Increased tumor innervation in turn regulates angiogenesis, immune and inflammatory and oncogene activation through adrenergic signaling, promoting tumor growth and metastasis [138]. Not only that, tumor-associated neurons release the calcitonin gene-related peptide (CGRP), which directly increases the exhaustion of CD8+ T-cells and limits their ability to eliminate melanoma [139]. In contrast, sensory neuron ablation reverses these phenomena [140]. The data suggest that tumor nerve infiltration represents a new feature of cancer. And interfering with nerve signaling in TME through surgical or pharmacological approaches offers a promising new strategy for cancer treatment.
Treatment for cancer patients with chronic stress
Epidemiological studies have shown that depression is related to cancer recurrence and mortality and it plays a key role as an independent predictor of cancer recurrence and prognosis [42, 141]. Obviously, the stress perception of cancer patients is affected by the physical and psychological burden of the disease. In recent years, based on studies in animal models, the endocrine, cellular and molecular mechanisms by which stress can promote cancer progression have been elucidated, including direct effects on the malignancy of cancer cells, anti-tumor immune activity, and indirect regulation of peritumor nerves. However, in clinic, there is lack of effective treatment for chronic stress in cancer patients [13]. We will discuss the opportunities and challenges of potential pharmacological and non-pharmacological treatment based on preliminary clinical data (Table 1) and retrospective analysis (Table 2).
Pharmacological treatment
Inhibiting tumor growth by regulating the nervous system
As mentioned above, chronic stress affects the occurrence and development of tumors mainly through ARs/GRs pathway. The β-adrenergic blockers, a class of neuromodulator drugs which blocks catecholamines from binding to β-ARs, have potential in improving life quality, anxiety, and depression in cancer patients. Moreover, it also reduces distant metastasis, recurrence of cancer, and cancer-specific mortality [142,143,144,145,146,147,148,149,150]. In clinical trials, BBs are also used as anti-hypertensive agents to prevent chemotherapy-induced cardiac injury [151]. However, part retrospective analyses of BBs show (Table 2) that there are conflicting claims about whether BBs alone can improve OS and progression-free survival (PFS) in patients [152,153,154,155,156,157,158,159,160,161]. The potential benefits of BBs may be offset by anxiety and depression [159]. Notably, β-ARs have different roles across tumor types and there is selective heterogeneity in the BBs [160]. It is also possible that the impact of GRs is being ignored [7]. There are fewer clinical trials and retrospective analyses supporting the use of GRs antagonists alone to improve patient survival. To be sure, mifepristone cannot be as a single agent to treat breast cancer [162]. However, low-dose of mifepristone improves life quality and reduces tumor size in patients with uterine fibroids (NCT00133705) [163].
Major clinical challenges to the use of neuromodulator drugs may include lacking tumor specificity and potential side effects at effective doses for cancer treatment. Additional randomized controlled trials (RCTs) are required to ascertain whether these drugs will improve clinical outcomes of cancer.
Combating treatment resistance by disrupting stress adaptation
The key role of chronic stress in influencing clinical treatment is to enhance the radiation resistance and drug resistance of tumor cells through β-ARs, thereby blocking apoptosis. Ongoing clinical trials tend to combine BBs/GRs antagonists with radiotherapy/chemotherapy (Table 1) to counteract treatment resistance due to chronic stress. A completed clinical trial shows that BBs delay the resistance of tumor cells to EGFR tyrosine kinase inhibitors, and PFS is improved in lung cancer patients [164]. Retrospective analyses have shown (Table 2) that the combination of BBs with anti-angiogenic drugs, immunotherapy, radiotherapy, or chemotherapy prolongs overall patient survival [165,166,167,168,169,170,171,172], which provides a direction for anti-cancer treatment resistance.
Combined with immunotherapy to enhance anti-tumor immunity
Preclinical studies show that psychological stress regulates the immune microenvironment of tumors by remodeling immune cells in the body. New evidence suggests that BBs may enhance the efficacy of cytotoxic therapies by modulating immune responses [92]. Retrospective analysis shows that combining immunotherapy with BBs improves the OS of patients with metastatic melanoma (Table 2) [166, 170]. BBs also prevent immune escape induced by chronic stress and improve survival in pancreatic cancer [173]. This finding may have important implications for immune checkpoint inhibitor-silenced tumors (e.g., pancreatic cancer, prostate cancer) [174]. Therefore, the combination therapy of BBs and immune checkpoint for these tumors deserves further investigation. Given that many neurotropic drugs are already used for other indications, there may be some promise in combining these drugs with cytotoxic therapies, immune checkpoint blockade, or cancer vaccines.
Non-pharmacological treatment
Many complementary and integrative therapies are used to reduce stress and improve life quality of cancer patients. Complementary and alternative medicine bring some benefits to patients beyond surgery and medications by improving life quality, boosting the immune system, and relieving symptoms caused by conventional therapies [175].
Psychological interventions
Recent findings support that multiple psychological interventions may be effective in relieving stress and strengthening social support [176, 177]. Mindfulness interventions typically focus on fostering greater self-awareness for reducing responsiveness under stress. Mindfulness interventions help cancer patients relieve the anxiety associated with cancer diagnosis, treatment and fear of recurrence, and provide psychological support [178]. A 6-week mindfulness-based stress reduction (MBSR) program, which includes meditation, relaxation, yoga, and daily practices, shows that participation in MBSR programs improves life quality and reduces stress and mood disorders in breast cancer patients [179]. This is manifested by reduced levels of cortisol and proinflammatory cytokines and the recovery of NK cell activity [180, 181]. Randomized trials also show that MBSR improves cancer patients’ fatigue, insomnia, life quality, and biological markers of health [182, 183]. However, most studies focus on breast cancer patients, so the success of psychological interventions in other types of cancer may not be generalizable.
Exercise
Exercise interventions during and after treatment have great potential to improve survival and reduce recurrence in cancer patients [184]. The potential effects of adding exercise to conventional treatment to improve psychosocial, cognitive function, and depressive symptoms in cancer patients is being explored recently. Several RCTs have shown that physical activity during cancer treatment results in improved fatigue in cancer patients [185,186,187]. Exercise also improves treatment-related inflammatory markers levels in cancer patients [188], increases the CD8+ T cells activity [189], and promotes favorable immune environment restoration. Yoga is a form of aerobic exercise which is commonly used in mind–body therapy for breast cancer patients [190, 191]. In an RCT of breast cancer survivors, yoga reduces fatigue and inflammation and improves mood and life quality [192].
Taken together, physical exercise help to improve psychological stress, fatigue, and depression in cancer patients. We believe that trials with larger samples and longer follow-up periods should be conducted to assess the effects of exercise in patients with different malignancies and to determine whether this translates into a survival advantage.
Dietary strategies, herbs and nutritional products
Epidemiological data support the idea that dietary strategies have the potential to influence biological pathways associated with carcinogenesis [193]. Following a traditional dietary pattern, such as the Mediterranean, Norwegian or Japanese diets, and increasing the intake of fruits, vegetables, whole grains, nuts and seeds have anti-cancer and anti-depressive effects [194, 195]. RCTs illustrate that low-calorie diet combines with exercise promote the mental and physical health of depressed patients [196, 197].
The most frequently used complementary and alternative medicine are vitamins and herbal products [198]. Vitamin and mineral supplementation are beneficial to improve stress, anxiety and fatigue. A prospective study of breast cancer indicates that Withania somnifera improve cancer-related fatigue and life quality [199]. Withania somnifera is an herb that prevents oxidation, inflammation, cancer and stress.
However, vitamins, minerals and herbs may have a negative impact on treatment. A recent ancillary study shows that any anti-oxidant (vitamins A, C and E, coenzyme Q10) during treatment is correlated with an increased risk of recurrence and mortality [200]. Iron and vitamin B12 supplementation before and during chemotherapy are associated with an increased risk of death [200]. Given the present data, although the intake of natural products may relieve cancer-related stress and improve life quality, vitamins or minerals, especially during chemotherapy, ought to be used with caution by patients and clinicians.
Conclusions and perspectives
There is strong evidence that chronic stress contributes to cancer development by modulating most features of cancer. Molecular and systemic mechanisms mediating these effects have been identified. Chronic stress affects the occurrence of various important events that promote cancer progression through ARs/GRs, including maintenance of stem cell-like traits, metastasis, angiogenesis, DNA damage accumulation, apoptosis resistance, immune escape, and metabolic reprogramming. These findings provide a basis for treating stress-related tumors. Stress signaling reprograms immune cells through glycometabolism, disrupts immune checkpoints, and regulates the distribution of immune cells in TME. Understanding the mechanisms by which chronic stress drives its immunosuppressive function may improve cancer immunotherapy, which could also be a potential breakthrough point for the current immunotherapy lack of effectiveness and help to discover new therapeutic approaches. Chronic stress also affects tumor-associated stromal cells to reshape TME. In addition, chronic stress affects tumor neural infiltration, which provides a potential mechanism for how stress hormones enter the TME. This reminds us to focus on the impact of chronic stress in cancer treatment, and timely take psychological intervention to relieve the chronic stress of patients.
Epidemiological and clinical studies have shown that stress affects cancer recurrence and the effectiveness of cancer treatments. And the use of synthetic glucocorticoids as anti-inflammatory and analgesic adjuvant drugs in cancer treatment affects the effectiveness of immunotherapy, which is also currently one of the problems that need to be solved in clinical practice. By modulating the nervous system and disrupting stress adaptation mechanisms, the use of BBs in synergy with anti-angiogenic drugs, immunotherapy, radiotherapy, or chemotherapy might be effective in treatment, but RCTs are needed to examine this hypothesis. Meanwhile, complementary and alternative medicine helps cancer patients improve their life quality from various aspects of psychological interventions, exercise, dietary strategies, herbs and nutritional additives and herbs and nutritional additives, relieving cancer patients' anxiety, then alleviating tumor progression. These pharmacological and non-pharmacological approaches against chronic stress provide novel interventional treatments for cancer.
Availability of data and materials
Not applicable.
Abbreviations
- ACTH:
-
Adrenocorticotropic hormone
- ARs:
-
Adrenergic receptors
- BBs:
-
β-Adrenergic blockers
- BDNF:
-
Brain-derived neurotrophic factor
- CAFs:
-
Cancer-associated fibroblasts
- CCL2:
-
C-C motif chemokine ligand 2
- CCR2:
-
C-C motif chemokine receptor 2
- CDK:
-
Cyclin-dependent kinase
- CRH:
-
Corticotropin-releasing hormone
- CNS:
-
Central nervous system
- COX-2:
-
Cyclooxygenase2
- CSCs:
-
Cancer stem cells
- DCs:
-
Dendritic cells
- Dex:
-
Dexamethasone
- EGFR:
-
Epidermal growth factor receptor
- FAK:
-
Focal adhesion kinase
- FAO:
-
Fatty acid oxidation
- GILZ:
-
Glucocorticoid-induced leucine zipper
- GLUT1:
-
Glucose transporter protein 1
- GRs:
-
Glucocorticoid receptors
- HDAC2:
-
Histone deacetylase 2
- hTERT:
-
Human telomerase reverse transcriptase
- HPA:
-
Hypothalamic–pituitary–adrenal
- LDHA:
-
Lactate dehydrogenase A
- MACC1:
-
Metastasis‐associated in colon cancer 1
- MBSR:
-
Mindfulness-based stress reduction
- MDSCs:
-
Myeloid-derived suppressor cells
- MMP:
-
Matrix metalloproteinase
- NGF:
-
Nerve growth factor
- NSCLC:
-
Non-small cell lung cancer
- OS:
-
Overall survival
- PFC:
-
Prefrontal cortex
- PFS:
-
Progression-free survival
- PGE2:
-
Prostaglandin E2
- PPARγ:
-
Peroxisome proliferator-activated receptor γ
- RCTs:
-
Randomized controlled trial
- ROR1:
-
Receptor-tyrosine-kinase-like orphan receptor
- SGK1:
-
Serum glucocorticoid-induced kinase 1
- SNS:
-
Sympathetic nervous system
- SYP:
-
Synaptophysin
- TAMs:
-
Tumor-associated macrophages
- TEAD4:
-
TEA domain transcription factor 4
- TILs:
-
Tumor-infiltrating lymphocytes
- TME:
-
Tumor microenvironment
- TrkB:
-
Tyrosine kinase 2
- USP28:
-
Ubiquitin-specific protease 28
- VEGF:
-
Vascular endothelial growth factor
References
Yuan K, Zheng YB, Wang YJ, Sun YK, Gong YM, Huang YT, Chen X, Liu XX, Zhong Y, Su SZ, et al. A systematic review and meta-analysis on prevalence of and risk factors associated with depression, anxiety and insomnia in infectious diseases, including COVID-19: a call to action. Mol Psychiatry. 2022. https://doi.org/10.1038/s41380-022-01638-z.
Russell G, Lightman S. The human stress response. Nat Rev Endocrinol. 2019;15(9):525–34.
Cole SW, Nagaraja AS, Lutgendorf SK, Green PA, Sood AK. Sympathetic nervous system regulation of the tumour microenvironment. Nat Rev Cancer. 2015;15(9):563–72.
Sloan EK, Capitanio JP, Tarara RP, Cole SW. Social temperament and lymph node innervation. Brain Behav Immun. 2008;22(5):717–26.
Cui B, Peng F, Lu J, He B, Su Q, Luo H, Deng Z, Jiang T, Su K, Huang Y, et al. Cancer and stress: NextGen strategies. Brain Behav Immun. 2021;93:368–83.
Lutgendorf SK, DeGeest K, Dahmoush L, Farley D, Penedo F, Bender D, Goodheart M, Buekers TE, Mendez L, Krueger G, et al. Social isolation is associated with elevated tumor norepinephrine in ovarian carcinoma patients. Brain Behav Immun. 2011;25(2):250–5.
Yang H, Xia L, Chen J, Zhang S, Martin V, Li Q, Lin S, Chen J, Calmette J, Lu M, et al. Stress-glucocorticoid-TSC22D3 axis compromises therapy-induced antitumor immunity. Nat Med. 2019;25(9):1428–41.
Mitchell AJ, Chan M, Bhatti H, Halton M, Grassi L, Johansen C, Meader N. Prevalence of depression, anxiety, and adjustment disorder in oncological, haematological, and palliative-care settings: a meta-analysis of 94 interview-based studies. Lancet Oncol. 2011;12(2):160–74.
Hartung TJ, Brahler E, Faller H, Harter M, Hinz A, Johansen C, Keller M, Koch U, Schulz H, Weis J, et al. The risk of being depressed is significantly higher in cancer patients than in the general population: prevalence and severity of depressive symptoms across major cancer types. Eur J Cancer. 2017;72:46–53.
Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017;387:61–8.
Gysler SM, Drapkin R. Tumor innervation: peripheral nerves take control of the tumor microenvironment. J Clin Invest. 2021;131(11): e147276.
Monje M, Borniger JC, D’Silva NJ, Deneen B, Dirks PB, Fattahi F, Frenette PS, Garzia L, Gutmann DH, Hanahan D, et al. Roadmap for the emerging field of cancer neuroscience. Cell. 2020;181(2):219–22.
Eckerling A, Ricon-Becker I, Sorski L, Sandbank E, Ben-Eliyahu S. Stress and cancer: mechanisms, significance and future directions. Nat Rev Cancer. 2021;21(12):767–85.
Zhang L, Pan J, Chen W, Jiang J, Huang J. Chronic stress-induced immune dysregulation in cancer: implications for initiation, progression, metastasis, and treatment. Am J Cancer Res. 2020;10(5):1294–307.
Cole SW, Sood AK. Molecular pathways: beta-adrenergic signaling in cancer. Clin Cancer Res. 2012;18(5):1201–6.
Eng JW, Reed CB, Kokolus KM, Pitoniak R, Utley A, Bucsek MJ, Ma WW, Repasky EA, Hylander BL. Housing temperature-induced stress drives therapeutic resistance in murine tumour models through beta2-adrenergic receptor activation. Nat Commun. 2015;6:6426.
Garcia-Mayea Y, Mir C, Masson F, Paciucci R, LLeonart ME. Insights into new mechanisms and models of cancer stem cell multidrug resistance. Semin Cancer Biol. 2020;60:166–80.
Cui B, Luo Y, Tian P, Peng F, Lu J, Yang Y, Su Q, Liu B, Yu J, Luo X, et al. Stress-induced epinephrine enhances lactate dehydrogenase A and promotes breast cancer stem-like cells. J Clin Invest. 2019;129(3):1030–46.
Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, Zhang G, Wang X, Dong Z, Chen F, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020;5(1):8.
Banerjee J, Papu John AM, Schuller HM. Regulation of nonsmall-cell lung cancer stem cell like cells by neurotransmitters and opioid peptides. Int J Cancer. 2015;137(12):2815–24.
Zheng Y, Zhang J, Huang W, Zhong LLD, Wang N, Wang S, Yang B, Wang X, Pan B, Situ H, et al. Sini San inhibits chronic psychological stress-induced breast cancer stemness by suppressing cortisol-mediated GRP78 activation. Front Pharmacol. 2021;12: 714163.
Ma W, Liu P, Zheng J, Lu J, Zhao Q, Li D, Guo Y, Qian L, Wang Q, Miao X, et al. Immune and nonimmune mechanisms mediate the mental stress-induced tumor growth in a xenograft model of breast cancer. Cell Death Dis. 2021;12(11):987.
He L, Yuan L, Sun Y, Wang P, Zhang H, Feng X, Wang Z, Zhang W, Yang C, Zeng YA, et al. Glucocorticoid receptor signaling activates TEAD4 to promote breast cancer progression. Cancer Res. 2019;79(17):4399–411.
Massague J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016;529(7586):298–306.
Chang A, Le CP, Walker AK, Creed SJ, Pon CK, Albold S, Carroll D, Halls ML, Lane JR, Riedel B, et al. beta2-Adrenoceptors on tumor cells play a critical role in stress-enhanced metastasis in a mouse model of breast cancer. Brain Behav Immun. 2016;57:106–15.
Armaiz-Pena GN, Allen JK, Cruz A, Stone RL, Nick AM, Lin YG, Han LY, Mangala LS, Villares GJ, Vivas-Mejia P, et al. Src activation by beta-adrenoreceptors is a key switch for tumour metastasis. Nat Commun. 2013;4:1403.
Choi MJ, Cho KH, Lee S, Bae YJ, Jeong KJ, Rha SY, Choi EJ, Park JH, Kim JM, Lee JS, et al. hTERT mediates norepinephrine-induced Slug expression and ovarian cancer aggressiveness. Oncogene. 2015;34(26):3402–12.
Bu S, Wang Q, Sun J, Li X, Gu T, Lai D. Melatonin suppresses chronic restraint stress-mediated metastasis of epithelial ovarian cancer via NE/AKT/beta-catenin/SLUG axis. Cell Death Dis. 2020;11(8):644.
Du P, Zeng H, Xiao Y, Zhao Y, Zheng B, Deng Y, Liu J, Huang B, Zhang X, Yang K, et al. Chronic stress promotes EMT-mediated metastasis through activation of STAT3 signaling pathway by miR-337-3p in breast cancer. Cell Death Dis. 2020;11(9):761.
Cheng Y, Gao XH, Li XJ, Cao QH, Zhao DD, Zhou JR, Wu HX, Wang Y, You LJ, Yang HB, et al. Depression promotes prostate cancer invasion and metastasis via a sympathetic-cAMP-FAK signaling pathway. Oncogene. 2018;37(22):2953–66.
Li J, Yang XM, Wang YH, Feng MX, Liu XJ, Zhang YL, Huang S, Wu Z, Xue F, Qin WX, et al. Monoamine oxidase A suppresses hepatocellular carcinoma metastasis by inhibiting the adrenergic system and its transactivation of EGFR signaling. J Hepatol. 2014;60(6):1225–34.
Pan C, Wu J, Zheng S, Sun H, Fang Y, Huang Z, Shi M, Liang L, Bin J, Liao Y, et al. Depression accelerates gastric cancer invasion and metastasis by inducing a neuroendocrine phenotype via the catecholamine/beta2 -AR/MACC1 axis. Cancer Commun. 2021;41(10):1049–70.
Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475(7355):222–5.
Chen H, Liu D, Guo L, Cheng X, Guo N, Shi M. Chronic psychological stress promotes lung metastatic colonization of circulating breast cancer cells by decorating a pre-metastatic niche through activating beta-adrenergic signaling. J Pathol. 2018;244(1):49–60.
Obradovic MMS, Hamelin B, Manevski N, Couto JP, Sethi A, Coissieux MM, Munst S, Okamoto R, Kohler H, Schmidt A, et al. Glucocorticoids promote breast cancer metastasis. Nature. 2019;567(7749):540–4.
Lansinger OM, Biedermann S, He Z, Colevas AD. Do steroids matter? A retrospective review of premedication for taxane chemotherapy and hypersensitivity reactions. J Clin Oncol. 2021;39(32):3583–90.
Janowitz T, Kleeman S, Vonderheide RH. Reconsidering dexamethasone for antiemesis when combining chemotherapy and immunotherapy. Oncologist. 2021;26(4):269–73.
Yang R, Yu Y. Glucocorticoids are double-edged sword in the treatment of COVID-19 and cancers. Int J Biol Sci. 2021;17(6):1530–7.
Zhang Y, Shi G, Zhang H, Xiong Q, Cheng F, Wang H, Luo J, Zhang Y, Shi P, Xu J, et al. Dexamethasone enhances the lung metastasis of breast cancer via a PI3K-SGK1-CTGF pathway. Oncogene. 2021;40(35):5367–78.
Tian D, Tian M, Han G, Li JL. Increased glucocorticoid receptor activity and proliferation in metastatic colon cancer. Sci Rep. 2019;9(1):11257.
Horowitz M, Neeman E, Sharon E, Ben-Eliyahu S. Exploiting the critical perioperative period to improve long-term cancer outcomes. Nat Rev Clin Oncol. 2015;12(4):213–26.
Wang X, Wang N, Zhong L, Wang S, Zheng Y, Yang B, Zhang J, Lin Y, Wang Z. Prognostic value of depression and anxiety on breast cancer recurrence and mortality: a systematic review and meta-analysis of 282,203 patients. Mol Psychiatry. 2020;25(12):3186–97.
Levi B, Matzner P, Goldfarb Y, Sorski L, Shaashua L, Melamed R, Rosenne E, Page GG, Ben-Eliyahu S. Stress impairs the efficacy of immune stimulation by CpG-C: potential neuroendocrine mediating mechanisms and significance to tumor metastasis and the perioperative period. Brain Behav Immun. 2016;56:209–20.
Viallard C, Larrivee B. Tumor angiogenesis and vascular normalization: alternative therapeutic targets. Angiogenesis. 2017;20(4):409–26.
Hisano Y, Hla T. Bioactive lysolipids in cancer and angiogenesis. Pharmacol Ther. 2019;193:91–8.
Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C, Jennings NB, Armaiz-Pena G, Bankson JA, Ravoori M, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006;12(8):939–44.
Landen CN Jr, Lin YG, Armaiz Pena GN, Das PD, Arevalo JM, Kamat AA, Han LY, Jennings NB, Spannuth WA, Thaker PH, et al. Neuroendocrine modulation of signal transducer and activator of transcription-3 in ovarian cancer. Cancer Res. 2007;67(21):10389–96.
Hulsurkar M, Li Z, Zhang Y, Li X, Zheng D, Li W. Beta-adrenergic signaling promotes tumor angiogenesis and prostate cancer progression through HDAC2-mediated suppression of thrombospondin-1. Oncogene. 2017;36(11):1525–36.
Zhou J, Liu Z, Zhang L, Hu X, Wang Z, Ni H, Wang Y, Qin J. Activation of beta2-adrenergic receptor promotes growth and angiogenesis in breast cancer by down-regulating PPARgamma. Cancer Res Treat. 2020;52(3):830–47.
Deng GH, Liu J, Zhang J, Wang Y, Peng XC, Wei YQ, Jiang Y. Exogenous norepinephrine attenuates the efficacy of sunitinib in a mouse cancer model. J Exp Clin Cancer Res. 2014;33(1):21.
MacDonald CR, Bucsek MJ, Qiao G, Chen M, Evans L, Greenberg DJ, Uccello TP, Battaglia NG, Hylander BL, Singh AK, et al. Adrenergic receptor signaling regulates the response of tumors to ionizing radiation. Radiat Res. 2019;191(6):585–9.
Zhang Y, Zanos P, Jackson IL, Zhang X, Zhu X, Gould T, Vujaskovic Z. Psychological stress enhances tumor growth and diminishes radiation response in preclinical model of lung cancer. Radiother Oncol. 2020;146:126–35.
Hassan S, Karpova Y, Baiz D, Yancey D, Pullikuth A, Flores A, Register T, Cline JM, D’Agostino R Jr, Danial N, et al. Behavioral stress accelerates prostate cancer development in mice. J Clin Invest. 2013;123(2):874–86.
Sood AK, Armaiz-Pena GN, Halder J, Nick AM, Stone RL, Hu W, Carroll AR, Spannuth WA, Deavers MT, Allen JK, et al. Adrenergic modulation of focal adhesion kinase protects human ovarian cancer cells from anoikis. J Clin Invest. 2010;120(5):1515–23.
Liu D, Yang Z, Wang T, Yang Z, Chen H, Hu Y, Hu C, Guo L, Deng Q, Liu Y, et al. beta2-AR signaling controls trastuzumab resistance-dependent pathway. Oncogene. 2016;35(1):47–58.
Zhang X, Zhang Y, He Z, Yin K, Li B, Zhang L, Xu Z. Chronic stress promotes gastric cancer progression and metastasis: an essential role for ADRB2. Cell Death Dis. 2019;10(11):788.
Zhang D, Ma Q, Wang Z, Zhang M, Guo K, Wang F, Wu E. beta2-adrenoceptor blockage induces G1/S phase arrest and apoptosis in pancreatic cancer cells via Ras/Akt/NFkappaB pathway. Mol Cancer. 2011;10:146.
Wu W, Chaudhuri S, Brickley DR, Pang D, Karrison T, Conzen SD. Microarray analysis reveals glucocorticoid-regulated survival genes that are associated with inhibition of apoptosis in breast epithelial cells. Cancer Res. 2004;64(5):1757–64.
Petrella A, Ercolino SF, Festa M, Gentilella A, Tosco A, Conzen SD, Parente L. Dexamethasone inhibits TRAIL-induced apoptosis of thyroid cancer cells via Bcl-xL induction. Eur J Cancer. 2006;42(18):3287–93.
Celentano A, McCullough M, Cirillo N. Glucocorticoids reduce chemotherapeutic effectiveness on OSCC cells via glucose-dependent mechanisms. J Cell Physiol. 2019;234(3):2013–20.
An W, Yao S, Sun X, Hou Z, Lin Y, Su L, Liu X. Glucocorticoid modulatory element-binding protein 1 (GMEB1) interacts with the de-ubiquitinase USP40 to stabilize CFLARL and inhibit apoptosis in human non-small cell lung cancer cells. J Exp Clin Cancer Res. 2019;38(1):181.
Skor MN, Wonder EL, Kocherginsky M, Goyal A, Hall BA, Cai Y, Conzen SD. Glucocorticoid receptor antagonism as a novel therapy for triple-negative breast cancer. Clin Cancer Res. 2013;19(22):6163–72.
Yawata A, Adachi M, Okuda H, Naishiro Y, Takamura T, Hareyama M, Takayama S, Reed JC, Imai K. Prolonged cell survival enhances peritoneal dissemination of gastric cancer cells. Oncogene. 1998;16(20):2681–6.
Grossmann J. Molecular mechanisms of “detachment-induced apoptosis–Anoikis.” Apoptosis. 2002;7(3):247–60.
Mason JA, Davison-Versagli CA, Leliaert AK, Pape DJ, McCallister C, Zuo J, Durbin SM, Buchheit CL, Zhang S, Schafer ZT. Oncogenic Ras differentially regulates metabolism and anoikis in extracellular matrix-detached cells. Cell Death Differ. 2016;23(8):1271–82.
Mason JA, Cockfield JA, Pape DJ, Meissner H, Sokolowski MT, White TC, Valentin Lopez JC, Liu J, Liu X, Martinez-Reyes I, et al. SGK1 signaling promotes glucose metabolism and survival in extracellular matrix detached cells. Cell Rep. 2021;34(11): 108821.
Banimohamad-Shotorbani B, Kahroba H, Sadeghzadeh H, Wilson DM 3rd, Maadi H, Samadi N, Hejazi MS, Farajpour H, Onari BN, Sadeghi MR. DNA damage repair response in mesenchymal stromal cells: from cellular senescence and aging to apoptosis and differentiation ability. Ageing Res Rev. 2020;62: 101125.
Feng Z, Liu L, Zhang C, Zheng T, Wang J, Lin M, Zhao Y, Wang X, Levine AJ, Hu W. Chronic restraint stress attenuates p53 function and promotes tumorigenesis. Proc Natl Acad Sci USA. 2012;109(18):7013–8.
Hara MR, Kovacs JJ, Whalen EJ, Rajagopal S, Strachan RT, Grant W, Towers AJ, Williams B, Lam CM, Xiao K, et al. A stress response pathway regulates DNA damage through beta2-adrenoreceptors and beta-arrestin-1. Nature. 2011;477(7364):349–53.
Flaherty RL, Owen M, Fagan-Murphy A, Intabli H, Healy D, Patel A, Allen MC, Patel BA, Flint MS. Glucocorticoids induce production of reactive oxygen species/reactive nitrogen species and DNA damage through an iNOS mediated pathway in breast cancer. Breast Cancer Res. 2017;19(1):35.
Reeder A, Attar M, Nazario L, Bathula C, Zhang A, Hochbaum D, Roy E, Cooper KL, Oesterreich S, Davidson NE, et al. Stress hormones reduce the efficacy of paclitaxel in triple negative breast cancer through induction of DNA damage. Br J Cancer. 2015;112(9):1461–70.
Antoni MH, Dhabhar FS. The impact of psychosocial stress and stress management on immune responses in patients with cancer. Cancer. 2019;125(9):1417–31.
Bader JE, Voss K, Rathmell JC. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol Cell. 2020;78(6):1019–33.
Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140(6):883–99.
Kim JH, Almuwaqqat Z, Hammadah M, Liu C, Ko YA, Lima B, Sullivan S, Alkhoder A, Abdulbaki R, Ward L, et al. Peripheral vasoconstriction during mental stress and adverse cardiovascular outcomes in patients with coronary artery disease. Circ Res. 2019;125(10):874–83.
Devi S, Alexandre YO, Loi JK, Gillis R, Ghazanfari N, Creed SJ, Holz LE, Shackleford D, Mackay LK, Heath WR, et al. Adrenergic regulation of the vasculature impairs leukocyte interstitial migration and suppresses immune responses. Immunity. 2021;54(6):1219–30.
Rytelewski M, Haryutyunan K, Nwajei F, Shanmugasundaram M, Wspanialy P, Zal MA, Chen CH, El Khatib M, Plunkett S, Vinogradov SA, et al. Merger of dynamic two-photon and phosphorescence lifetime microscopy reveals dependence of lymphocyte motility on oxygen in solid and hematological tumors. J Immunother Cancer. 2019;7(1):78.
Kumar S. Natural killer cell cytotoxicity and its regulation by inhibitory receptors. Immunology. 2018;154(3):383–93.
Glasner A, Avraham R, Rosenne E, Benish M, Zmora O, Shemer S, Meiboom H, Ben-Eliyahu S. Improving survival rates in two models of spontaneous postoperative metastasis in mice by combined administration of a beta-adrenergic antagonist and a cyclooxygenase-2 inhibitor. J Immunol. 2010;184(5):2449–57.
Neeman E, Ben-Eliyahu S. Surgery and stress promote cancer metastasis: new outlooks on perioperative mediating mechanisms and immune involvement. Brain Behav Immun. 2013;30(Suppl):S32–40.
Theorell J, Gustavsson AL, Tesi B, Sigmundsson K, Ljunggren HG, Lundback T, Bryceson YT. Immunomodulatory activity of commonly used drugs on Fc-receptor-mediated human natural killer cell activation. Cancer Immunol Immunother. 2014;63(6):627–41.
Rosenne E, Sorski L, Shaashua L, Neeman E, Matzner P, Levi B, Ben-Eliyahu S. In vivo suppression of NK cell cytotoxicity by stress and surgery: glucocorticoids have a minor role compared to catecholamines and prostaglandins. Brain Behav Immun. 2014;37:207–19.
Herve J, Dubreil L, Tardif V, Terme M, Pogu S, Anegon I, Rozec B, Gauthier C, Bach JM, Blancou P. beta2-Adrenoreceptor agonist inhibits antigen cross-presentation by dendritic cells. J Immunol. 2013;190(7):3163–71.
Sommershof A, Scheuermann L, Koerner J, Groettrup M. Chronic stress suppresses anti-tumor T(CD8+) responses and tumor regression following cancer immunotherapy in a mouse model of melanoma. Brain Behav Immun. 2017;65:140–9.
Mueller M, Schlosser E, Gander B, Groettrup M. Tumor eradication by immunotherapy with biodegradable PLGA microspheres—an alternative to incomplete Freund’s adjuvant. Int J Cancer. 2011;129(2):407–16.
Paijens ST, Vledder A, de Bruyn M, Nijman HW. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell Mol Immunol. 2021;18(4):842–59.
Qiao G, Bucsek MJ, Winder NM, Chen M, Giridharan T, Olejniczak SH, Hylander BL, Repasky EA. beta-Adrenergic signaling blocks murine CD8(+) T-cell metabolic reprogramming during activation: a mechanism for immunosuppression by adrenergic stress. Cancer Immunol Immunother. 2019;68(1):11–22.
Nissen MD, Sloan EK, Mattarollo SR. beta-Adrenergic signaling impairs antitumor CD8(+) T-cell responses to B-cell lymphoma immunotherapy. Cancer Immunol Res. 2018;6(1):98–109.
Qiao G, Chen M, Mohammadpour H, MacDonald CR, Bucsek MJ, Hylander BL, Barbi JJ, Repasky EA. Chronic adrenergic stress contributes to metabolic dysfunction and an exhausted phenotype in T cells in the tumor microenvironment. Cancer Immunol Res. 2021;9(6):651–64.
Kamiya A, Hayama Y, Kato S, Shimomura A, Shimomura T, Irie K, Kaneko R, Yanagawa Y, Kobayashi K, Ochiya T. Genetic manipulation of autonomic nerve fiber innervation and activity and its effect on breast cancer progression. Nat Neurosci. 2019;22(8):1289–305.
Zhang Y, Zhang Z. The history and advances in cancer immunotherapy: understanding the characteristics of tumor-infiltrating immune cells and their therapeutic implications. Cell Mol Immunol. 2020;17(8):807–21.
Bucsek MJ, Qiao G, MacDonald CR, Giridharan T, Evans L, Niedzwecki B, Liu H, Kokolus KM, Eng JW, Messmer MN, et al. beta-Adrenergic signaling in mice housed at standard temperatures suppresses an effector phenotype in CD8(+) T cells and undermines checkpoint inhibitor therapy. Cancer Res. 2017;77(20):5639–51.
Liu C, Yang Y, Chen C, Li L, Li J, Wang X, Chu Q, Qiu L, Ba Q, Li X, et al. Environmental eustress modulates beta-ARs/CCL2 axis to induce anti-tumor immunity and sensitize immunotherapy against liver cancer in mice. Nat Commun. 2021;12(1):5725.
Scott SC, Pennell NA. Early use of systemic corticosteroids in patients with advanced NSCLC treated with nivolumab. J Thorac Oncol. 2018;13(11):1771–5.
Arbour KC, Mezquita L, Long N, Rizvi H, Auclin E, Ni A, Martinez-Bernal G, Ferrara R, Lai WV, Hendriks LEL, et al. Impact of baseline steroids on efficacy of programmed cell death-1 and programmed death-ligand 1 blockade in patients with non-small-cell lung cancer. J Clin Oncol. 2018;36(28):2872–8.
Borst J, Ahrends T, Babala N, Melief CJM, Kastenmuller W. CD4(+) T cell help in cancer immunology and immunotherapy. Nat Rev Immunol. 2018;18(10):635–47.
Knochelmann HM, Dwyer CJ, Bailey SR, Amaya SM, Elston DM, Mazza-McCrann JM, Paulos CM. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell Mol Immunol. 2018;15(5):458–69.
Riether C, Kavelaars A, Wirth T, Pacheco-Lopez G, Doenlen R, Willemen H, Heijnen CJ, Schedlowski M, Engler H. Stimulation of beta(2)-adrenergic receptors inhibits calcineurin activity in CD4(+) T cells via PKA-AKAP interaction. Brain Behav Immun. 2011;25(1):59–66.
McAlees JW, Smith LT, Erbe RS, Jarjoura D, Ponzio NM, Sanders VM. Epigenetic regulation of beta2-adrenergic receptor expression in T(H)1 and T(H)2 cells. Brain Behav Immun. 2011;25(3):408–15.
Kumar P, Bhattacharya P, Prabhakar BS. A comprehensive review on the role of co-signaling receptors and Treg homeostasis in autoimmunity and tumor immunity. J Autoimmun. 2018;95:77–99.
Bereshchenko O, Coppo M, Bruscoli S, Biagioli M, Cimino M, Frammartino T, Sorcini D, Venanzi A, Di Sante M, Riccardi C. GILZ promotes production of peripherally induced Treg cells and mediates the crosstalk between glucocorticoids and TGF-beta signaling. Cell Rep. 2014;7(2):464–75.
Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. 2018;17(12):887–904.
Cheng Y, Tang XY, Li YX, Zhao DD, Cao QH, Wu HX, Yang HB, Hao K, Yang Y. Depression-induced neuropeptide Y secretion promotes prostate cancer growth by recruiting myeloid cells. Clin Cancer Res. 2019;25(8):2621–32.
Cao M, Huang W, Chen Y, Li G, Liu N, Wu Y, Wang G, Li Q, Kong D, Xue T, et al. Chronic restraint stress promotes the mobilization and recruitment of myeloid-derived suppressor cells through beta-adrenergic-activated CXCL5-CXCR2-Erk signaling cascades. Int J Cancer. 2021;149(2):460–72.
Jiang W, Li Y, Li ZZ, Sun J, Li JW, Wei W, Li L, Zhang C, Huang C, Yang SY, et al. Chronic restraint stress promotes hepatocellular carcinoma growth by mobilizing splenic myeloid cells through activating beta-adrenergic signaling. Brain Behav Immun. 2019;80:825–38.
Armaiz-Pena GN, Gonzalez-Villasana V, Nagaraja AS, Rodriguez-Aguayo C, Sadaoui NC, Stone RL, Matsuo K, Dalton HJ, Previs RA, Jennings NB, et al. Adrenergic regulation of monocyte chemotactic protein 1 leads to enhanced macrophage recruitment and ovarian carcinoma growth. Oncotarget. 2015;6(6):4266–73.
Wu Y, Luo X, Zhou Q, Gong H, Gao H, Liu T, Chen J, Liang L, Kurihara H, Li YF, et al. The disbalance of LRP1 and SIRPalpha by psychological stress dampens the clearance of tumor cells by macrophages. Acta Pharm Sin B. 2022;12(1):197–209.
Niraula A, Wang Y, Godbout JP, Sheridan JF. Corticosterone production during repeated social defeat causes monocyte mobilization from the bone marrow, glucocorticoid resistance, and neurovascular adhesion molecule expression. J Neurosci. 2018;38(9):2328–40.
Muthuswamy R, Okada NJ, Jenkins FJ, McGuire K, McAuliffe PF, Zeh HJ, Bartlett DL, Wallace C, Watkins S, Henning JD, et al. Epinephrine promotes COX-2-dependent immune suppression in myeloid cells and cancer tissues. Brain Behav Immun. 2017;62:78–86.
Mohammadpour H, MacDonald CR, McCarthy PL, Abrams SI, Repasky EA. beta2-Adrenergic receptor signaling regulates metabolic pathways critical to myeloid-derived suppressor cell function within the TME. Cell Rep. 2021;37(4): 109883.
Biswas SK. Metabolic reprogramming of immune cells in cancer progression. Immunity. 2015;43(3):435–49.
Kalinski P. Regulation of immune responses by prostaglandin E2. J Immunol. 2012;188(1):21–8.
Ben-Shaanan TL, Schiller M, Azulay-Debby H, Korin B, Boshnak N, Koren T, Krot M, Shakya J, Rahat MA, Hakim F, et al. Modulation of anti-tumor immunity by the brain’s reward system. Nat Commun. 2018;9(1):2723.
Qin JF, Jin FJ, Li N, Guan HT, Lan L, Ni H, Wang Y. Adrenergic receptor beta2 activation by stress promotes breast cancer progression through macrophages M2 polarization in tumor microenvironment. BMB Rep. 2015;48(5):295–300.
Xia Y, Wei Y, Li ZY, Cai XY, Zhang LL, Dong XR, Zhang S, Zhang RG, Meng R, Zhu F, et al. Catecholamines contribute to the neovascularization of lung cancer via tumor-associated macrophages. Brain Behav Immun. 2019;81:111–21.
Le CP, Nowell CJ, Kim-Fuchs C, Botteri E, Hiller JG, Ismail H, Pimentel MA, Chai MG, Karnezis T, Rotmensz N, et al. Chronic stress in mice remodels lymph vasculature to promote tumour cell dissemination. Nat Commun. 2016;7:10634.
Wissmann C, Detmar M. Pathways targeting tumor lymphangiogenesis. Clin Cancer Res. 2006;12(23):6865–8.
Houthuijzen JM, Jonkers J. Cancer-associated fibroblasts as key regulators of the breast cancer tumor microenvironment. Cancer Metastasis Rev. 2018;37(4):577–97.
Nagaraja AS, Dood RL, Armaiz-Pena G, Kang Y, Wu SY, Allen JK, Jennings NB, Mangala LS, Pradeep S, Lyons Y, et al. Adrenergic-mediated increases in INHBA drive CAF phenotype and collagens. JCI Insight. 2017;2(16): e93076.
Zhang H, Fredericks T, Xiong G, Qi Y, Rychahou PG, Li JD, Pihlajaniemi T, Xu W, Xu R. Membrane associated collagen XIII promotes cancer metastasis and enhances anoikis resistance. Breast Cancer Res. 2018;20(1):116.
Wu Q, Li B, Li Z, Li J, Sun S, Sun S. Cancer-associated adipocytes: key players in breast cancer progression. J Hematol Oncol. 2019;12(1):95.
Zhao C, Wu M, Zeng N, Xiong M, Hu W, Lv W, Yi Y, Zhang Q, Wu Y. Cancer-associated adipocytes: emerging supporters in breast cancer. J Exp Clin Cancer Res. 2020;39(1):156.
Avril P, Vidal L, Barille-Nion S, Le Nail LR, Redini F, Layrolle P, Pinault M, Chevalier S, Perrot P, Trichet V. Epinephrine infiltration of adipose tissue impacts MCF7 breast cancer cells and total lipid content. Int J Mol Sci. 2019;20(22):5626.
Bussard KM, Mutkus L, Stumpf K, Gomez-Manzano C, Marini FC. Tumor-associated stromal cells as key contributors to the tumor microenvironment. Breast Cancer Res. 2016;18(1):84.
Zahalka AH, Arnal-Estape A, Maryanovich M, Nakahara F, Cruz CD, Finley LWS, Frenette PS. Adrenergic nerves activate an angio-metabolic switch in prostate cancer. Science. 2017;358(6361):321–6.
Lutgendorf SK, DeGeest K, Sung CY, Arevalo JM, Penedo F, Lucci J 3rd, Goodheart M, Lubaroff D, Farley DM, Sood AK, et al. Depression, social support, and beta-adrenergic transcription control in human ovarian cancer. Brain Behav Immun. 2009;23(2):176–83.
Magnon C, Hall SJ, Lin J, Xue X, Gerber L, Freedland SJ, Frenette PS. Autonomic nerve development contributes to prostate cancer progression. Science. 2013;341(6142):1236361.
Hayakawa Y, Sakitani K, Konishi M, Asfaha S, Niikura R, Tomita H, Renz BW, Tailor Y, Macchini M, Middelhoff M, et al. Nerve growth factor promotes gastric tumorigenesis through aberrant cholinergic signaling. Cancer Cell. 2017;31(1):21–34.
Ayala GE, Dai H, Powell M, Li R, Ding Y, Wheeler TM, Shine D, Kadmon D, Thompson T, Miles BJ, et al. Cancer-related axonogenesis and neurogenesis in prostate cancer. Clin Cancer Res. 2008;14(23):7593–603.
Allen JK, Armaiz-Pena GN, Nagaraja AS, Sadaoui NC, Ortiz T, Dood R, Ozcan M, Herder DM, Haemmerle M, Gharpure KM, et al. Sustained adrenergic signaling promotes intratumoral innervation through BDNF induction. Cancer Res. 2018;78(12):3233–42.
Renz BW, Takahashi R, Tanaka T, Macchini M, Hayakawa Y, Dantes Z, Maurer HC, Chen X, Jiang Z, Westphalen CB, et al. beta2 Adrenergic-neurotrophin feedforward loop promotes pancreatic cancer. Cancer Cell. 2018;33(1):75-90.e77.
Amit M, Takahashi H, Dragomir MP, Lindemann A, Gleber-Netto FO, Pickering CR, Anfossi S, Osman AA, Cai Y, Wang R, et al. Loss of p53 drives neuron reprogramming in head and neck cancer. Nature. 2020;578(7795):449–54.
Madeo M, Colbert PL, Vermeer DW, Lucido CT, Cain JT, Vichaya EG, Grossberg AJ, Muirhead D, Rickel AP, Hong Z, et al. Cancer exosomes induce tumor innervation. Nat Commun. 2018;9(1):4284.
Lapuk AV, Wu C, Wyatt AW, McPherson A, McConeghy BJ, Brahmbhatt S, Mo F, Zoubeidi A, Anderson S, Bell RH, et al. From sequence to molecular pathology, and a mechanism driving the neuroendocrine phenotype in prostate cancer. J Pathol. 2012;227(3):286–97.
Mauffrey P, Tchitchek N, Barroca V, Bemelmans AP, Firlej V, Allory Y, Romeo PH, Magnon C. Progenitors from the central nervous system drive neurogenesis in cancer. Nature. 2019;569(7758):672–8.
Adriaenssens E, Vanhecke E, Saule P, Mougel A, Page A, Romon R, Nurcombe V, Le Bourhis X, Hondermarck H. Nerve growth factor is a potential therapeutic target in breast cancer. Cancer Res. 2008;68(2):346–51.
Au CW, Siu MK, Liao X, Wong ES, Ngan HY, Tam KF, Chan DC, Chan QK, Cheung AN. Tyrosine kinase B receptor and BDNF expression in ovarian cancers—effect on cell migration, angiogenesis and clinical outcome. Cancer Lett. 2009;281(2):151–61.
Li X, Peng X, Yang S, Wei S, Fan Q, Liu J, Yang L, Li H. Targeting tumor innervation: premises, promises, and challenges. Cell Death Discov. 2022;8(1):131.
Balood M, Ahmadi M, Eichwald T, Ahmadi A, Majdoubi A, Roversi K, Roversi K, Lucido CT, Restaino AC, Huang S, et al. Nociceptor neurons affect cancer immunosurveillance. Nature. 2022;611(7935):405–12.
Vats K, Kruglov O, Sahoo B, Soman V, Zhang J, Shurin GV, Chandran UR, Skums P, Shurin MR, Zelikovsky A, et al. Sensory nerves impede the formation of tertiary lymphoid structures and development of protective antimelanoma immune responses. Cancer Immunol Res. 2022;10(9):1141–54.
Pinquart M, Duberstein PR. Depression and cancer mortality: a meta-analysis. Psychol Med. 2010;40(11):1797–810.
Botteri E, Munzone E, Rotmensz N, Cipolla C, De Giorgi V, Santillo B, Zanelotti A, Adamoli L, Colleoni M, Viale G, et al. Therapeutic effect of beta-blockers in triple-negative breast cancer postmenopausal women. Breast Cancer Res Treat. 2013;140(3):567–75.
Powe DG, Voss MJ, Zanker KS, Habashy HO, Green AR, Ellis IO, Entschladen F. Beta-blocker drug therapy reduces secondary cancer formation in breast cancer and improves cancer specific survival. Oncotarget. 2010;1(7):628–38.
Jansen L, Hoffmeister M, Arndt V, Chang-Claude J, Brenner H. Stage-specific associations between beta blocker use and prognosis after colorectal cancer. Cancer. 2014;120(8):1178–86.
De Giorgi V, Gandini S, Grazzini M, Benemei S, Marchionni N, Geppetti P. Effect of beta-blockers and other antihypertensive drugs on the risk of melanoma recurrence and death. Mayo Clin Proc. 2013;88(11):1196–203.
De Giorgi V, Grazzini M, Benemei S, Marchionni N, Botteri E, Pennacchioli E, Geppetti P, Gandini S. Propranolol for off-label treatment of patients with melanoma: results from a cohort study. JAMA Oncol. 2018;4(2): e172908.
Aydiner A, Ciftci R, Karabulut S, Kilic L. Does beta-blocker therapy improve the survival of patients with metastatic non-small cell lung cancer? Asian Pac J Cancer Prev. 2013;14(10):6109–14.
Al-Niaimi A, Dickson EL, Albertin C, Karnowski J, Niemi C, Spencer R, Shahzad MM, Uppal S, Saha S, Rice L, et al. The impact of perioperative beta blocker use on patient outcomes after primary cytoreductive surgery in high-grade epithelial ovarian carcinoma. Gynecol Oncol. 2016;143(3):521–5.
Watkins JL, Thaker PH, Nick AM, Ramondetta LM, Kumar S, Urbauer DL, Matsuo K, Squires KC, Coleman RL, Lutgendorf SK, et al. Clinical impact of selective and nonselective beta-blockers on survival in patients with ovarian cancer. Cancer. 2015;121(19):3444–51.
Udumyan R, Montgomery S, Fang F, Almroth H, Valdimarsdottir U, Ekbom A, Smedby KE, Fall K. Beta-blocker drug use and survival among patients with pancreatic adenocarcinoma. Cancer Res. 2017;77(13):3700–7.
Tadic M, Cuspidi C, Belyavskiy E, Grassi G. Intriguing relationship between antihypertensive therapy and cancer. Pharmacol Res. 2019;141:501–11.
Sakellakis M, Kostaki A, Starakis I, Koutras A. beta-Blocker use and risk of recurrence in patients with early breast cancer. Chemotherapy. 2014;60(5–6):288–9.
Shah SM, Carey IM, Owen CG, Harris T, Dewilde S, Cook DG. Does beta-adrenoceptor blocker therapy improve cancer survival? Findings from a population-based retrospective cohort study. Br J Clin Pharmacol. 2011;72(1):157–61.
Holmes S, Griffith EJ, Musto G, Minuk GY. Antihypertensive medications and survival in patients with cancer: a population-based retrospective cohort study. Cancer Epidemiol. 2013;37(6):881–5.
Sorensen GV, Ganz PA, Cole SW, Pedersen LA, Sorensen HT, Cronin-Fenton DP, Garne JP, Christiansen PM, Lash TL, Ahern TP. Use of beta-blockers, angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers, and risk of breast cancer recurrence: a Danish nationwide prospective cohort study. J Clin Oncol. 2013;31(18):2265–72.
Weberpals J, Jansen L, Haefeli WE, Hoffmeister M, Wolkewitz M, Herk-Sukel M, Vissers PAJ, Brenner H. Pre- and post-diagnostic beta-blocker use and lung cancer survival: a population-based cohort study. Sci Rep. 2017;7(1):2911.
Cata JP, Villarreal J, Keerty D, Thakar DR, Liu DD, Sood AK, Gottumukkala V. Perioperative beta-blocker use and survival in lung cancer patients. J Clin Anesth. 2014;26(2):106–17.
Yang P, Deng W, Han Y, Shi Y, Xu T, Shi J, Elhalawani H, Zhao Y, Xie X, Lou F, et al. Analysis of the correlation among hypertension, the intake of beta-blockers, and overall survival outcome in patients undergoing chemoradiotherapy with inoperable stage III non-small cell lung cancer. Am J Cancer Res. 2017;7(4):946–54.
Huang T, Townsend MK, Dood RL, Sood AK, Tworoger SS. Antihypertensive medication use and ovarian cancer survival. Gynecol Oncol. 2021;163(2):342–7.
Heitz F, Hengsbach A, Harter P, Traut A, Ataseven B, Schneider S, Prader S, Kurzeder C, Sporkmann M, du Bois A. Intake of selective beta blockers has no impact on survival in patients with epithelial ovarian cancer. Gynecol Oncol. 2017;144(1):181–6.
Parker WP, Lohse CM, Zaid HB, Cheville JC, Boorjian SA, Leibovich BC, Thompson RH. Evaluation of beta-blockers and survival among hypertensive patients with renal cell carcinoma. Urol Oncol. 2017;35(1):36.e31-36.e36.
Perrault D, Eisenhauer EA, Pritchard KI, Panasci L, Norris B, Vandenberg T, Fisher B. Phase II study of the progesterone antagonist mifepristone in patients with untreated metastatic breast carcinoma: a National Cancer Institute of Canada Clinical Trials Group study. J Clin Oncol. 1996;14(10):2709–12.
Fiscella K, Eisinger SH, Meldrum S, Feng C, Fisher SG, Guzick DS. Effect of mifepristone for symptomatic leiomyomata on quality of life and uterine size: a randomized controlled trial. Obstet Gynecol. 2006;108(6):1381–7.
Nilsson MB, Sun H, Diao L, Tong P, Liu D, Li L, Fan Y, Poteete A, Lim SO, Howells K, et al. Stress hormones promote EGFR inhibitor resistance in NSCLC: implications for combinations with beta-blockers. Sci Transl Med. 2017;9(415): eaao4307.
Chaudhary KR, Yan SX, Heilbroner SP, Sonett JR, Stoopler MB, Shu C, Halmos B, Wang TJC, Hei TK, Cheng SK. Effects of beta-adrenergic antagonists on chemoradiation therapy for locally advanced non-small cell lung cancer. J Clin Med. 2019;8(5):575.
Gandhi S, Pandey MR, Attwood K, Ji W, Witkiewicz AK, Knudsen ES, Allen C, Tario JD, Wallace PK, Cedeno CD, et al. Phase I clinical trial of combination propranolol and pembrolizumab in locally advanced and metastatic melanoma: safety, tolerability, and preliminary evidence of antitumor activity. Clin Cancer Res. 2021;27(1):87–95.
Melhem-Bertrandt A, Chavez-Macgregor M, Lei X, Brown EN, Lee RT, Meric-Bernstam F, Sood AK, Conzen SD, Hortobagyi GN, Gonzalez-Angulo AM. Beta-blocker use is associated with improved relapse-free survival in patients with triple-negative breast cancer. J Clin Oncol. 2011;29(19):2645–52.
Wang HM, Liao ZX, Komaki R, Welsh JW, O’Reilly MS, Chang JY, Zhuang Y, Levy LB, Lu C, Gomez DR. Improved survival outcomes with the incidental use of beta-blockers among patients with non-small-cell lung cancer treated with definitive radiation therapy. Ann Oncol. 2013;24(5):1312–9.
Diaz ES, Karlan BY, Li AJ. Impact of beta blockers on epithelial ovarian cancer survival. Gynecol Oncol. 2012;127(2):375–8.
Kokolus KM, Zhang Y, Sivik JM, Schmeck C, Zhu J, Repasky EA, Drabick JJ, Schell TD. Beta blocker use correlates with better overall survival in metastatic melanoma patients and improves the efficacy of immunotherapies in mice. Oncoimmunology. 2018;7(3): e1405205.
Fiala O, Ostasov P, Sorejs O, Liska V, Buchler T, Poprach A, Finek J. Incidental use of beta-blockers is associated with outcome of metastatic colorectal cancer patients treated with bevacizumab-based therapy: a single-institution retrospective analysis of 514 patients. Cancers. 2019;11(12):1856.
Ramondetta LM, Hu W, Thaker PH, Urbauer DL, Chisholm GB, Westin SN, Sun Y, Ramirez PT, Fleming N, Sahai SK, et al. Prospective pilot trial with combination of propranolol with chemotherapy in patients with epithelial ovarian cancer and evaluation on circulating immune cell gene expression. Gynecol Oncol. 2019;154(3):524–30.
Partecke LI, Speerforck S, Kading A, Seubert F, Kuhn S, Lorenz E, Schwandke S, Sendler M, Kessler W, Trung DN, et al. Chronic stress increases experimental pancreatic cancer growth, reduces survival and can be antagonised by beta-adrenergic receptor blockade. Pancreatology. 2016;16(3):423–33.
Shi DD, Guo JA, Hoffman HI, Su J, Mino-Kenudson M, Barth JL, Schenkel JM, Loeffler JS, Shih HA, Hong TS, et al. Therapeutic avenues for cancer neuroscience: translational frontiers and clinical opportunities. Lancet Oncol. 2022;23(2):e62–74.
Yarney J, Donkor A, Opoku SY, Yarney L, Agyeman-Duah I, Abakah AC, Asampong E. Characteristics of users and implications for the use of complementary and alternative medicine in Ghanaian cancer patients undergoing radiotherapy and chemotherapy: a cross-sectional study. BMC Complement Altern Med. 2013;13:16.
Yang M, Zhang Z, Nice EC, Wang C, Zhang W, Huang C. Psychological intervention to treat distress: an emerging frontier in cancer prevention and therapy. Biochim Biophys Acta Rev Cancer. 2022;1877(1): 188665.
Carlson LE, Doll R, Stephen J, Faris P, Tamagawa R, Drysdale E, Speca M. Randomized controlled trial of mindfulness-based cancer recovery versus supportive expressive group therapy for distressed survivors of breast cancer. J Clin Oncol. 2013;31(25):3119–26.
Carlson LE. Mindfulness-based interventions for coping with cancer. Ann N Y Acad Sci. 2016;1373(1):5–12.
Lengacher CA, Reich RR, Paterson CL, Ramesar S, Park JY, Alinat C, Johnson-Mallard V, Moscoso M, Budhrani-Shani P, Miladinovic B, et al. Examination of broad symptom improvement resulting from mindfulness-based stress reduction in breast cancer survivors: a randomized controlled trial. J Clin Oncol. 2016;34(24):2827–34.
Carlson LE, Speca M, Faris P, Patel KD. One year pre-post intervention follow-up of psychological, immune, endocrine and blood pressure outcomes of mindfulness-based stress reduction (MBSR) in breast and prostate cancer outpatients. Brain Behav Immun. 2007;21(8):1038–49.
WitekJanusek L, Tell D, Mathews HL. Mindfulness based stress reduction provides psychological benefit and restores immune function of women newly diagnosed with breast cancer: a randomized trial with active control. Brain Behav Immun. 2019;80:358–73.
Haller H, Winkler MM, Klose P, Dobos G, Kummel S, Cramer H. Mindfulness-based interventions for women with breast cancer: an updated systematic review and meta-analysis. Acta Oncol. 2017;56(12):1665–76.
Carlson LE, Beattie TL, Giese-Davis J, Faris P, Tamagawa R, Fick LJ, Degelman ES, Speca M. Mindfulness-based cancer recovery and supportive-expressive therapy maintain telomere length relative to controls in distressed breast cancer survivors. Cancer. 2015;121(3):476–84.
Cannioto RA, Hutson A, Dighe S, McCann W, McCann SE, Zirpoli GR, Barlow W, Kelly KM, DeNysschen CA, Hershman DL, et al. Physical activity before, during, and after chemotherapy for high-risk breast cancer: relationships with survival. J Natl Cancer Inst. 2021;113(1):54–63.
Steindorf K, Schmidt ME, Klassen O, Ulrich CM, Oelmann J, Habermann N, Beckhove P, Owen R, Debus J, Wiskemann J, et al. Randomized, controlled trial of resistance training in breast cancer patients receiving adjuvant radiotherapy: results on cancer-related fatigue and quality of life. Ann Oncol. 2014;25(11):2237–43.
Travier N, Velthuis MJ, Steins Bisschop CN, van den Buijs B, Monninkhof EM, Backx F, Los M, Erdkamp F, Bloemendal HJ, Rodenhuis C, et al. Effects of an 18-week exercise programme started early during breast cancer treatment: a randomised controlled trial. BMC Med. 2015;13:121.
Kampshoff CS, Chinapaw MJ, Brug J, Twisk JW, Schep G, Nijziel MR, van Mechelen W, Buffart LM. Randomized controlled trial of the effects of high intensity and low-to-moderate intensity exercise on physical fitness and fatigue in cancer survivors: results of the resistance and endurance exercise after chemotherapy (REACT) study. BMC Med. 2015;13:275.
Schmidt ME, Meynkohn A, Habermann N, Wiskemann J, Oelmann J, Hof H, Wessels S, Klassen O, Debus J, Potthoff K, et al. Resistance exercise and inflammation in breast cancer patients undergoing adjuvant radiation therapy: mediation analysis from a randomized, controlled intervention trial. Int J Radiat Oncol Biol Phys. 2016;94(2):329–37.
Gomes-Santos IL, Amoozgar Z, Kumar AS, Ho WW, Roh K, Talele NP, Curtis H, Kawaguchi K, Jain RK, Fukumura D. Exercise training improves tumor control by increasing CD8(+) T-cell Infiltration via CXCR3 signaling and sensitizes breast cancer to immune checkpoint blockade. Cancer Immunol Res. 2021;9(7):765–78.
Cramer H, Lauche R, Klose P, Lange S, Langhorst J, Dobos GJ. Yoga for improving health-related quality of life, mental health and cancer-related symptoms in women diagnosed with breast cancer. Cochrane Database Syst Rev. 2017;1(1):CD010802.
Chandwani KD, Perkins G, Nagendra HR, Raghuram NV, Spelman A, Nagarathna R, Johnson K, Fortier A, Arun B, Wei Q, et al. Randomized, controlled trial of yoga in women with breast cancer undergoing radiotherapy. J Clin Oncol. 2014;32(10):1058–65.
Kiecolt-Glaser JK, Bennett JM, Andridge R, Peng J, Shapiro CL, Malarkey WB, Emery CF, Layman R, Mrozek EE, Glaser R. Yoga’s impact on inflammation, mood, and fatigue in breast cancer survivors: a randomized controlled trial. J Clin Oncol. 2014;32(10):1040–9.
Bortolato B, Hyphantis TN, Valpione S, Perini G, Maes M, Morris G, Kubera M, Kohler CA, Fernandes BS, Stubbs B, et al. Depression in cancer: the many biobehavioral pathways driving tumor progression. Cancer Treat Rev. 2017;52:58–70.
Opie RS, Itsiopoulos C, Parletta N, Sanchez-Villegas A, Akbaraly TN, Ruusunen A, Jacka FN. Dietary recommendations for the prevention of depression. Nutr Neurosci. 2017;20(3):161–71.
Herr I, Buchler MW. Dietary constituents of broccoli and other cruciferous vegetables: implications for prevention and therapy of cancer. Cancer Treat Rev. 2010;36(5):377–83.
Saxton JM, Scott EJ, Daley AJ, Woodroofe M, Mutrie N, Crank H, Powers HJ, Coleman RE. Effects of an exercise and hypocaloric healthy eating intervention on indices of psychological health status, hypothalamic-pituitary-adrenal axis regulation and immune function after early-stage breast cancer: a randomised controlled trial. Breast Cancer Res. 2014;16(2):R39.
Sanchez-Villegas A, Martinez-Gonzalez MA, Estruch R, Salas-Salvado J, Corella D, Covas MI, Aros F, Romaguera D, Gomez-Gracia E, Lapetra J, et al. Mediterranean dietary pattern and depression: the PREDIMED randomized trial. BMC Med. 2013;11:208.
Naing A, Stephen SK, Frenkel M, Chandhasin C, Hong DS, Lei X, Falchook G, Wheler JJ, Fu S, Kurzrock R. Prevalence of complementary medicine use in a phase 1 clinical trials program: the MD Anderson cancer center experience. Cancer. 2011;117(22):5142–50.
Biswal BM, Sulaiman SA, Ismail HC, Zakaria H, Musa KI. Effect of Withania somnifera (Ashwagandha) on the development of chemotherapy-induced fatigue and quality of life in breast cancer patients. Integr Cancer Ther. 2013;12(4):312–22.
Ambrosone CB, Zirpoli GR, Hutson AD, McCann WE, McCann SE, Barlow WE, Kelly KM, Cannioto R, Sucheston-Campbell LE, Hershman DL, et al. Dietary supplement use during chemotherapy and survival outcomes of patients with breast cancer enrolled in a cooperative group clinical trial (SWOG S0221). J Clin Oncol. 2020;38(8):804–14.
Funding
This work was supported by the National Natural Science Foundation of China (81972479, U2004118 and 82072899), Natural Science Foundation of Guangdong province (2019A1515011100 and 2021A1515012576), Henan Natural Science Foundation (202300410359) and Henan Medical Research Program (SBGJ2020002081), Guangzhou High-Level Clinical Key Specialty Construction Project: Clinical Key Specialty Construction Project of Guangzhou Medical University (202005), the Innovation Project of Universities in Guangdong Province (NO. 2021KTSCX026). Special project of South China Normal University for foreign exchange in Guangdong- Hong Kong-Macao Great Bay Area in 2022.
Author information
Authors and Affiliations
Contributions
ZZZ, YJJ design the study and drafted the manuscript. LMH, HXY, and LHS collected the related references. ZZZ, CYB, and LQ revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yan, J., Chen, Y., Luo, M. et al. Chronic stress in solid tumor development: from mechanisms to interventions. J Biomed Sci 30, 8 (2023). https://doi.org/10.1186/s12929-023-00903-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-023-00903-9